
Quantum Biochemistry Screening and In Vitro Evaluation of Leishmania Metalloproteinase Inhibitors

 ,
,  , , , ,
, , , ,  ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Results
2.1. Identification of Potential Metalloprotease Inhibitors
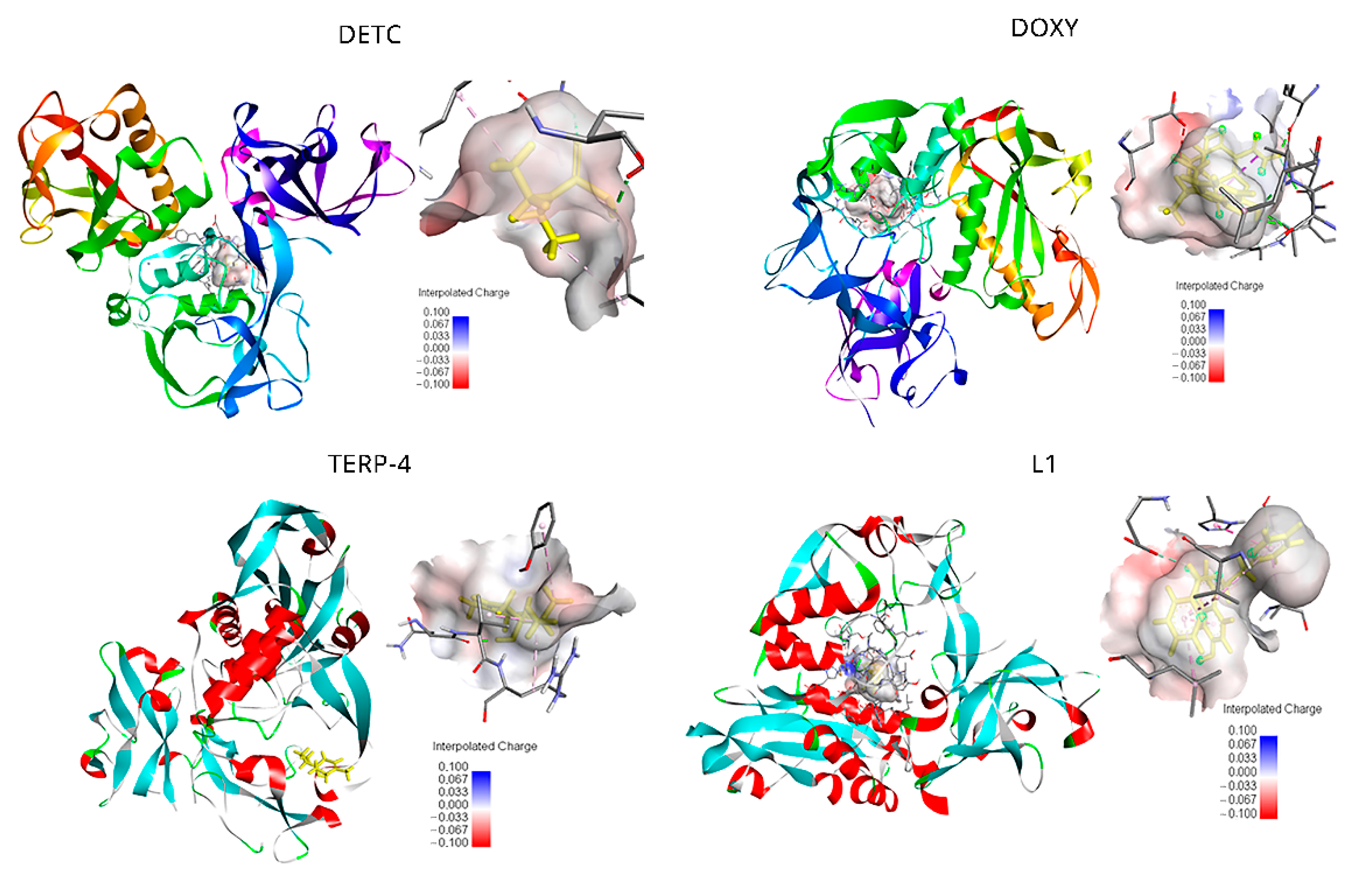
2.2. Quantum Energy Calculation between the Ligands and gp63 Protein
2.3. L1 Molecule as a Potential Inhibitor against Leishmania amazonensis
2.4. Morphological and Structural Changes of Leishmania amazonensis Promastigotes in the Presence of Potential Inhibitors
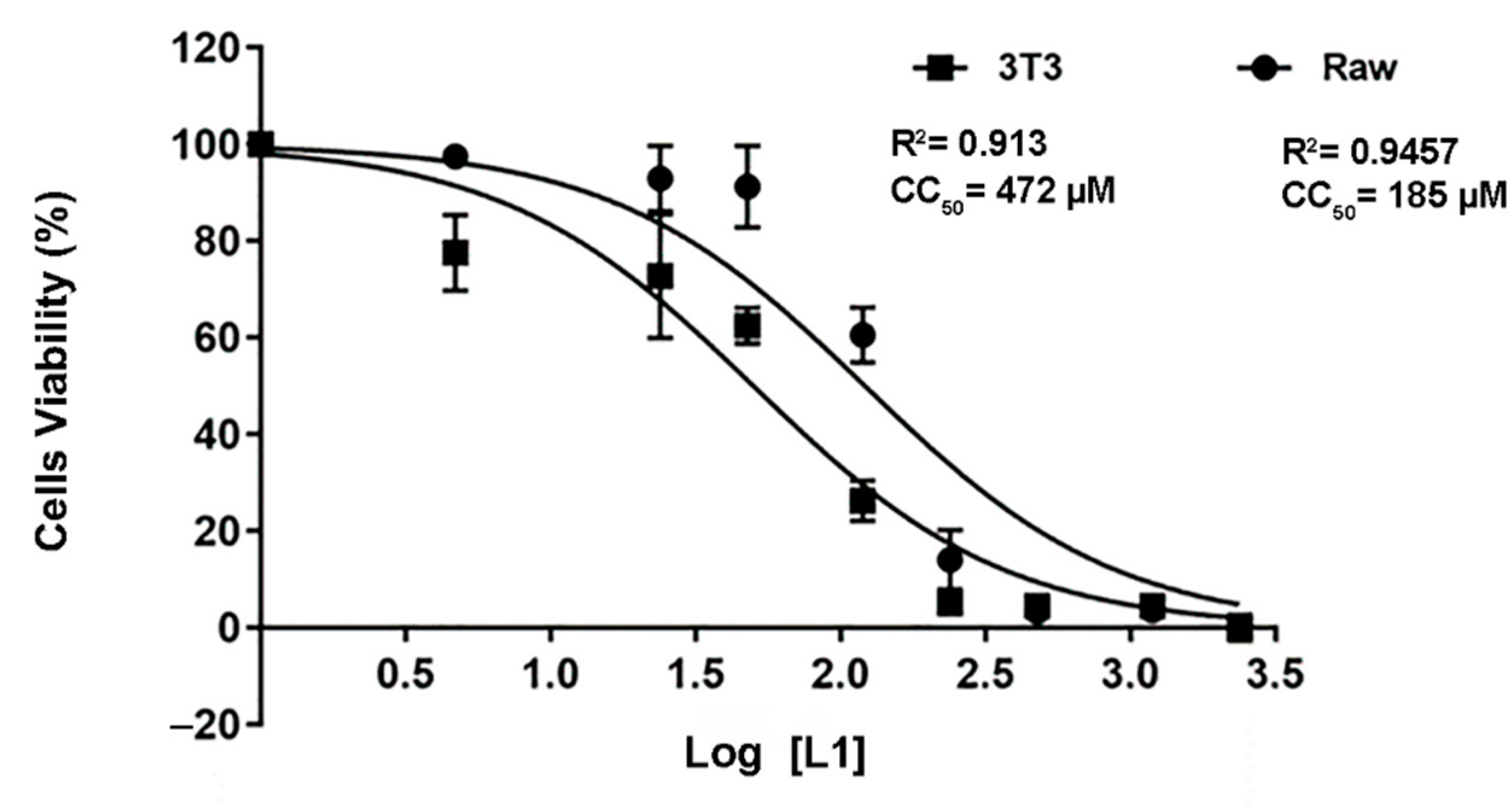
2.5. L1 Molecule Demonstrated a Well-Tolerated Profile against the Cells Lines
3. Discussion
4. Materials and Methods
4.1. Leishmania 63-kDa Glycoprotein Structures
4.2. Ligand Structures
4.3. Docking
4.4. Molecular Fractionation Conjugate Caps (MFCC)
4.5. Antiparasitic Activity of In Silico Selected Compounds
4.5.1. Compounds
4.5.2. Parasites
4.5.3. Anti-Parasitic Activity Determined by Microscope Counting and Resazurin Assay
4.5.4. Analysis of Cell Membrane Integrity
4.5.5. Morphological Analysis Using Simultaneous Scanning Electron Microscopy (SEM) and Energy Dispersive X-ray Spectroscopy (EDS)
4.6. Cell Lines and Cell Culture
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Akhoundi, M.; Kuhls, K.; Cannet, A.; Votypka, J.; Marty, P.; Delaunay, P.; Sereno, D. A Historical Overview of the Classification, Evolution, and Dispersion of Leishmania Parasites and Sandflies. PLoS Negl. Trop. Dis. 2016, 10, e0004349. [Google Scholar] [CrossRef] [PubMed]
- Azeredo-Coutinho, R.; Conceicao-Silva, F.; Schubach, A.; Cupolillo, E.; Quintella, L.; Madeira, M.; Pacheco, R.; Valete-Rosalino, C.; Mendonça, S. First report of diffuse cutaneous leishmaniasis and Leishmania amazonensis infection in Rio de Janeiro State, Brazil. Trans. R. Soc. Trop. Med. Hyg. 2007, 101, 735–737. [Google Scholar] [CrossRef] [PubMed]
- Silva-Jardim, I.; Thiemann, O.H.; Anibal, F.F. Leishmaniasis and Chagas Disease Chemotherapy: A Critical Review. J. Braz. Chem. Soc. 2014. [Google Scholar] [CrossRef]
