
The Screening of Therapeutic Peptides for Anti-Inflammation through Phage Display Technology


Abstract
:1. Introduction
2. How to Use the Phage Display to Screen Peptides
3. The Peptides Obtained according to Different Targets Related to Inflammation
3.1. TNFR1
3.2. CD40
3.3. IL-17
3.4. IFN-α
3.5. MMP
3.6. Complement Component 3a (C3a)
3.7. GPR1
3.8. CD14
3.9. Cell
3.10. Others
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eltzschig, H.K.; Carmeliet, P. Mechanisms of Disease: Hypoxia and Inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Paul, A.T.; Gohil, V.M.; Bhutani, K.K. Modulating TNF-alpha signaling with natural products. Drug Discov. Today 2006, 11, 725–732. [Google Scholar] [CrossRef]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharmacol. 2020, 180, 21. [Google Scholar] [CrossRef]
- Hamley, I.W. Small Bioactive Peptides for Biomaterials Design and Therapeutics. Chem. Rev. 2017, 117, 14015–14041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, B.H. Therapeutic peptides for CNS indications: Progress and challenges. Bioorg. Med. Chem. 2018, 26, 2859–2862. [Google Scholar] [CrossRef] [PubMed]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Han, L.; Wang, F.; Petrenko, V.A.; Liu, A.H. Gold nanoprobe functionalized with specific fusion protein selection from phage display and its application in rapid, selective and sensitive colorimetric biosensing of Staphylococcus aureus. Biosens. Bioelectron. 2016, 82, 195–203. [Google Scholar] [CrossRef]
- Smith, G.P.; Petrenko, V.A. Phage display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef]
- Koivunen, E.; Arap, W.; Rajotte, D.; Lahdenranta, J.; Pasqualini, R. Identification of receptor ligands with phage display peptide libraries. J. Nucl. Med. 1999, 40, 883–888. [Google Scholar]
- Sclavons, C.; Burtea, C.; Boutry, S.; Laurent, S.; Elst, L.V.; Muller, R.N. Phage Display Screening for Tumor Necrosis Factor- alpha -Binding Peptides: Detection of Inflammation in a Mouse Model of Hepatitis. Int. J. Pept. 2013, 2013, 348409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirinos-Rojas, C.L.; Steward, M.W.; Partidos, C.D. A peptidomimetic antagonist of TNF-alpha-mediated cytotoxicity identified from a phage-displayed random peptide library. J. Immunol. 1998, 161, 5621–5626. [Google Scholar]
- Smith, G.P. Filamentous fusion phage-novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Ercan, I.; Tufekci, K.U.; Karaca, E.; Genc, S.; Genc, K. Peptide Derivatives of Erythropoietin in the Treatment of Neuroinflammation and Neurodegeneration. In Therapeutic Proteins and Peptides; Donev, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 112, pp. 309–357. [Google Scholar]
- Tan, Y.Y.; Tian, T.; Liu, W.L.; Zhu, Z.; Yang, C.Y.J. Advance in phage display technology for bioanalysis. Biotechnol. J. 2016, 11, 732–745. [Google Scholar] [CrossRef]
- Danner, S.; Belasco, J.G. T7 phage display: A novel genetic selection system for cloning RNA-binding proteins from cDNA libraries. Proc. Natl. Acad. Sci. USA 2001, 98, 12954–12959. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.Y.; Wang, L.; You, X.L.; Dai, P.; Zeng, Y.H. Advances in the T7 phage display system. Mol. Med. Rep. 2018, 17, 714–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homola, J.; Yee, S.S.; Gauglitz, G. Surface plasmon resonance sensors: Review. Sens. Actuator B Chem. 1999, 54, 3–15. [Google Scholar] [CrossRef]
- Green, R.J.; Frazier, R.A.; Shakesheff, K.M.; Davies, M.C.; Roberts, C.J.; Tendler, S.J.B. Surface plasmon resonance analysis of dynamic biological interactions with biomaterials. Biomaterials 2000, 21, 1823–1835. [Google Scholar] [CrossRef]
- Patil, P.O.; Pandey, G.R.; Patil, A.G.; Borse, V.B.; Deshmukh, P.K.; Patil, D.R.; Tade, R.S.; Nangare, S.N.; Khan, Z.G.; Patil, A.M.; et al. Graphene-based nanocomposites for sensitivity enhancement of surface plasmon resonance sensor for biological and chemical sensing: A review. Biosens. Bioelectron. 2019, 139, 22. [Google Scholar] [CrossRef]
- Boominathan, R.; Parimaladevi, B.; Mandal, S.C.; Ghoshal, S.K. Anti-inflammatory evaluation of Ionidium suffruticosam Ging. in rats. J. Ethnopharmacol. 2004, 91, 367–370. [Google Scholar] [CrossRef]
