
Abnormal mTOR Activity in Pediatric Autoimmune Neuropsychiatric and MIA-Associated Autism Spectrum Disorders


 and
and
Abstract
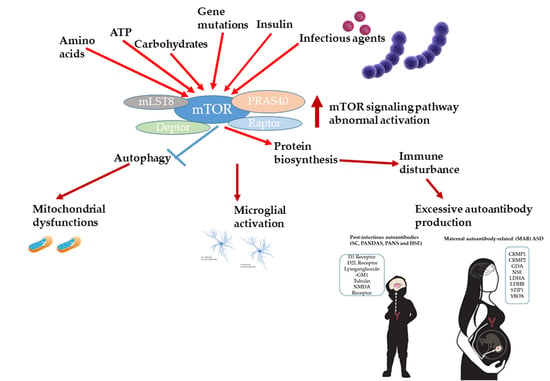
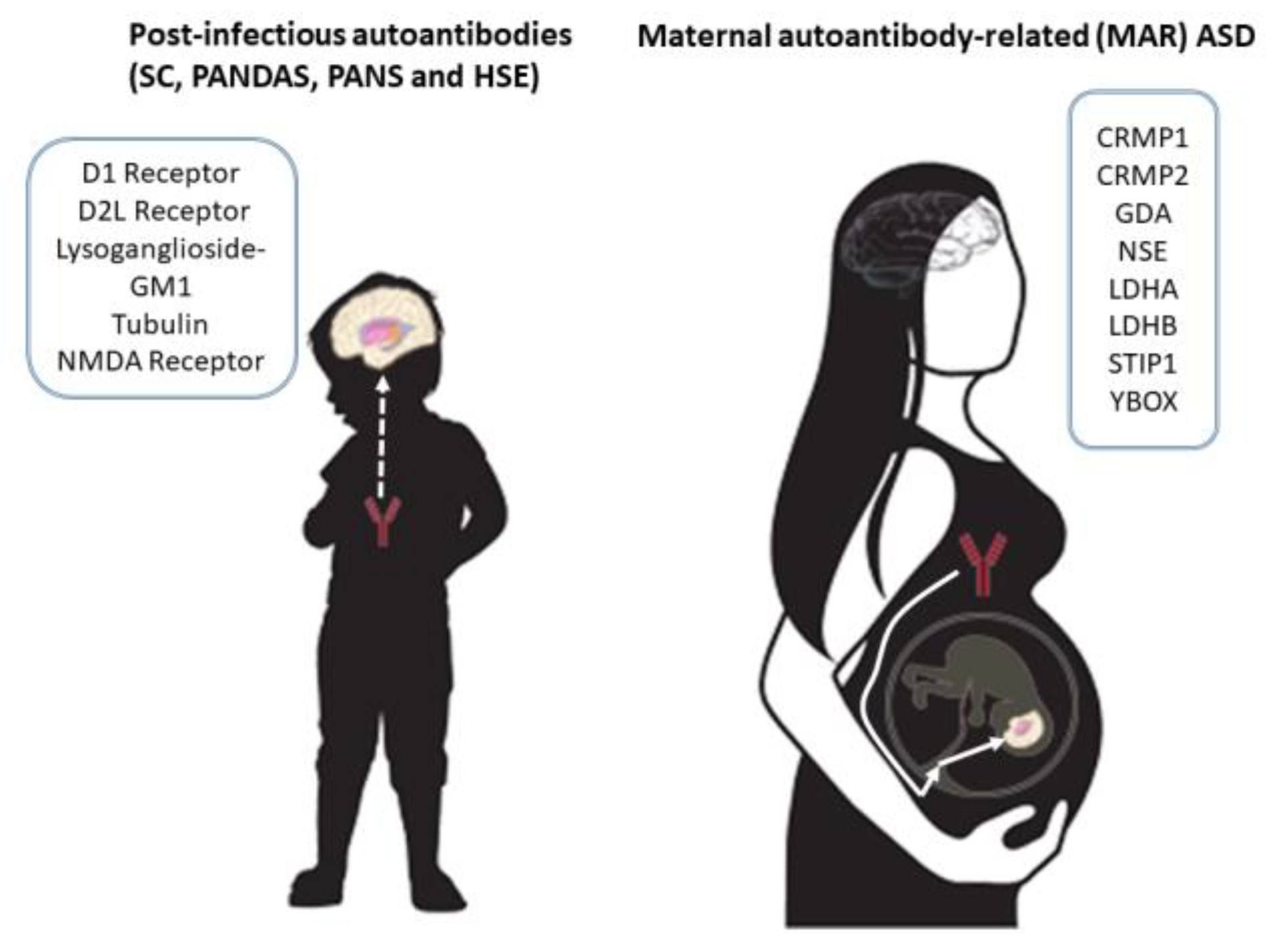
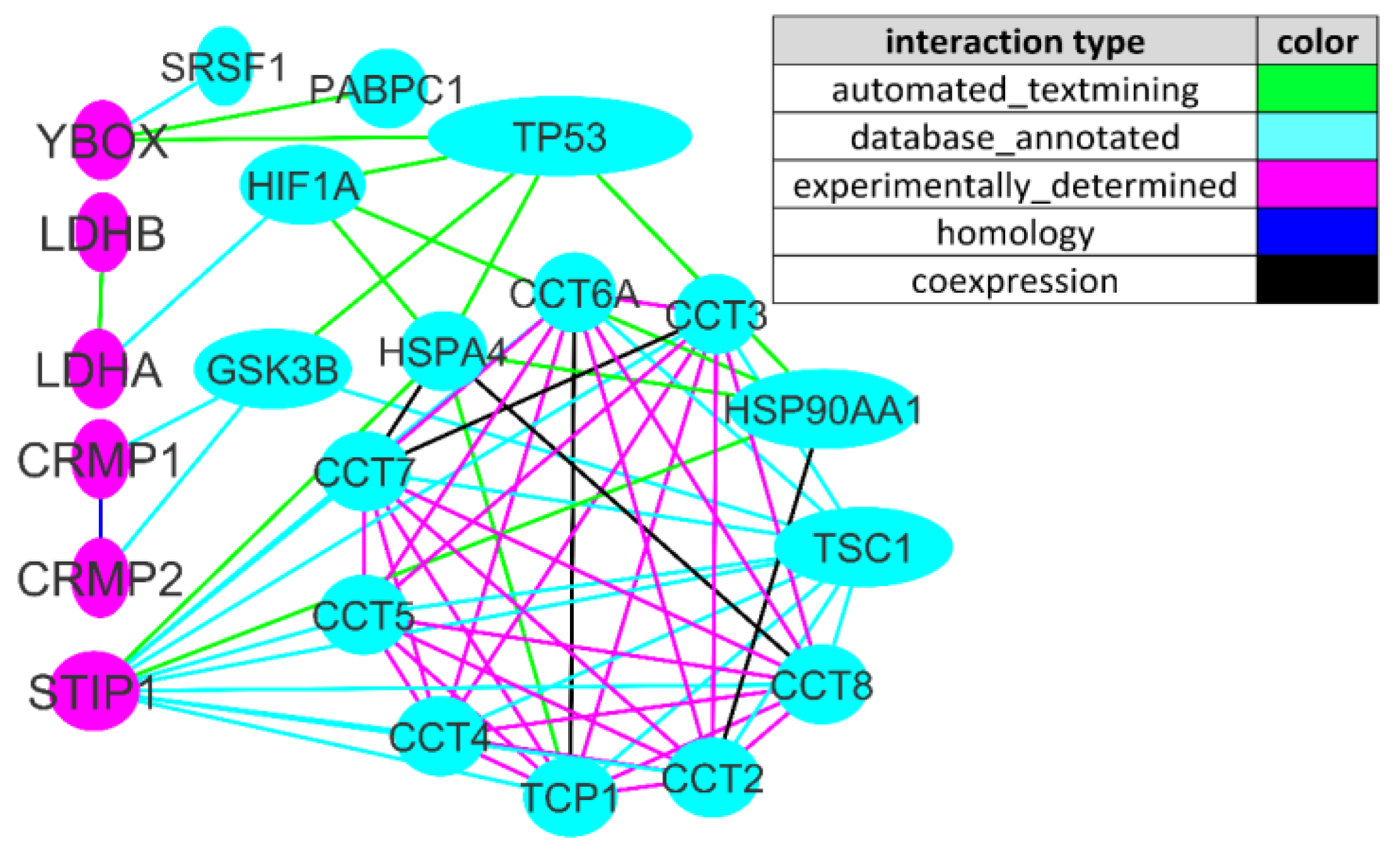
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Maternal Immune Activation (MIA)-Associated ASD
3. Post-Infectious Autoimmune Neuropsychiatric Disorders: Sydenham’s Chorea, PANDAS, PANS, and Herpes Simplex Encephalitis
4. mTOR Signaling as a Key Player in Immune, Microbiome, and Behavior Abnormalities in ASD
5. Challenges, Limitations and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Center for Disease Control and Prevention. Available online: https://www.cdc.gov/ncbddd/autism/data.html (accessed on 25 September 2020).
