
Immunometabolism of Immune Cells in Mucosal Environment Drives Effector Responses against Mycobacterium tuberculosis


{kind=link}
{kind=link}
Abstract
:1. Introduction
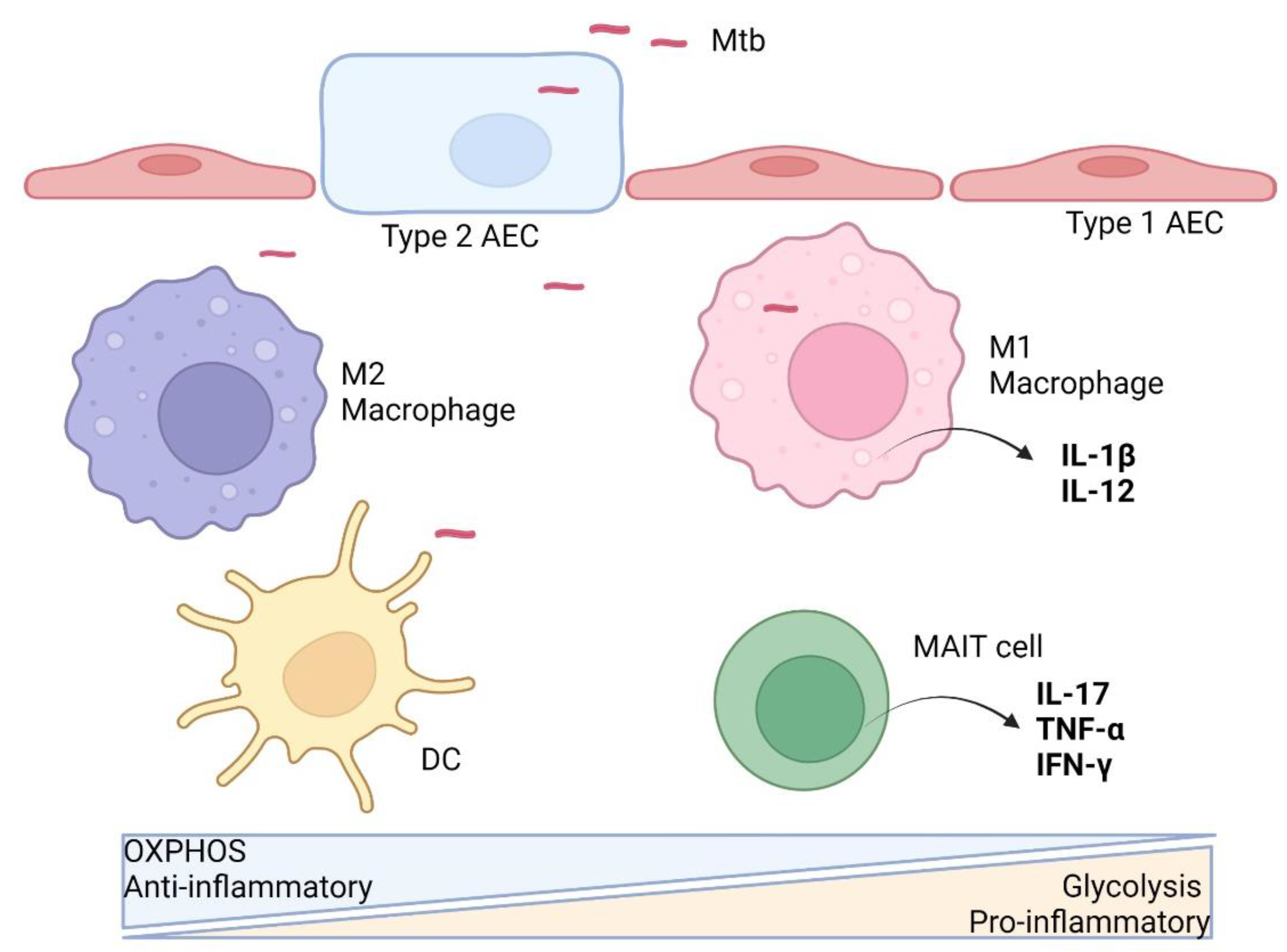
2. Immunometabolism during Early Phase of Mtb Infection
2.1. Alveolar Epithelial Cells
2.2. Macrophages
2.3. Dendritic Cells
2.4. Neutrophils
2.5. MAIT Cells
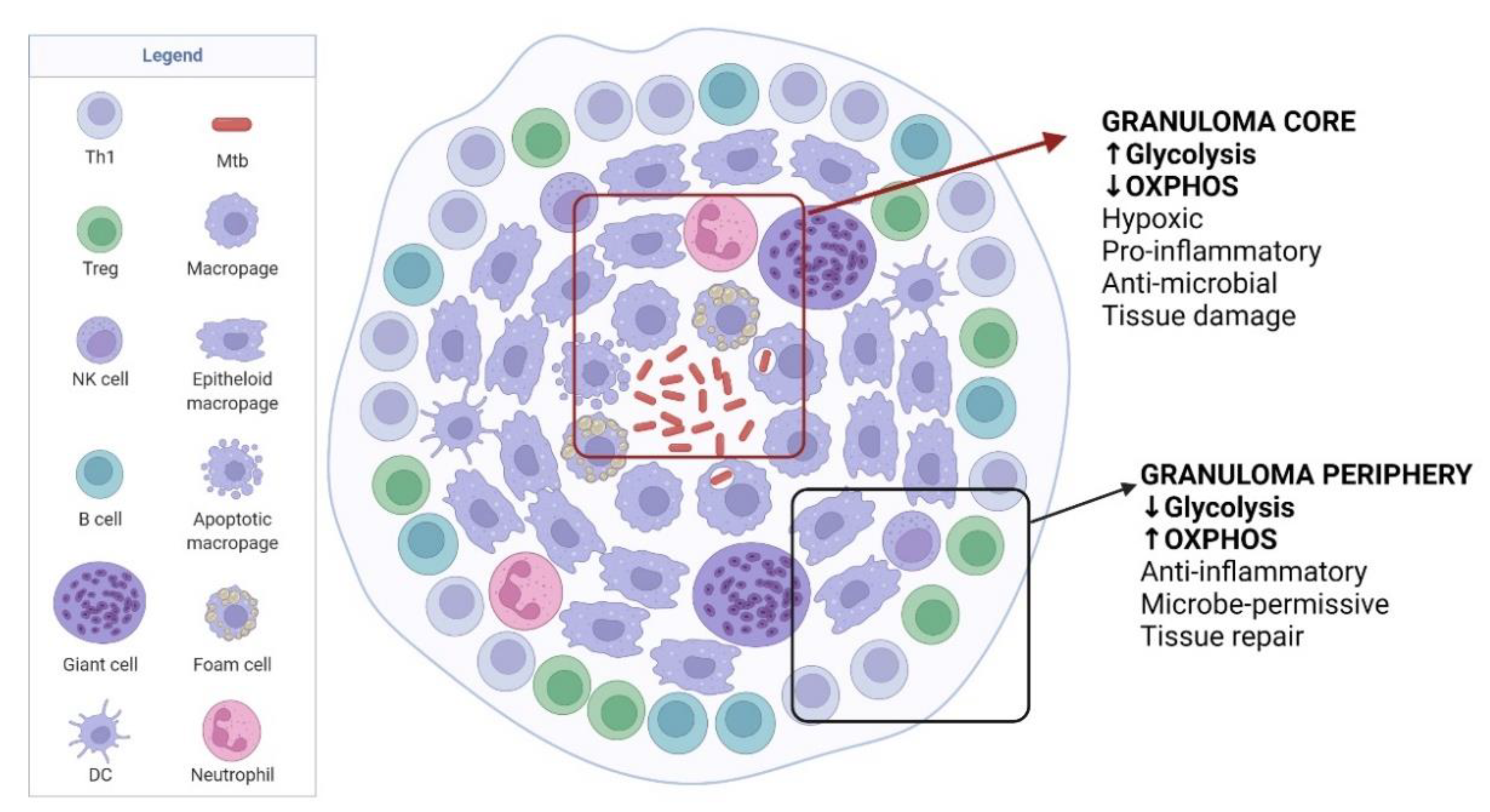
3. Immunometabolism during Chronic Phase of TB
3.1. Granuloma Formation
3.2. Foamy Macrophages
3.3. T Cells
3.4. Modulation of Granuloma for Mtb Dormancy
4. Modulation of Immunometabolism as Host Directed Therapy for TB Patients and Vaccination Strategy
5. A Possible Gut-Lung Axis in TB Protection
6. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AEC | Alveolar epithelial cell |
| AM | Alveolar macrophage |
| AMP | Adenosine monophosphate |
| CTLA-4 | Cytotoxic T-lymphocyte associated protein 4 |
| DC | Dendritic cell |
| FAO | Fatty acid oxidation |
| HIF-1α | Hypoxia-inducible factor 1α |
| IM | Interstitial macrophage |
| LPS | Lipopolysaccharide |
| M1 | Classically activated macrophage (pro-inflammatory phenotype) |
| M2 | Alternatively activated macrophage (anti-inflammatory phenotype) |
| MAIT | Mucosal-associated invariant T |
| Mtb | Mycobacterium tuberculosis |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| OXPHOS | Oxidative phosphorylation |
| TCR | T cell receptor |
References
- World Health Organization Global Tuberculosis Report 2021. Available online: https://www.who.int/publications/i/item/9789240037021 (accessed on 9 April 2022).
- Jeremiah, C.; Petersen, E.; Nantanda, R.; Mungai, B.N.; Migliori, G.B.; Amanullah, F.; Lungu, P.; Ntoumi, F.; Kumarasamy, N.; Maeurer, M.; et al. The WHO Global Tuberculosis 2021 Report—Not so Good News and Turning the Tide Back to End TB. Int. J. Infect. Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- Houben, R.M.G.J.; Dodd, P.J. The Global Burden of Latent Tuberculosis Infection: A Re-Estimation Using Mathematical Modelling. PLoS Med. 2016, 13, e1002152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; To, K.K.-W.; Wong, Y.-C.; Liu, L.; Zhou, B.; Li, X.; Huang, H.; Mo, Y.; Luk, T.-Y.; Lau, T.T.-K.; et al. Acute SARS-CoV-2 Infection Impairs Dendritic Cell and T Cell Responses. Immunity 2020, 53, 864–877.e5. [Google Scholar] [CrossRef]