- Patil, K.R.; Mahajan, U.B.; Unger, B.S.; Goyal, S.N.; Belemkar, S.; Surana, S.J.; Ojha, S.; Patil, C.R. Animal Models of Inflammation for Screening of Anti-inflammatory Drugs: Implications for the Discovery and Development of Phytopharmaceuticals. Int. J. Mol. Sci. 2019, 20, 4367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.R.; Liu, Y.; Tang, Y.Z. Screening of TNFR1 Binding Peptides from Deinagkistrodon acutus Venom through Phage Display. Toxins 2022, 14, 155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Guo, S.; Wang, J.; Li, A.; Sun, K.; Qiu, L.; Li, J.; Wang, S.; Ma, X.; Lu, Y. Anti-Inflammatory Activity and Mechanism of Hydrostatin-SN1 From Hydrophis cyanocinctus in Interleukin-10 Knockout Mice. Front. Pharmacol. 2020, 11, 930. [Google Scholar] [CrossRef]
- Bernatchez, P.N.; Tao, B.; Bradshaw, R.A.; Eveleth, D.; Sessa, W.C. Characterization of a Novel Caveolin Modulator That Reduces Vascular Permeability and Ocular Inflammation. Transl. Vis. Sci. Technol. 2021, 10, 9. [Google Scholar] [CrossRef]
- Zapi-Colin, L.A.; Gutierrez-Gonzalez, G.; Rodriguez-Martinez, S.; Cancino-Diaz, J.C.; Mendez-Tenorio, A.; Perez-Tapia, S.M.; Gomez-Chavez, F.; Cedillo-Pelaez, C.; Cancino-Diaz, M.E. A peptide derived from phage-display limits psoriasis—Like lesions in mice. Heliyon 2020, 6, 10. [Google Scholar] [CrossRef]
- Xiao, H.; Wang, M.H.; Fan, X.B.; Xu, W.; Zhang, R.; Wu, G.Q. A novel peptide binding to the C-terminal domain of connective tissue growth factor for the treatment of bleomycin-induced pulmonary fibrosis. Int. J. Biol. Macromol. 2020, 156, 1464–1473. [Google Scholar] [CrossRef]
- Vazquez-Sanchez, E.A.; Mendoza-Figueroa, J.S.; Gutierrez-Gonzalez, G.; Zapi-Colin, L.A.; Torales-Cardena, A.; Briseno-Lugo, P.E.; Diaz-Toala, I.; Cancino-Diaz, J.C.; Perez-Tapia, S.M.; Cancino-Diaz, M.E.; et al. Heptapeptide HP3 acts as a potent inhibitor of experimental imiquimod-induced murine psoriasis and impedes the trans-endothelial migration of mononuclear cells. Mol. Med. Rep. 2020, 22, 507–515. [Google Scholar] [CrossRef]
- Sohn, Y.-K.; Son, S.; Choi, Y.; Hwang, D.-E.; Seo, H.-D.; Lee, J.-J.; Kim, H.-S. Effective inhibition of C3a-mediated pro-inflammatory response by a human C3a-specific protein binder. Biotechnol. Bioeng. 2020, 117, 1904–1908. [Google Scholar] [CrossRef]
- Huang, C.; Dai, X.-Y.; Cai, J.-X.; Chen, J.; Wang, B.B.; Zhu, W.; Wang, E.; Wei, W.; Zhang, J.V. A Screened GPR1 Peptide Exerts Antitumor Effects on Triple-Negative Breast Cancer. Mol. Ther. Oncolytics 2020, 18, 602–612. [Google Scholar] [CrossRef]
- Shoari, A.; Rasaee, M.J.; Kanavi, M.R.; Daraei, B. Functional mimetic peptide discovery isolated by phage display interacts selectively to fibronectin domain and inhibits gelatinase. J. Cell. Biochem. 2019, 120, 19699–19711. [Google Scholar] [CrossRef]
- Maola, K.; Wilbs, J.; Touati, J.; Sabisz, M.; Kong, X.D.; Baumann, A.; Deyle, K.; Heinis, C. Engineered Peptide Macrocycles Can Inhibit Matrix Metalloproteinases with High Selectivity. Angew. Chem. Int. Edit. 2019, 58, 11801–11805. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, L.; Wang, J.; Yang, Y.; Wu, Y.; Jiang, Q.; Yang, Y.; Ma, D.; Zhang, R.; Huang, N.; et al. Alternaria B Cell Mimotope Immunotherapy Alleviates Allergic Responses in a Mouse Model. J. Immunol. 2019, 203, 31–38. [Google Scholar] [CrossRef]
- Gomez-Soler, M.; Gehring, M.P.; Lechtenberg, B.C.; Zapata-Mercado, E.; Hristova, K.; Pasquale, E.B. Engineering nanomolar peptide ligands that differentially modulate EphA2 receptor signaling. J. Biol. Chem. 2019, 294, 8791–8805. [Google Scholar] [CrossRef] [Green Version]