- Stockdale, L.; Newton, R. A Review of Preventative Methods against Human Leishmaniasis Infection. PLoS Negl. Trop. Dis. 2013, 7, e2278. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C.D.; Ezzati, M.; Lopez, A.D. Measuring the Burden of Neglected Tropical Diseases: The Global Burden of Disease Framework. PLoS Negl. Trop. Dis. 2007, 1, e114. [Google Scholar] [CrossRef]
- Ready, P.D. Leishmaniasis emergence in Europe. Eurosurveillance 2010, 15, 19505. [Google Scholar] [CrossRef]
- Mitra, A.K.; Mawson, A.R. Neglected Tropical Diseases: Epidemiology and Global Burden. Trop. Med. Infect. Dis. 2017, 2, 36. [Google Scholar] [CrossRef]
- Cecílio, P.; Pérez-Cabezas, B.; Santarém, N.; Maciel, J.; Rodrigues, V.; Cordeiro da Silva, A. Deception and Manipulation: The Arms of Leishmania, a Successful Parasite. Front. Immunol. 2014, 5, 480. [Google Scholar] [CrossRef]
- Podinovskaia, M.; Descoteaux, A. Leishmania and the macrophage: A multifaceted interaction. Futur. Microbiol. 2015, 10, 111–129. [Google Scholar] [CrossRef]
- Yao, C. Major Surface Protease (MSP, or GP63) of Trypanosomatids, One Size Fits All? Infect. Immun. 2010, 78, 22–31. [Google Scholar] [CrossRef]
- Moreno, C.J.G.; Torres, T.; Silva, M.S. Variable Surface Glycoprotein from Trypanosoma brucei Undergoes Cleavage by Matrix Metalloproteinases: An in silico Approach. Pathogens 2019, 8, 178. [Google Scholar] [CrossRef]
- Geurts, N.; Opdenakker, G.; Steen, P.E.V.D. Matrix metalloproteinases as therapeutic targets in protozoan parasitic infections. Pharmacol. Ther. 2012, 133, 257–279. [Google Scholar] [CrossRef]
- Murase, L.S.; de Souza, J.V.P.; Neto, Q.A.D.L.; de Mello, T.F.P.; Cardoso, B.M.; Lera-Nonose, D.S.S.L.; Teixeira, J.J.V.; Lonardoni, M.V.C.; Demarchi, I.G. The role of metalloproteases in Leishmania species infection in the New World: A systematic review. Parasitology 2018, 145, 1499–1509. [Google Scholar] [CrossRef]
- McGwire, B.S.; Chang, K.-P.; Engman, D.M. Migration through the Extracellular Matrix by the Parasitic Protozoan Leishmania Is Enhanced by Surface Metalloprotease gp63. Infect. Immun. 2003, 71, 1008–1010. [Google Scholar] [CrossRef]