- Winheimid, E.; Rinkeid, L.; Lutzid, K.; Reischer, A.; Leutbecher, A.; Wolfram, L.; Rausch, L.; Kranichid, J.; Wratil, P.R.; Huber, J.E.; et al. Impaired Function and Delayed Regeneration of Dendritic Cells in COVID-19. PLoS Pathog. 2021, 17, e1009742. [Google Scholar] [CrossRef]
- O’Garra, A.; Redford, P.S.; McNab, F.W.; Bloom, C.I.; Wilkinson, R.J.; Berry, M.P.R. The Immune Response in Tuberculosis. Annu. Rev. Immunol. 2013, 31, 475–527. [Google Scholar] [CrossRef] [PubMed]
- Pai, M.; Behr, M. Latent Mycobacterium tuberculosis Infection and Interferon-Gamma Release Assays. Microbiol. Spectr. 2016, 4, 4–5. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A Guide to Immunometabolism for Immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Mason, R.J. Biology of Alveolar Type II Cells. Respirology 2006, 11, S12–S15. [Google Scholar] [CrossRef]
- Ryndak, M.B.; Laal, S. Mycobacterium tuberculosis Primary Infection and Dissemination: A Critical Role for Alveolar Epithelial Cells. Front. Cell. Infect. Microbiol. 2019, 9, 299. [Google Scholar] [CrossRef]
- Scordo, J.M.; Knoell, D.L.; Torrelles, J.B. Alveolar Epithelial Cells in Mycobacterium tuberculosis Infection: Active Players or Innocent Bystanders? J. Innate Immun. 2016, 8, 3–14. [Google Scholar] [CrossRef]
- Ryndak, M.B.; Singh, K.K.; Peng, Z.; Laal, S. Transcriptional Profile of Mycobacterium tuberculosis Replicating in Type II Alveolar Epithelial Cells. PLoS ONE 2015, 10, e0123745. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Summer, R. Cellular Metabolism in Lung Health and Disease. Annu. Rev. Physiol. 2019, 81, 403. [Google Scholar] [CrossRef]
- Stallings, C.L.; Thacker, V.V.; Dhar, N.; Sharma, K.; Barrile, R.; Karalis, K.; McKinney, J.D. A Lung-on-Chip Model of Early Mycobacterium tuberculosis Infection Reveals an Essential Role for Alveolar Epithelial Cells in Controlling Bacterial Growth. eLife 2020, 9, e59961. [Google Scholar] [CrossRef]
- Takahashi, K.; Naito, M.; Takeya, M. Development and Heterogeneity of Macrophages and Their Related Cells through Their Differentiation Pathways. Pathol. Int. 1996, 46, 473–485. [Google Scholar] [CrossRef]