- Koolpe, M.; Dail, M.; Pasquale, E.B. An ephrin mimetic peptide that selectively targets the EphA2 receptor. J. Biol. Chem. 2002, 277, 46974–46979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.Q.; Sha, J.C.; Wang, H.; An, L.F.; Liu, T.; Li, L. P-FN12, an H4R-Based Epitope Vaccine Screened by Phage Display, Regulates the Th1/Th2 Balance in Rat Allergic Rhinitis. Mol. Ther. Methods Clin. Dev. 2018, 11, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Dwyer, R.; Kovaleva, M.; Zhang, J.; Steven, J.; Cummins, E.; Luxenberg, D.; Darmanin-Sheehan, A.; Carvalho, M.F.; Whitters, M.; Saunders, K.; et al. Anti-ICOSL New Antigen Receptor Domains Inhibit T Cell Proliferation and Reduce the Development of Inflammation in the Collagen-Induced Mouse Model of Rheumatoid Arthritis. J. Immunol. Res. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Issa, E.; Moss, A.J.; Fischer, M.; Kang, M.; Ahmed, S.; Farah, H.; Bate, N.; Giakomidi, D.; Brindle, N.P.J. Development of an Orthogonal Tie2 Ligand Resistant to Inhibition by Ang2. Mol. Pharm. 2018, 15, 3962–3968. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.; Wald, H.; Vaizel-Ohayon, D.; Grabovsky, V.; Oren, Z.; Karni, A.; Weiss, L.; Galun, E.; Peled, A.; Eizenberg, O. Development of novel Promiscuous anti-chemokine Peptibodies for Treating autoimmunity and inflammation. Front. Immunol. 2017, 8, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Desharnais, J.; Sahasrabudhe, P.V.; Jin, P.; Li, W.; Oates, B.D.; Shanker, S.; Banker, M.E.; Chrunyk, B.A.; Song, X.; et al. Inhibiting complex IL-17A and IL-17RA interactions with a linear peptide. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Burtea, C.; Laurent, S.; Sanli, T.; Fanfone, D.; Devalckeneer, A.; Sauvage, S.; Beckers, M.-C.; Rorive, S.; Salmon, I.; Elst, L.V.; et al. Screening for peptides targeted to IL-7R alpha for molecular imaging of rheumatoid arthritis synovium. Arthritis Res. Ther. 2016, 18, 1–19. [Google Scholar] [CrossRef]
- Vaz, E.R.; Fujimura, P.T.; Araujo, G.R.; Silva, C.A.T.D.; Silva, R.L.; Cunha, T.M.; Lopes-Ferreira, M.; Lima, C.; Ferreira, M.J.; Cunha-Junior, J.P.; et al. A Short Peptide That Mimics the Binding Domain of TGF-beta 1 Presents Potent Anti-Inflammatory Activity. PLoS ONE 2015, 10, e0136116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.W.; Lu, S.; Su, Y.J.; Xue, D.; Yu, X.L.; Wang, S.W.; Zhang, H.; Xu, P.X.; Xie, X.X.; Liu, R.T. Decreasing oxidative stress and neuroinflammation with a multifunctional peptide rescues memory deficits in mice with Alzheimer disease. Free Radic. Biol. Med. 2014, 74, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Xu, Z.; Wang, G.S.; Ji, F.Y.; Mei, C.X.; Liu, J.; Wu, G.M. Directed Evolution of an LBP/CD14 Inhibitory Peptide and Its Anti-Endotoxin Activity. PLoS ONE 2014, 9, e101406. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Segers, F.; Sliedregt-Bol, K.; Bot, I.; Woltman, A.M.; Boross, P.; Verbeek, S.; Overkleeft, H.; Marel, G.A.V.D.; Kooten, C.V.; et al. Identification of a novel CD40 ligand for targeted imaging of inflammatory plaques by phage display. FASEB J. 2013, 27, 4136–4146. [Google Scholar] [CrossRef] [PubMed]
- Houimel, M.; Mazzucchelli, L. Chemokine CCR3 ligands-binding peptides derived from a random phage-epitope library. Immunol. Lett. 2013, 149, 19–29. [Google Scholar] [CrossRef]
- Tolg, C.; Hamilton, S.R.; Zalinska, E.; McCulloch, L.; Amin, R.; Akentieva, N.; Winnik, F.; Savani, R.; Bagli, D.J.; Luyt, L.G.; et al. A RHAMM Mimetic Peptide Blocks Hyaluronan Signaling and Reduces Inflammation and Fibrogenesis in Excisional Skin Wounds. Am. J. Pathol. 2012, 181, 1250–1270. [Google Scholar] [CrossRef] [Green Version]
- Stanger, K.; Steffek, M.; Zhou, L.; Pozniak, C.D.; Quan, C.; Franke, Y.; Tom, J.; Tam, C.; Elliott, J.M.; Lewcock, J.W.; et al. Allosteric peptides bind a caspase zymogen and mediate caspase tetramerization. Nat. Chem. Biol. 2012, 8, 655–660. [Google Scholar] [CrossRef]
- Shanmugam, A.; Rajoria, S.; George, A.L.; Mittelman, A.; Suriano, R.; Tiwari, R.K. Synthetic Toll Like Receptor-4 (TLR-4) Agonist Peptides as a Novel Class of Adjuvants. PLoS ONE 2012, 7, e30839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segers, F.M.E.; Yu, H.; Molenaar, T.J.M.; Prince, P.; Tanaka, T.; Berkel, T.J.C.V.; Biessen, E.A.L. Design and Validation of a Specific Scavenger Receptor Class AI Binding Peptide for Targeting the Inflammatory Atherosclerotic Plaque. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 971–978. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Sun, H.-X.; Li, X.-Y.; Mo, X.-M.; Zhang, G. Screening for a Peptide That Inhibits Expression of a Broad-spectrum of Chemokines Using Models of Endotoxin Tolerance and LIPS-induced Pro-inflammation. Prog. Biochem. Biophys. 2013, 40, 461–470. [Google Scholar]