- Eisnard, A.; Shio, M.T.; Eolivier, M. Impact of Leishmania metalloprotease GP63 on macrophage signaling. Front. Cell. Infect. Microbiol. 2012, 2, 72. [Google Scholar] [CrossRef]
- Yao, C.; Donelson, J.E.; Wilson, M.E. The major surface protease (MSP or GP63) of Leishmania sp. Biosynthesis, regulation of expression, and function. Mol. Biochem. Parasitol. 2003, 132, 1–16. [Google Scholar] [CrossRef]
- McKerrow, J.H.; Caffrey, C.; Kelly, B.; Loke, P.; Sajid, M. PROTEASES IN PARASITIC DISEASES. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 497–536. [Google Scholar] [CrossRef]
- Bangs, J.D.; Ransom, D.A.; Nimick, M.; Christie, G.; Hooper, N. In vitro cytocidal effects on Trypanosoma brucei and inhibition of Leishmania major GP63 by peptidomimetic metalloprotease inhibitors. Mol. Biochem. Parasitol. 2001, 114, 111–117. [Google Scholar] [CrossRef]
- Alvarez, V.E.; Niemirowicz, G.T.; Cazzulo, J.J. The peptidases of Trypanosoma cruzi: Digestive enzymes, virulence factors, and mediators of autophagy and programmed cell death. Biochim. et Biophys. Acta (BBA) Proteins Proteom. 2012, 1824, 195–206. [Google Scholar] [CrossRef]
- Zhang, D.W.; Zhang, J.Z.H. Molecular fractionation with conjugate caps for full quantum mechanical calculation of protein–molecule interaction energy. J. Chem. Phys. 2003, 119, 3599–3605. [Google Scholar] [CrossRef]
- He, X.; Zhang, J.Z.H. The generalized molecular fractionation with conjugate caps/molecular mechanics method for direct calculation of protein energy. J. Chem. Phys. 2006, 124, 184703. [Google Scholar] [CrossRef]
- Gordon, M.S.; Fedorov, D.G.; Pruitt, S.R.; Slipchenko, L.V. Fragmentation Methods: A Route to Accurate Calculations on Large Systems. Chem. Rev. 2011, 112, 632–672. [Google Scholar] [CrossRef]
- Vannier-Santos, M.; De Castro, S. Electron Microscopy in Antiparasitic Chemotherapy: A (Close) View to a Kill. Curr. Drug Targets 2009, 10, 246–260. [Google Scholar] [CrossRef]
- Vannier-Santos, M.A.; Lins, U. Cytochemical techniques and energy-filtering transmission electron microscopy applied to the study of parasitic protozoa. Biol. Proced. Online 2001, 3, 8–18. [Google Scholar] [CrossRef]