- Bowden, D.H. The Alveolar Macrophage. Environ. Health Perspect. 1984, 55, 327–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbi, N.; Lambrecht, B.N. Location, Function, and Ontogeny of Pulmonary Macrophages during the Steady State. Pflugers Arch. Eur. J. Physiol. 2017, 469, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The Integrin Alpha v Beta 6 Binds and Activates Latent TGF Beta 1: A Mechanism for Regulating Pulmonary Inflammation and Fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Coleman, M.M.; Ruane, D.; Moran, B.; Dunne, P.J.; Keane, J.; Mills, K.H.G. Alveolar Macrophages Contribute to Respiratory Tolerance by Inducing FoxP3 Expression in Naive T Cells. Am. J. Respir. Cell Mol. Biol. 2013, 48, 773–780. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Divangahi, M. Targeting Immunometabolism in Host Defence against Mycobacterium tuberculosis. Immunology 2021, 162, 145–159. [Google Scholar] [CrossRef]
- Chai, Q.; Wang, X.; Qiang, L.; Zhang, Y.; Ge, P.; Lu, Z.; Zhong, Y.; Li, B.; Wang, J.; Zhang, L.; et al. A Mycobacterium tuberculosis Surface Protein Recruits Ubiquitin to Trigger Host Xenophagy. Nat. Commun. 2019, 10, 1973. [Google Scholar] [CrossRef]
- Chandra, P.; He, L.; Zimmerman, M.; Yang, G.; Köster, S.; Ouimet, M.; Wang, H.; Moore, K.J.; Dartois, V.; Schilling, J.D.; et al. Inhibition of Fatty Acid Oxidation Promotes Macrophage Control of Mycobacterium tuberculosis. mBio 2020, 11, e01139-20. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Eugenin, E.A.; Subbian, S. Immunometabolism in Tuberculosis. Front. Immunol. 2016, 7, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleeson, L.E.; Sheedy, F.J.; Palsson-McDermott, E.M.; Triglia, D.; O’Leary, S.M.; O’Sullivan, M.P.; O’Neill, L.A.J.; Keane, J. Cutting Edge: Mycobacterium tuberculosis Induces Aerobic Glycolysis in Human Alveolar Macrophages That Is Required for Control of Intracellular Bacillary Replication. J. Immunol. 2016, 196, 2444–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackett, E.E.; Charles-Messance, H.; O’Leary, S.M.; Gleeson, L.E.; Muñoz-Wolf, N.; Case, S.; Wedderburn, A.; Johnston, D.G.W.; Williams, M.A.; Smyth, A.; et al. Mycobacterium tuberculosis Limits Host Glycolysis and IL-1β by Restriction of PFK-M via MicroRNA-21. Cell Rep. 2020, 30, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Cumming, B.M.; Addicott, K.W.; Pacl, H.T.; Russell, S.L.; Nargan, K.; Naidoo, T.; Ramdial, P.K.; Adamson, J.H.; Wang, R.; et al. Hydrogen Sulfide Dysregulates the Immune Response by Suppressing Central Carbon Metabolism to Promote Tuberculosis. Proc. Natl. Acad. Sci. USA 2020, 117, 6663–6674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Singh, P.; Kolloli, A.; Shi, L.; Bushkin, Y.; Tyagi, S.; Subbian, S. Immunometabolism of Phagocytes During Mycobacterium tuberculosis Infection. Front. Mol. Biosci. 