- Shibata, H.; Yoshioka, Y.; Ohkawa, A.; Minowa, K.; Mukai, Y.; Abe, Y.; Taniai, M.; Nomura, T.; Kayamuro, H.; Nabeshi, H.; et al. Creation and X-ray structure analysis of the tumor necrosis factor receptor-1-selective mutant of a tumor necrosis factor-alpha antagonist. J. Biol. Chem. 2008, 283, 998–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, H.; Yoshioka, Y.; Ohkawa, A.; Abe, Y.; Nomura, T.; Mukai, Y.; Nakagawa, S.; Taniai, M.; Ohta, T.; Mayumi, T.; et al. The therapeutic effect of TNFR1-selective antagonistic mutant TNF-alpha in murine hepatitis models. Cytokine 2008, 44, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Yoshioka, Y.; Abe, Y.; Ohkawa, A.; Nomura, T.; Minowa, K.; Mukai, Y.; Nakagawa, S.; Taniai, M.; Ohta, T.; et al. The treatment of established murine collagen-induced arthritis with a TNFR1-selective antagonistic mutant TNF. Biomaterials 2009, 30, 6638–6647. [Google Scholar] [CrossRef]
- Takagi, T.; Arisawa, T.; Yamamoto, K.; Hirata, I.; Nakano, H.; Sawada, M. Identification of ligands binding specifically to inflammatory intestinal mucosa using phage display. Clin. Exp. Pharmacol. Physiol. 2007, 34, 286–289. [Google Scholar] [CrossRef]
- Molenaar, T.J.M.; Appeldoorn, C.C.M.; Haas, S.A.M.D.; Michon, N.N.; Bonnefoy, A.; Hoylaerts, M.F.; Pannekoek, H.; Berkel, T.J.C.V.; Kuiper, J.; Biessen, E.A.L. Specific inhibition of P-selectin-mediated cell adhesion by phage display-derived peptide antagonists. Blood 2002, 100, 3570–3577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminska, B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy—From molecular mechanisms to therapeutic benefits. BBA Proteins Proteom. 2005, 1754, 253–262. [Google Scholar] [CrossRef]
- Hendrayani, S.F.; Al-Harbi, B.; Al-Ansari, M.M.; Silva, G.; Aboussekhra, A. The inflammatory/cancer-related IL-6/STAT3/NF-kappa B positive feedback loop includes AUF1 and maintains the active state of breast myofibroblasts. Oncotarget 2016, 7, 41974–41985. [Google Scholar] [CrossRef] [Green Version]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [Green Version]
- Henriquez-Olguin, C.; Altamirano, F.; Valladares, D.; Lopez, J.R.; Allen, P.D.; Jaimovich, E. Altered ROS production, NF-kappa B activation and interleukin-6 gene expression induced by electrical stimulation in dystrophic mdx skeletal muscle cells. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 1410–1419. [Google Scholar] [CrossRef] [Green Version]
- Kollias, G.; Douni, E.; Kassiotis, G.; Kontoyiannis, D. On the role of tumor necrosis factor and receptors in models of multiorgan failure, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Immunol. Rev. 1999, 169, 175–194. [Google Scholar] [CrossRef]
- Annibaldi, A.; Meier, P. Checkpoints in TNF-Induced Cell Death: Implications in Inflammation and Cancer. Trends Mol. Med. 2018, 24, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Jarosz-Griffiths, H.H.; Holbrook, J.; Lara-Reyna, S.; McDermott, M.F. TNF receptor signalling in autoinflammatory diseases. Int. Immunol. 2019, 31, 639–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, M.B.; Shelby, S.A.; Veatch, S.L. Super-Resolution Microscopy: Shedding Light on the Cellular Plasma Membrane. Chem. Rev. 2017, 117, 7457–7477. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar] [CrossRef] [Green Version]
- Weinelt, N.; Karathanasis, C.; Smith, S.; Medler, J.; Malkusch, S.; Fulda, S.; Wajant, H.; Heilemann, M.; Wijk, S.J.L.V. Quantitative single-molecule imaging of TNFR1 reveals zafirlukast as antagonist of TNFR1 clustering and TNFα-induced NF-ĸB. J. Leukoc. Biol. 2021, 109, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Efimov, G.A.; Kruglov, A.A.; Tillib, S.V.; Kuprash, D.V.; Nedospasov, S.A. Tumor Necrosis Factor and the consequences of its ablation in vivo. Mol. Immunol. 2009, 47, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Antoni, C.; Braun, J. Side effects of anti-TNF therapy: Current knowledge. Clin. Exp. Rheumatol. 2002, 20, S152–S157. [Google Scholar]
- Medler, J.; Wajant, H. Tumor necrosis factor receptor-2 (TNFR2): An overview of an emerging drug target. Expert Opin. Ther. Targets 2019, 23, 295–307. [Google Scholar] [CrossRef]