- Mann, S.; Frasca, K.; Scherrer, S.; Henao-Martínez, A.F.; Newman, S.; Ramanan, P.; Suarez, J.A. A Review of Leishmaniasis: Current Knowledge and Future Directions. Curr. Trop. Med. Rep. 2021, 8, 121–132. [Google Scholar] [CrossRef]
- De Menezes, J.P.B.; Guedes, C.E.S.; Petersen, A.L.D.O.A.; Fraga, D.B.M.; Veras, P.S.T. Advances in Development of New Treatment for Leishmaniasis. BioMed Res. Int. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Caridha, D.; Vesely, B.; Van Bocxlaer, K.; Arana, B.; Mowbray, C.; Rafati, S.; Uliana, S.; Reguera, R.; Kreishman-Deitrick, M.; Sciotti, R.; et al. Route map for the discovery and pre-clinical development of new drugs and treatments for cutaneous leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2019, 11, 106–117. [Google Scholar] [CrossRef]
- Saldívar-González, F.; Prieto-Martínez, F.D.; Medina-Franco, J.L. Descubrimiento y desarrollo de fármacos: Un enfoque computacional. Educación Química 2017, 28, 51–58. [Google Scholar] [CrossRef]
- Lee, J.; Freddolino, P.L.; Zhang, Y. Ab Initio Protein Structure Prediction. In From Protein Structure to Function with Bioinformatics; Springer: Berlin, Germany, 2017; pp. 3–35. [Google Scholar] [CrossRef]
- Tsai, C.S. An Introduction to Computational Biochemistry; Wiley: Hoboken, NJ, USA, 2002. [Google Scholar] [CrossRef]
- Celes, F.S.; Trovatti, E.; Khouri, R.; Van Weyenbergh, J.; Ribeiro, S.J.L.; Borges, V.M.; Barud, H.S.; de Oliveira, C.I. DETC-based bacterial cellulose bio-curatives for topical treatment of cutaneous leishmaniasis. Sci. Rep. 2016, 6, 38330. [Google Scholar] [CrossRef]
- Khouri, R.; Novais, F.; Santana, G.; de Oliveira, C.I.; dos Santos, M.A.V.; Barral, A.; Barral-Netto, M.; Van Weyenbergh, J. DETC Induces Leishmania Parasite Killing in Human In Vitro and Murine In Vivo Models: A Promising Therapeutic Alternative in Leishmaniasis. PLoS ONE 2010, 5, e14394. [Google Scholar] [CrossRef]
- Pinger, J.; Chowdhury, S.; Papavasiliou, F.N. Variant surface glycoprotein density defines an immune evasion threshold for African trypanosomes undergoing antigenic variation. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhang, J.Z.H. A new method for direct calculation of total energy of protein. J. Chem. Phys. 2005, 122, 031103. [Google Scholar] [CrossRef] [PubMed]