2019, 6, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, S.; Murphy, A.N.; Brown, J.H. Akt Mediates Mitochondrial Protection in Cardiomyocytes through Phosphorylation of Mitochondrial Hexokinase-II. Cell Death Differ. 2008, 15, 521–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrofsky, M.; Bermudez, L.E. Neutrophils from Mycobacterium Avium-Infected Mice Produce TNF-Alpha, IL-12, and IL-1 Beta and Have a Putative Role in Early Host Response. Clin. Immunol. 1999, 91, 354–358. [Google Scholar] [CrossRef]
- Seiler, P.; Aichele, P.; Bandermann, S.; Hauser, A.E.; Lu, B.; Gerard, N.P.; Gerard, C.; Ehlers, S.; Mollenkopf, H.J.; Kaufmann, S.H.E. Early Granuloma Formation after Aerosol Mycobacterium Tuberculosis Infection Is Regulated by Neutrophils via CXCR3-Signaling Chemokines. Eur. J. Immunol. 2003, 33, 2676–2686. [Google Scholar] [CrossRef]
- Ong, C.W.M.; Fox, K.; Ettorre, A.; Elkington, P.T.; Friedland, J.S. Hypoxia Increases Neutrophil-Driven Matrix Destruction after Exposure to Mycobacterium tuberculosis. Sci. Rep. 2018, 8, 11475. [Google Scholar] [CrossRef]
- Steinwede, K.; Maus, R.; Bohling, J.; Voedisch, S.; Braun, A.; Ochs, M.; Schmiedl, A.; Länger, F.; Gauthier, F.; Roes, J.; et al. Cathepsin G and Neutrophil Elastase Contribute to Lung-Protective Immunity against Mycobacterial Infections in Mice. J. Immunol. 2012, 188, 4476–4487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ussher, J.E.; Klenerman, P.; Willberg, C.B. Mucosal-Associated Invariant T-Cells: New Players in Anti-Bacterial Immunity. Front. Immunol. 2014, 5, 450. [Google Scholar] [CrossRef] [PubMed]
- Vorkas, C.K.; Wipperman, M.F.; Li, K.; Bean, J.; Bhattarai, S.K.; Adamow, M.; Wong, P.; Aubé, J.; Juste, M.A.J.; Bucci, V.; et al. Mucosal-Associated Invariant and Γδ T Cell Subsets Respond to Initial Mycobacterium tuberculosis Infection. JCI Insight 2018, 3, e121899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nel, I.; Bertrand, L.; Toubal, A.; Lehuen, A. MAIT Cells, Guardians of Skin and Mucosa? Mucosal Immunol. 2021, 14, 803–814. [Google Scholar] [CrossRef]