- Medler, J.; Nelke, J.; Weisenberger, D.; Steinfatt, T.; Rothaug, M.; Berr, S.; Hunig, T.; Beilhack, A.; Wajant, H. TNFRSF receptor-specific antibody fusion proteins with targeting controlled Fc gamma R-independent agonistic activity. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Kontermann, R.E.; Pfizenmaier, K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front. Cell. Dev. Biol. 2020, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.H.; Schaaf, T.M.; Grant, B.D.; Lim, C.K.W.; Bawaskar, P.; Aldrich, C.C.; Thomas, D.D.; Sachs, J.N. Noncompetitive inhibitors of TNFR1 probe conformational activation states. Sci. Signal. 2019, 12, eaav5637. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.H.; Vunnam, N.; Lewis, A.K.; Chiu, T.L.; Brummel, B.E.; Schaaf, T.M.; Grant, B.D.; Bawaskar, P.; Thomas, D.D.; Sachs, J.N. An Innovative High-Throughput Screening Approach for Discovery of Small Molecules That Inhibit TNF Receptors. SLAS Discov. 2017, 22, 950–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Jiang, H.; Huang, Y.; Wang, J.; Qiu, L.; Hu, Z.; Ma, X.; Lu, Y. Screening of an anti-inflammatory peptide from Hydrophis cyanocinctus and analysis of its activities and mechanism in DSS-induced acute colitis. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.S.; Wang, J.J.; Luo, P.F.; Li, A.; Tian, S.; Jiang, H.L.; Zheng, Y.J.; Zhu, F.; Lu, Y.M.; Xia, Z.F. Hydrostatin-SN1, a Sea Snake-Derived Bioactive Peptide, Reduces Inflammation in a Mouse Model of Acute Lung Injury. Front. Pharmacol. 2017, 8, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Wu, H.; Xiang, Y. Selection of peptide ligands for TNF receptor imaging. Chin. J. Nucl. Med. 2005, 25, 43–45. [Google Scholar]
- Fu, H.; Wu, H.; Zhang, X.; Huang, J.; He, X.; Chen, L.; Guo, W.; Guo, X.; Hao, B.; Li, Y. Pre-clinical study of a TNFR1-targeted F-18 probe for PET imaging of breast cancer. Amino Acids 2018, 50, 409–419. [Google Scholar] [CrossRef]
- Schonbeck, U.; Libby, P. The CD40/CD154 receptor/ligand dyad. Cell. Mol. Life Sci. 2001, 58, 4–43. [Google Scholar]
- Kooten, C.V.; Banchereau, J. CD40-CD40 ligand: A multifunctional receptor-ligand pair. In Advances in Immunology; Dixon, F.J., Ed.; Elsevier: Amsterdam, The Netherlands, 1996; Volume 61, pp. 1–77. [Google Scholar]
- Clark, L.B.; Foy, T.M.; Noelle, R.J. CD40 and its ligand. In Advances in Immunology; Dixon, F.J., Ed.; Elsevier: Amsterdam, The Netherlands, 1996; Volume 63, pp. 43–78. [Google Scholar]
- Richards, D.M.; Sefrin, J.P.; Gieffers, C.; Hill, O.; Merz, C. Concepts for agonistic targeting of CD40 in immuno-oncology. Hum. Vaccines Immunother. 2020, 16, 377–387. [Google Scholar] [CrossRef]
- Berger, A. Science commentary: Th1 and Th2 responses: What are they? Br. Med. J. 2000, 321, 424. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. 2 types of murine helper T-cell clone. I. definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.Y.; Kim, J.Y.; Kim, K.W.; Park, M.K.; Moon, Y.; Kim, W.U.; Kim, H.Y. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappa B- and PI3-kinase/Akt-dependent pathways. Arthritis Res. Ther. 2004, 6, R120–R128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onishi, R.M.; Gaffen, S.L. Interleukin-17 and its target genes: Mechanisms of interleukin-17 function in disease. Immunol. 2010, 129, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.H.G. Induction, function and regulation of IL-17-producing T cells. Eur. J. Immunol. 2008, 38, 2636–2649. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Xu, J.; Zhang, C.Y.; Jiang, C.Y.; Ma, Y.F.; He, H.J.; Wu, Y.; Devriendt, B.; Cox, E.; Zhang, H.B. Toll-like receptor 5-mediated IL-17C expression in intestinal epithelial cells enhances epithelial host defense against F4+ ETEC infection. Vet. Res. 2019, 50, 14. [Google Scholar] [CrossRef] [Green Version]
- Crowley, J.J. Humanized monoclonal antibody demonstrating dual inhibition of IL-17A and IL-17F Treatment of psoriasis and psoriatic arthritis. Drugs Future 2018, 43, 483–487. [Google Scholar] [CrossRef]
- Raychaudhuri, S.P.; Raychaudhuri, S.K. Mechanistic rationales for targeting interleukin-17A in spondyloarthritis. Arthritis Res. Ther. 2017, 19, 9. [Google Scholar] [CrossRef] [Green Version]