- Masmoudi, A.; Dammak, A.; Chaaben, H.; Maalej, N.; Akrout, F.; Turki, H. Doxycycline for the treatment of cutaneous leishmaniasis. Dermatol. Online J. 2008, 14. [Google Scholar] [CrossRef]
- Paris, C.; Loiseau, P.M.; Bories, C.; Bréard, J. Miltefosine Induces Apoptosis-Like Death in Leishmania donovani Promastigotes. Antimicrob. Agents Chemother. 2004, 48, 852–859. [Google Scholar] [CrossRef]
- Verma, N.K.; Singh, G.; Dey, C.S. Miltefosine induces apoptosis in arsenite-resistant Leishmania donovani promastigotes through mitochondrial dysfunction. Exp. Parasitol. 2007, 116, 1–13. [Google Scholar] [CrossRef]
- Olivier, M.; Atayde, V.D.; Isnard, A.; Hassani, K.; Shio, M.T. Leishmania virulence factors: Focus on the metalloprotease GP63. Microbes Infect. 2012, 14, 1377–1389. [Google Scholar] [CrossRef]
- Schlagenhauf, E.; Etges, R.; Metcalf, P. The crystal structure of the Leishmania major surface proteinase leishmanolysin (gp63). Structure 1998, 6, 1035–1046. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Mota, K.; Neto, J.L.; Costa, A.L.; Oliveira, J.; Bezerra, K.; Albuquerque, E.; Caetano, E.; Freire, V.; Fulco, U. A quantum biochemistry model of the interaction between the estrogen receptor and the two antagonists used in breast cancer treatment. Comput. Theor. Chem. 2016, 1089, 21–27. [Google Scholar] [CrossRef]
- Swamy, K.; Kim, M.-J.; Jeon, H.-R.; Jung, J.-Y.; Yoon, J.-Y. New 7-Hydroxycoumarin-Based Fluorescent Chemosensors for Zn(II) and Cd(II). Bull. Korean Chem. Soc. 2010, 31, 3611–3616. [Google Scholar] [CrossRef][Green Version]
- Rolón, M.; Vega, C.; Escario, J.A.; Gómez-Barrio, A. Development of resazurin microtiter assay for drug sensibility testing of Trypanosoma cruzi epimastigotes. Parasitol. Res. 2006, 99, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.A.; Azevedo, C.D.S.; Motta, F.N.; dos Santos, M.L.; Silva, C.L.; de Santana, J.M.; Bastos, I.M.D. Essential oils: In vitro activity against Leishmania amazonensis, cytotoxicity and chemical composition. BMC Complement. Altern. Med. 2016, 16, 444. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands | Structures | EFE (kcal/mol) | FIE (kcal/mol) |