- Wong, E.B.; Gold, M.C.; Meermeier, E.W.; Xulu, B.Z.; Khuzwayo, S.; Sullivan, Z.A.; Mahyari, E.; Rogers, Z.; Kløverpris, H.; Sharma, P.K.; et al. TRAV1-2+ CD8+ T-Cells Including Oligoconal Expansions of MAIT Cells Are Enriched in the Airways in Human Tuberculosis. Commun. Biol. 2019, 2, 203. [Google Scholar] [CrossRef]
- Balfour, A.; Schutz, C.; Goliath, R.; Wilkinson, K.A.; Sayed, S.; Sossen, B.; Kanyik, J.P.; Ward, A.; Ndzhukule, R.; Gela, A.; et al. Functional and Activation Profiles of Mucosal-Associated Invariant T Cells in Patients with Tuberculosis and HIV in a High Endemic Setting. Front. Immunol. 2021, 12, 648216. [Google Scholar] [CrossRef]
- Vorkas, C.K.; Levy, O.; Skular, M.; Li, K.; Aubé, J.; Glickman, M.S. Efficient 5-OP-RU-Induced Enrichment of Mucosa-Associated Invariant t Cells in the Murine Lung Does Not Enhance Control of Aerosol Mycobacterium tuberculosis Infection. Infect. Immun. 2021, 89, e00524-20. [Google Scholar] [CrossRef]
- Zinser, M.E.; Highton, A.J.; Kurioka, A.; Kronsteiner, B.; Hagel, J.; Leng, T.; Marchi, E.; Phetsouphanh, C.; Willberg, C.B.; Dunachie, S.J.; et al. Human MAIT Cells Show Metabolic Quiescence with Rapid Glucose-Dependent Upregulation of Granzyme B upon Stimulation. Immunol. Cell Biol. 2018, 96, 666–674. [Google Scholar] [CrossRef] [Green Version]
- Amini, A.; Pang, D.; Hackstein, C.-P.; Klenerman, P. MAIT Cells in Barrier Tissues: Lessons from Immediate Neighbors. Front. Immunol. 2020, 11, 584521. [Google Scholar] [CrossRef]
- Lu, B.; Liu, M.; Wang, J.; Fan, H.; Yang, D.; Zhang, L.; Gu, X.; Nie, J.; Chen, Z.; Corbett, A.J.; et al. IL-17 Production by Tissue-Resident MAIT Cells Is Locally Induced in Children with Pneumonia. Mucosal Immunol. 2020, 13, 824–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, D.O. The Granulomatous Inflammatory Response. A Review. Am. J. Pathol. 1976, 84, 164–192. [Google Scholar]
- Petersen, H.J.; Smith, A.M. The Role of the Innate Immune System in Granulomatous Disorders. Front. Immunol. 2013, 4, 120. [Google Scholar] [CrossRef] [Green Version]
- McClean, C.M.; Tobin, D.M. Macrophage Form, Function, and Phenotype in Mycobacterial Infection: Lessons from Tuberculosis and Other Diseases. Pathog. Dis. 2016, 74, ftw068. [Google Scholar] [CrossRef] [Green Version]
- Ehlers, S.; Schaible, U.E. The Granuloma in Tuberculosis: Dynamics of a Host-Pathogen Collusion. Front. Immunol. 2012, 3, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orme, I.M.; Basaraba, R.J. The Formation of the Granuloma in Tuberculosis Infection. Semin. Immunol. 2014, 26, 601–609. [Google Scholar] [CrossRef]