- Dubash, S.; McGonagle, D.; Marzo-Ortega, H. New advances in the understanding and treatment of axial spondyloarthritis: From chance to choice. Ther. Adv. Chronic Dis. 2018, 9, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.H.; Ye, X.Q.; Iwakura, Y. Interleukin-17 family members in health and disease. Int. Immunol. 2021, 33, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C.; Sen, G.C.; Uze, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at age 50: Past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Muskardin, T.L.W.; Niewold, T.B. Type I interferon in rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 214–228. [Google Scholar] [CrossRef]
- Mazewski, C.; Perez, R.E.; Fish, E.N.; Platanias, L.C. Type I Interferon (IFN)-Regulated Activation of Canonical and Non-Canonical Signaling Pathways. Front. Immunol. 2020, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Niewold, T.B. Interferon Alpha as a Primary Pathogenic Factor in Human Lupus. J. Interferon Cytokine Res. 2011, 31, 887–892. [Google Scholar] [CrossRef] [Green Version]
- Marketos, N.; Cinoku, I.; Rapti, A.; Mavragani, C.P. Type I interferon signature in Sjogren’s syndrome: Pathophysiological and clinical implications. Clin. Exp. Rheumatol. 2019, 37, 185–191. [Google Scholar]
- Skaug, B.; Assassi, S. Type I interferon dysregulation in Systemic Sclerosis. Cytokine 2020, 132, 154635. [Google Scholar] [CrossRef]
- Guo, X.; Higgs, B.W.; Rebelatto, M.; Zhu, W.; Greth, W.; Yao, Y.H.; Roskos, L.K.; White, W.I. Suppression of soluble T cell-associated proteins by an anti-interferon-alpha monoclonal antibody in adult patients with dermatomyositis or polymyositis. Rheumatology 2014, 53, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Ceuninck, F.D.; Duguet, F.; Aussy, A.; Laigle, L.; Moingeon, P. IFN-a: A key therapeutic target for multiple autoimmune rheumatic diseases. Drug Discov. Today 2021, 26, 2465–2473. [Google Scholar] [CrossRef]
- Agrawal, A.; Romero-Perez, D.; Jacobsen, J.A.; Villarreal, F.J.; Cohen, S.M. Zinc-binding groups modulate selective inhibition of MMPs. Chem. Med. Chem. 2008, 3, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Murphy, G.; Nagase, H. Progress in matrix metalloproteinase research. Mol. Asp. Med. 2008, 29, 290–308. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, M.M.; Khalil, R.A. Matrix metalloproteinase inhibitors as investigative tools in the pathogenesis and management of vascular disease. Exp. Suppl. 2012, 103, 209–279. [Google Scholar] [PubMed] [Green Version]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemper, C.; Verbsky, J.W.; Price, J.D.; Atkinson, J.P. T-cell stimulation and regulation: With complements from CD46. Immunol. Res. 2005, 32, 31–43. [Google Scholar] [CrossRef]
- Kennedy, A.J.; Davenport, A.P. International Union of Basic and Clinical Pharmacology CIII: Chemerin Receptors CMKLR1 (Chemerin(1)) and GPR1 (Chemerin(2)) Nomenclature, Pharmacology, and Function. Pharmacol. Rev. 2018, 70, 174–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goralski, K.B.; Jackson, A.E.; McKeown, B.T.; Sinal, C.J. More Than an Adipokine: The Complex Roles of Chemerin Signaling in Cancer. Int. J. Mol. Sci. 2019, 20, 4778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Nikolettos, K.; Dimitroulis, D.; Diamantis, E.; Farmaki, P.; Patsouras, A.; Voutyritsa, E.; Syllaios, A.; et al. Molecular Classification and Future Therapeutic Challenges of Triple-negative Breast Cancer. Vivo 2020, 34, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Erridge, C.; Bennett-Guerrero, E.; Poxton, I.R. Structure and function of lipopolysaccharides. Microbes Infect. 2002, 4, 837–851. [Google Scholar] [CrossRef]
- Bishop, R.E. Fundamentals of endotoxin structure and function. Contrib. Microbiol. 2005, 12, 1–27. [Google Scholar]
- Dauphinee, S.M.; Karsan, A. Lipopolysaccharide signaling in endothelial cells. Lab. Investig. 2006, 86, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Schumann, R.R. Function of lipopolysaccharide (LPS)-binding protein (LBP) and CD14, the receptor for LPS/LBP complexes—A short review. Res. Immunol. 1992, 143, 11–15. [Google Scholar] [CrossRef]