|---|---|---|---|
| DETC (ZINC03633221) |  | −4.45 | −5.64 |
| Doxycycline (ZINC21984014) |  | −5.46 | −7.54 |
| Terpinen-4-ol (ZINC03861537) |  | −5.04 | −5.64 |
| L1 |  | −8.07 | −9.86 |
| Compounds | IC50 (µM) |
|---|---|
| AmpB | 0.906 ± 0.06 |
| Doxycycline | 26.82 ± 0.1 |
| DETC | 4.97 ± 0.119 |
| L1 | 1.24 ± 0.08746 |
| Cells | IC50—CC50 (µM) | SI |
|---|---|---|
| L. amazonensis | 1.24 ± 0.0874 | - |
| 3T3 | 185 ± 0.0524 | 149.19 |
| RAW | 472 ± 0.0706 | 380.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno, C.J.G.; Farias, H.M.; Medeiros, R.; Brito, T.; Oliveira, J.; de Sousa, F.L., Jr.; Medeiros, M.J.C.d.; Amorim, B.; Santos-Gomes, G.; Pontes, D.; et al. Quantum Biochemistry Screening and In Vitro Evaluation of Leishmania Metalloproteinase Inhibitors. Int. J. Mol. Sci. 2022, 23, 8553. https://doi.org/10.3390/ijms23158553
Moreno CJG, Farias HM, Medeiros R, Brito T, Oliveira J, de Sousa FL Jr., Medeiros MJCd, Amorim B, Santos-Gomes G, Pontes D, et al. Quantum Biochemistry Screening and In Vitro Evaluation of Leishmania Metalloproteinase Inhibitors. International Journal of Molecular Sciences. 2022; 23(15):8553. https://doi.org/10.3390/ijms23158553
Chicago/Turabian StyleMoreno, Cláudia Jassica Gonçalves, Henriqueta Monalisa Farias, Rafael Medeiros, Talita Brito, Johny Oliveira, Francimar Lopes de Sousa, Jr., Mayara Jane Campos de Medeiros, Bruno Amorim, Gabriela Santos-Gomes, Daniel Pontes, and et al. 2022. "Quantum Biochemistry Screening and In Vitro Evaluation of Leishmania Metalloproteinase Inhibitors" International Journal of Molecular Sciences 23, no. 15: 8553. https://doi.org/10.3390/ijms23158553
APA StyleMoreno, C. J. G., Farias, H. M., Medeiros, R., Brito, T., Oliveira, J., de Sousa, F. L., Jr., Medeiros, M. J. C. d., Amorim, B., Santos-Gomes, G., Pontes, D., Rocha, H. A. O., Frazao, N. F., & Silva, M. S. (2022). Quantum Biochemistry Screening and In Vitro Evaluation of Leishmania Metalloproteinase Inhibitors. International Journal of Molecular Sciences, 23(15), 8553. https://doi.org/10.3390/ijms23158553