- Singh, V.; Kaur, C.; Chaudhary, V.K.; Rao, K.V.S.; Chatterjee, S.M. Tuberculosis Secretory Protein ESAT-6 Induces Metabolic Flux Perturbations to Drive Foamy Macrophage Differentiation. Sci. Rep. 2015, 5, 12906. [Google Scholar] [CrossRef]
- Mahajan, S.; Dkhar, H.K.; Chandra, V.; Dave, S.; Nanduri, R.; Janmeja, A.K.; Agrewala, J.N.; Gupta, P. Mycobacterium tuberculosis Modulates Macrophage Lipid-Sensing Nuclear Receptors PPARγ and TR4 for Survival. J. Immunol. 2012, 188, 5593–5603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer-Barber, K.D.; Sher, A. Cytokine and Lipid Mediator Networks in Tuberculosis. Immunol. Rev. 2015, 264, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T Cell Metabolism Drives Immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4 + T Cell Subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.L.; Lamprecht, D.A.; Mandizvo, T.; Jones, T.T.; Naidoo, V.; Addicott, K.W.; Moodley, C.; Ngcobo, B.; Crossman, D.K.; Wells, G.; et al. Compromised Metabolic Reprogramming Is an Early Indicator of CD8+ T Cell Dysfunction during Chronic Mycobacterium tuberculosis Infection. Cell Rep. 2019, 29, 3564–3579. [Google Scholar] [CrossRef] [Green Version]
- Marakalala, M.J.; Raju, R.M.; Sharma, K.; Zhang, Y.J.; Eugenin, E.A.; Prideaux, B.; Daudelin, I.B.; Chen, P.Y.; Booty, M.G.; Kim, J.H.; et al. Inflammatory Signaling in Human Tuberculosis Granulomas Is Spatially Organized. Nat. Med. 2016, 22, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate Is an Inflammatory Signal That Induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Shi, L.; Salamon, H.; Eugenin, E.A.; Pine, R.; Cooper, A.; Gennaro, M.L. Infection with Mycobacterium tuberculosis Induces the Warburg Effect in Mouse Lungs. Sci. Rep. 2015, 5, 18176. [Google Scholar] [CrossRef]
- Wayne, L.G. Dormancy of Mycobacterium tuberculosis and Latency of Disease. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 1994, 13, 908–914. [Google Scholar] [CrossRef]
- World Health Organization. WHO Consolidated Guidelines on Tuberculosis. Module 1: Prevention—Tuberculosis Preventive Treatment. Available online: https://apps.who.int/iris/bitstream/handle/10665/331170/9789240001503-eng.pdf (accessed on 3 May 2022).