- Xu, Z.; Qian, G.S.; Li, Q.; Feng, Q.J.; Wu, G.M.; Li, K.L. Screening of mimetic peptides for CD14 binding site with LBP and antiendotoxin activity of mimetic peptide in vivo and in vitro. Inflamm. Res. 2009, 58, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Ando, D.; Kamada, H.; Taki, S.; Niiyama, M.; Mukai, Y.; Tadokoro, T.; Maenaka, K.; Nakayama, T.; Kado, Y.; et al. A trimeric structural fusion of an antagonistic tumor necrosis factor-alpha mutant enhances molecular stability and enables facile modification. J. Biol. Chem. 2017, 292, 6438–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunath, J.; Delaroque, N.; Szardenings, M.; Neundorf, I.; Straub, R.H. Sympathetic nerve repulsion inhibited by designer molecules in vitro and role in experimental arthritis. Life Sci. 2017, 168, 47–53. [Google Scholar] [CrossRef]
- Parola, C.; Neumeier, D.; Reddy, S.T. Integrating high-throughput screening and sequencing for monoclonal antibody discovery and engineering. Immunology 2018, 153, 31–41. [Google Scholar] [CrossRef]
- Pattabiraman, V.R.; Bode, J.W. Rethinking amide bond synthesis. Nature 2011, 480, 471–479. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The Future of Peptide-based Drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Albericio, F.; Kruger, H.G. Therapeutic peptides foreword. Future Med. Chem. 2012, 4, 1527–1531. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, S. Peptides as ‘Drugs’: The Journey so Far. Int. J. Pept. Res. Ther. 2017, 23, 49–60. [Google Scholar] [CrossRef]
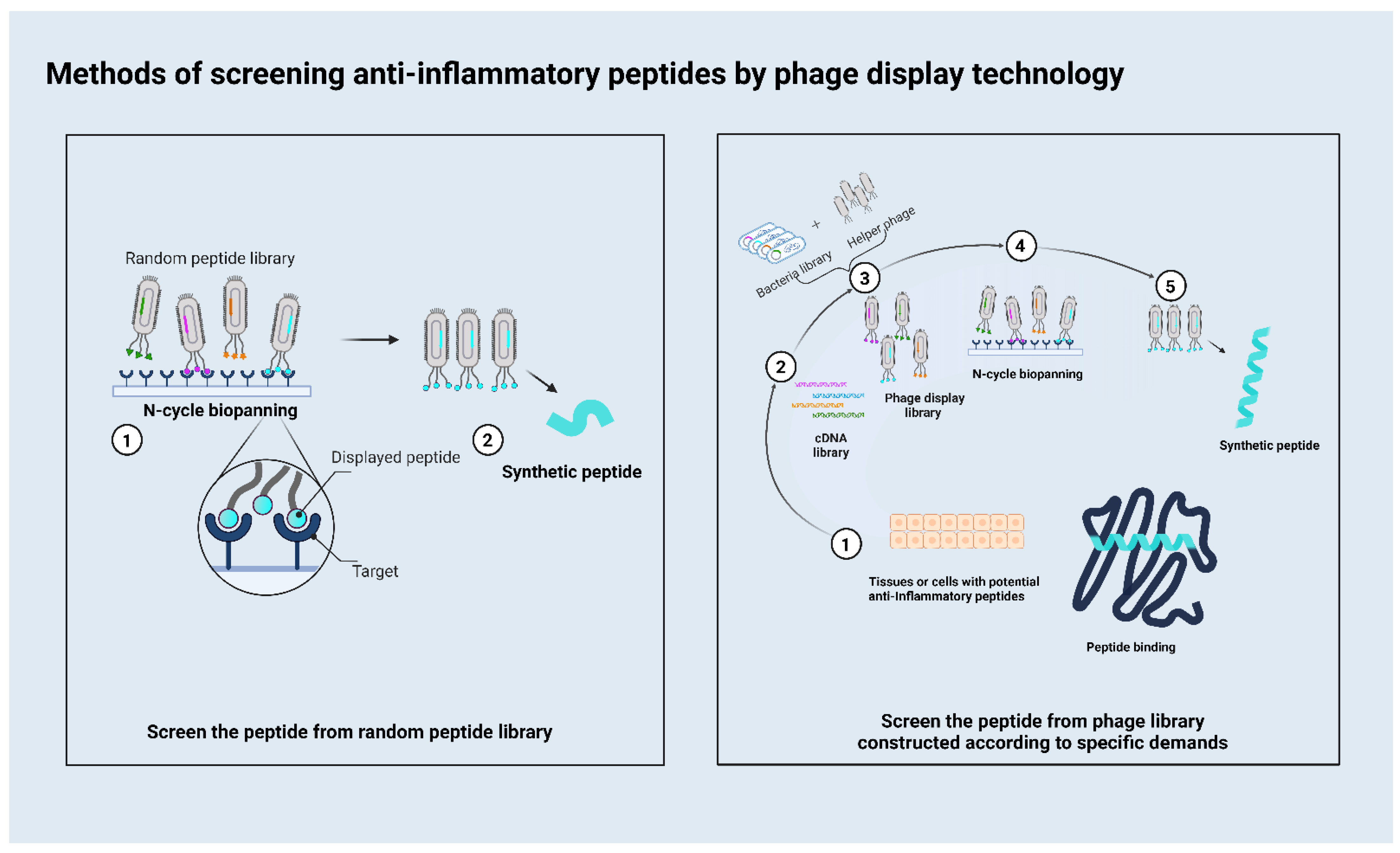

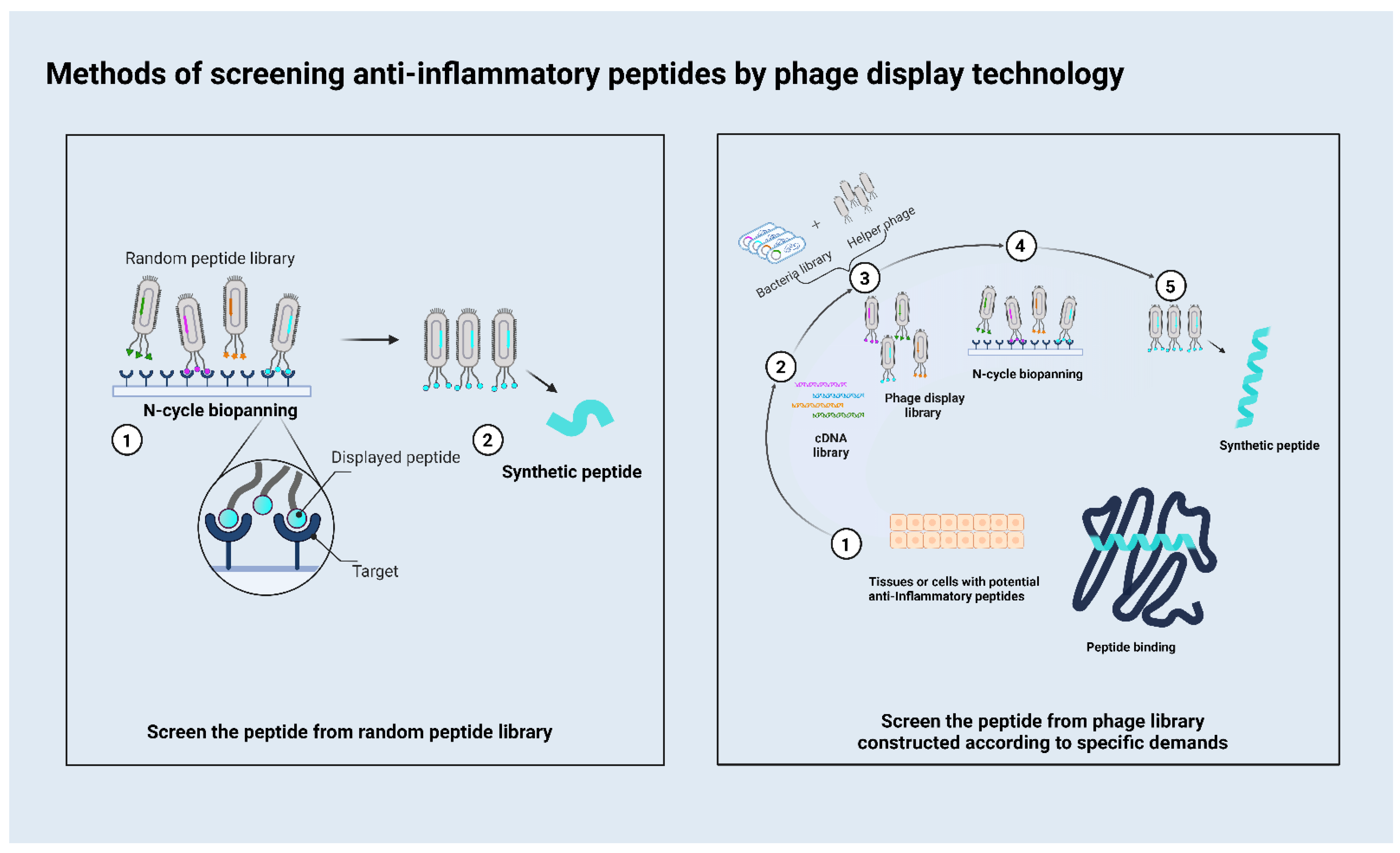{kind=link}
| Name/Sequence | Target | Phage Library Type | Properties | References |
|---|---|---|---|---|
| Davp-1 | Tumor necrosis factor receptor 1(TNFR1) | Venom gland T7 phage display library (Deinagkistrodon acutus) | Has the affinity with TNFR1. | [23] |
| Hydrostatin-SN1 | TNFR1 | Venom gland T7 phage display library (Hydrophis cyanocinctus) | Lowers the clinical parameters of acute colitis, including the disease activity index and histologic scores. Reduces inflammation in a mouse model of acute lung injury (ALI) with significant anti-inflammatory effects both in vitro and in vivo. | [24] |
| CVX51401 | Rat heart microvascular endothelial cells (RHMVEC) | Novagen T7 select phage display system | RRPPR is potent in blocking NO release. Fusing RRPPR with a minimal Cav inhibitory domain could dose-dependently block NO release, vascular endothelial growth factor (VEGF)-induced permeability, and retinal damage in a model of uveitis. | [25] |
| Phpep3D/Pep3D | Rabbit polyclonal antibody anti-human interferon α1 (IFNα1) | Ph.D.™-7 Phage Display Peptide Library | Limits psoriasis-like lesions in mice. | [26] |
| 810A | Thioredoxin-connective tissue growth factor (TrxA-CTGF) | Phage dodecapeptide peptide library | Alleviates fibrosis in the pulmonary index and inhibits inflammation. | [27] |
| HP3 | Peripheral blood mononuclear cells (PBMCs) | Ph.D.™-7 Phage Display Peptide Library | Inhibits the development of psoriatic lesions. | [28] |
| hC3a-specific protein binder | hC3 | Repebody library was constructed by introducing random mutations into six variable sites in nearby two modules, LRRV2 and LRRV3 | Suppresses the effect of pro-inflammatory responses in monocytes, by blocking the interaction between hC3a and its receptor. | [29] |
| LRH7-G5 | G protein-coupled receptor 1 (GPR1) | Ph.D.™-7 Phage Display Peptide Library | Suppresses triple-negative breast cancer (TNBC) tumor growth. | [30] |
| RSH-12 | Metalloproteinase 9 (MMP-9) | M13 phage display peptide library (Ph.D.-12) | Decreases the gelatin degradation by specifically preventing gelatin binding to MMP-9 and MMP-2. | [31] |
| M219hy | MMP-2 | [32] | ||
| MIT | B cell mimotopes | Random heptamer peptide library | Alleviates allergic responses in a mouse model. | [33] |
| YSA/SWL | EphA2 | M13 phage library displaying random 12-mer peptides | Has the affinity with EphA2. | [34,35] |
| P-FN12 | Anti-H4R antibody | A 12-mer random peptide library | Decreases the production of ovalbumin (OVA)-specific IgE, Th2 immunity, and tissue eosinophilia. | [36] |
| Anti- inducible T cell costimulatory ligand (ICOSL) variable domain (VNAR) | Antigen | A synthetic VNAR library | Decreases the inflammation of joints, delays overall disease progression, and reduces severity. | [37] |
| TSL1 | gTie2-ectodomain | Ph.D.™-7 Phage Display Peptide Library | Has the affinity with Ang2. | [38] |
| BKT120Fc and BKT130Fc | Chemokines CCL11, CXCL8, CXCL12, CXCL9, and CCL2 | Ph.D-12™ and Ph.D.™-7 Phage Display Peptide Library | Inhibits the ability of inflammatory chemokines to induce the adhesion and migration of immune cells. Inhibits disease progression in a variety of animal models of autoimmunity and inflammation. | [39] |
| HAP | Interleukin (IL)-17R-Fc | Cyclic and linear peptide libraries | Has the affinity with IL-17R-Fc. | [40] |
| P725 | IL-7Rα | Ph.D.™-7 Phage Display Peptide Library | Competes with IL-7 for IL-7Rα binding sites. | [41] |
| pm26TGF-β1 | Phages that were bound to receptors on the cell surfaces were competitively eluted with 10 ng/mL of recombinant TGF-β1. | Ph.D.™-7 Phage Display Peptide Library | Has direct inhibitory effects on neutrophil migration in a carrageenan-induced peritonitis model. | [42] |
| ZW1 | Aβ42 | Ph.D.™-7 Phage Display Peptide Library | Suppresses the inflammatory response by decreasing the release of proinflammatory cytokines, such as tumor necrosis factor α and interleukin 1β, in microglia and reducing microgliosis and astrogliosis in AD transgenic mice. | [43] |
| P1 | A cluster of differentiation 14 (CD14) | LPS-binding protein (LBP) mutants phage peptide library | Reduces the LPS-induced rat lung tissue injury. | [44] |
| NP31 | Human cluster of differentiation 40 (CD40)-murine IgG | pIF15 phage library containing randomized linear 15-mer amino acids peptide sequence | Allows targeted diagnosis of and intervention in inflammatory disorders such as atherosclerosis and autoimmune disease. | [45] |
| CKGERF and FERKGK | Human chemokine receptor C-C receptor 3 (hCCR3) | 6-mer linearpeptidelibrary | Has inhibitory effects on eosinophil chemotaxis in a murine model of mCCL11-induced peritoneal eosinophilia. | [46] |
| P15-1 | Hyaluronan (HA) oligosaccharides | 15mer phage display libraries | Attenuates proinflammatory, fibrotic repair by blocking hyaluronan oligosaccharide signaling. | [47] |
| pep419 | Caspase-6 | Linear and cyclic peptide phage libraries | Has the affinity with pep419. | [48] |
| LPS peptide mimics | LPS antibody | Ph.D.™-7 Phage Display Peptide Library | TLR-4 agonist adjuvants. | [49] |
| PP1 | Scavenger receptor A1(SR-AI) | pIF4 phage libraries | SR-AI antagonist. | [50] |
| CI-S5 | PBMCs | Ph.D.™-7 Phage Display Peptide Library | Broad-spectrum antagonist of pro-inflammatory chemokines through enhancing the expression of TTP to reduce chemokine mRNA expression. | [51] |
| R1antTN | Human TNFR1 Fc chimera | Tumor necrosis factor (TNF) variants in which six amino acid residues | Contains the clinically useful TNF-α antagonist used in hepatitis. | [52,53,54] |
| SQSHPRH | Inflamed bowel | Ph.D.™-7 Phage Display Peptide Library | Has affinity with inflammatory bowel. | [55] |
| TM11 | Fc-specific goat antihuman IgG | pComb8 phage-displayed peptide library CX15C (in which X is any amino acid and C is a fixed cysteine residue) | New class of small-calcium-dependent P-selectin antagonists based on single-letter amino acid code (EWVDV) core motifs. | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, K.; Tang, Y.; Chen, Q.; Liu, Y. The Screening of Therapeutic Peptides for Anti-Inflammation through Phage Display Technology. Int. J. Mol. Sci. 2022, 23, 8554. https://doi.org/10.3390/ijms23158554
Zhang K, Tang Y, Chen Q, Liu Y. The Screening of Therapeutic Peptides for Anti-Inflammation through Phage Display Technology. International Journal of Molecular Sciences. 2022; 23(15):8554. https://doi.org/10.3390/ijms23158554
Chicago/Turabian StyleZhang, Kangran, Yezhong Tang, Qin Chen, and Yang Liu. 2022. "The Screening of Therapeutic Peptides for Anti-Inflammation through Phage Display Technology" International Journal of Molecular Sciences 23, no. 15: 8554. https://doi.org/10.3390/ijms23158554
APA StyleZhang, K., Tang, Y., Chen, Q., & Liu, Y. (2022). The Screening of Therapeutic Peptides for Anti-Inflammation through Phage Display Technology. International Journal of Molecular Sciences, 23(15), 8554. https://doi.org/10.3390/ijms23158554