- Sterling, T.R.; Villarino, M.E.; Borisov, A.S.; Shang, N.; Gordin, F.; Bliven-Sizemore, E.; Hackman, J.; Hamilton, C.D.; Menzies, D.; Kerrigan, A.; et al. Three Months of Rifapentine and Isoniazid for Latent Tuberculosis Infection. N. Engl. J. Med. 2011, 365, 2155–2166. [Google Scholar] [CrossRef] [Green Version]
- Bliven-Sizemore, E.E.; Sterling, T.R.; Shang, N.; Benator, D.; Schwartzman, K.; Reves, R.; Drobeniuc, J.; Bock, N.; Villarino, M.E. Three Months of Weekly Rifapentine plus Isoniazid Is Less Hepatotoxic than Nine Months of Daily Isoniazid for LTBI. Int. J. Tuberc. Lung Dis. Off. J. Int. Union Against Tuberc. Lung Dis. 2015, 19, 1039–1044. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-Y.; Chiu, Y.-W.; Lu, P.-L.; Hwang, S.-J.; Chen, T.-C.; Hsieh, M.-H.; Chen, Y.-H. Three Months of Rifapentine and Isoniazid for Latent Tuberculosis Infection in Hemodialysis Patients: High Rates of Adverse Events. J. Microbiol. Immunol. Infect. 2019, 52, 158–162. [Google Scholar] [CrossRef]
- Kundu, M.; Basu, J. Applications of Transcriptomics and Proteomics for Understanding Dormancy and Resuscitation in Mycobacterium tuberculosis. Front. Microbiol. 2021, 12, 642487. [Google Scholar] [CrossRef]
- Phelan, J.J.; McQuaid, K.; Kenny, C.; Gogan, K.M.; Cox, D.J.; Basdeo, S.A.; O’Leary, S.; Tazoll, S.C.; Ó Maoldomhnaigh, C.; O’Sullivan, M.P.; et al. Desferrioxamine Supports Metabolic Function in Primary Human Macrophages Infected with Mycobacterium tuberculosis. Front. Immunol. 2020, 11, 836. [Google Scholar] [CrossRef]
- Cox, D.J.; Coleman, A.M.; Gogan, K.M.; Phelan, J.J.; Maoldomhnaigh, C.Ó.; Dunne, P.J.; Basdeo, S.A.; Keane, J. Inhibiting Histone Deacetylases in Human Macrophages Promotes Glycolysis, IL-1β, and T Helper Cell Responses to Mycobacterium tuberculosis. Front. Immunol. 2020, 11, 1609. [Google Scholar] [CrossRef]
- Böhme, J.; Martinez, N.; Li, S.; Lee, A.; Marzuki, M.; Tizazu, A.M.; Ackart, D.; Frenkel, J.H.; Todd, A.; Lachmandas, E.; et al. Metformin Enhances Anti-Mycobacterial Responses by Educating CD8+ T-Cell Immunometabolic Circuits. Nat. Commun. 2020, 11, 5225. [Google Scholar] [CrossRef]
- Hotez, P.J.; Brindley, P.J.; Bethony, J.M.; King, C.H.; Pearce, E.J.; Jacobson, J. Helminth Infections: The Great Neglected Tropical Diseases. J. Clin. Investig. 2008, 118, 1311–1321. [Google Scholar] [CrossRef] [Green Version]
- Babu, S.; Nutman, T.B. Helminth-Tuberculosis Co-Infection: An Immunologic Perspective. Trends Immunol. 2016, 37, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Toulza, F.; Tsang, L.; Ottenhoff, T.H.M.; Brown, M.; Dockrell, H.M. Mycobacterium tuberculosis-Specific CD4+ T-Cell Response Is Increased, and Treg Cells Decreased, in Anthelmintic-Treated Patients with Latent TB. Eur. J. Immunol. 2016, 46, 752–761. [Google Scholar] [CrossRef] [Green Version]
- Elias, D.; Britton, S.; Aseffa, A.; Engers, H.; Akuffo, H. Poor Immunogenicity of BCG in Helminth Infected Population Is Associated with Increased in Vitro TGF-Beta Production. Vaccine 2008, 26, 3897–3902. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Selva, D.; Fairfax, K. Schistosome and Intestinal Helminth Modulation of Macrophage Immunometabolism. Immunology 2021, 162, 123–134. [Google Scholar] [CrossRef]
- Dumas, A.; Corral, D.; Colom, A.; Levillain, F.; Peixoto, A.; Hudrisier, D.; Poquet, Y.; Neyrolles, O. The Host Microbiota Contributes to Early Protection against Lung Colonization by Mycobacterium tuberculosis. Front. Immunol. 2018, 9, 2656. [Google Scholar] [CrossRef]
- Qin, Z.; Yang, X.; Chen, G.; Park, C.; Liu, Z. Crosstalks between Gut Microbiota and Vibrio Cholerae. Front. Cell. Infect. Microbiol. 2020, 10, 582554. [Google Scholar] [CrossRef]
- Leung, D.T.; Bhuiyan, T.R.; Nishat, N.S.; Hoq, M.R.; Aktar, A.; Rahman, M.A.; Uddin, T.; Khan, A.I.; Chowdhury, F.; Charles, R.C.; et al. Circulating Mucosal Associated Invariant T Cells Are Activated in Vibrio Cholerae O1 Infection and Associated with Lipopolysaccharide Antibody Responses. PLoS Negl. Trop. Dis. 2014, 8, e3076. [Google Scholar] [CrossRef]
- Comas, I.; Coscolla, M.; Luo, T.; Borrell, S.; Holt, K.E.; Kato-Maeda, M.; Parkhill, J.; Malla, B.; Berg, S.; Thwaites, G.; et al. Out-of-Africa Migration and Neolithic Coexpansion of Mycobacterium tuberculosis with Modern Humans. Nat. Genet. 2013, 45, 1176–1182. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tukiman, M.H.; Norazmi, M.N. Immunometabolism of Immune Cells in Mucosal Environment Drives Effector Responses against Mycobacterium tuberculosis. Int. J. Mol. Sci. 2022, 23, 8531. https://doi.org/10.3390/ijms23158531
Tukiman MH, Norazmi MN. Immunometabolism of Immune Cells in Mucosal Environment Drives Effector Responses against Mycobacterium tuberculosis. International Journal of Molecular Sciences. 2022; 23(15):8531. https://doi.org/10.3390/ijms23158531
Chicago/Turabian StyleTukiman, Mohd Hatimi, and Mohd Nor Norazmi. 2022. "Immunometabolism of Immune Cells in Mucosal Environment Drives Effector Responses against Mycobacterium tuberculosis" International Journal of Molecular Sciences 23, no. 15: 8531. https://doi.org/10.3390/ijms23158531
APA StyleTukiman, M. H., & Norazmi, M. N. (2022). Immunometabolism of Immune Cells in Mucosal Environment Drives Effector Responses against Mycobacterium tuberculosis. International Journal of Molecular Sciences, 23(15), 8531. https://doi.org/10.3390/ijms23158531