- Taylor, M.J.; Rosenqvist, M.A.; Larsson, H.; Gillberg, C.; D’Onofrio, B.M.; Lichtenstein, P.; Lundström, S. Etiology of Autism Spectrum Disorders and Autistic Traits Over Time. JAMA Psychiatry 2020, 77, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, B.S.; Arking, D.E.; Campbell, D.B.; Meord, H.C.; Morrow, E.M.; Weiss, L.A.; Menashe, I.; Wadkins, T.; Banerjee-Basu, S.; Packer, A. SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 2013, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H. Genetics of autism spectrum disorder: Current status and possible clinical applications. Exp. Neurobiol. 2015, 24, 257–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winden, K.D.; Ebrahimi-Fakhari, D.; Sahin, M. Abnormal mTOR Activation in Autism. Annu. Rev. Neurosci. 2018, 41, 1–23. [Google Scholar] [CrossRef]
- Bockaert, J.; Marin, P. mTOR in Brain Physiology and Pathologies. Physiol. Rev. 2015, 95, 1157–1187. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Bear, M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, a009886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onore, C.; Yang, H.; van de Water, J.; Ashwood, P. Dynamic Akt/mTOR Signaling in Children with Autism Spectrum Disorder. Front. Pediatr. 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tylee, D.S.; Hess, J.L.; Quinn, T.P.; Barve, R.; Huang, H.; Zhang-James, Y.; Chang, J.; Stamova, B.S.; Sharp, F.R.; Hertz-Picciotto, I.; et al. Blood transcriptomic comparison of individuals with and without autism spectrum disorder: A combined-samples mega-analysis. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017, 174, 181–201. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.-Y.; Xu, L.-L.; Shao, L.; Xia, R.-M.; Yu, Z.-H.; Ling, Z.; Yang, F.; Deng, M.; Ruan, B. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain Behav. Immun. 2016, 58, 165–172. [Google Scholar] [CrossRef]
- Lee, B.K.; Magnusson, C.; Gardner, R.M.; Blomstrom, A.; Newschaffer, C.J.; Burstyn, I.; Karlsson, H.; Dalman, C. Maternal hospitalization with infection during pregnancy and risk of autism spectrum disorders. Brain Behav. Immun. 2015, 44, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Meyer, U. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol. Psychiatry 2014, 75, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Oskvig, D.B.; Elkahloun, A.G.; Johnson, K.R.; Phillips, T.M.; Herkenham, M. Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain Behav. Immun. 2012, 26, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin- 17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetto, M.C.; Belinson, H.; Tian, Y.; Freitas, B.C.; Fu, C.; Vadodaria, K.; Beltrão-Braga, P.; Trujillo, C.A.; Mendes, A.P.; Padmanabhan, K.; et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol. Psychiatry 2017, 22, 820–835. [Google Scholar] [CrossRef]
- Smith, S.E.; Elliott, R.M.; Anderson, M.P. Maternal immune activation increases neonatal mouse cortex thickness and cell density. J. Neuroimmune Pharmacol. 2012, 7, 529–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Belle, J.E.; Sperry, J.; Ngo, A.; Ghochani, Y.; Laks, D.R.; López-Aranda, M.; Silva, A.J.; Kornblum, H.I. Maternal inflammation contributes to brain overgrowth and autism-associated behaviors through altered redox signaling in stem and progenitor cells. Stem Cell Rep. 2014, 3, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovanoli, S.; Engler, H.; Engler, A.; Richetto, J.; Voget, M.; Willi, R.; Winter, C.; Riva, M.A.; Mortensen, P.B.; Feldon, J.; et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science 2013, 339, 1095–1099. [Google Scholar] [CrossRef] [Green Version]
- Richetto, J.; Calabrese, F.; Riva, M.A.; Meyer, U. Prenatal immune activation induces maturation-dependent alterations in the prefrontal GABAergic transcriptome. Schizophr. Bull. 2014, 40, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Weir, R.K.; Forghany, R.; Smith, S.E.; Patterson, P.H.; McAllister, A.K.; Schumann, C.M.; Bauman, M.D. Preliminary evidence of neuropathology in nonhuman primates prenatally exposed to maternal immune activation. Brain Behav. Immun. 2015, 48, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, M.V.; Moon, H.M.; Su, J.; Palmer, T.D.; Courchesne, E.; Pramparo, T. Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol. Psychiatry. 2018, 23, 1001–1013. [Google Scholar] [CrossRef] [Green Version]
- Ehninger, D.; Sano, Y.; De Vries, P.J.; Dies, K.; Franz, D.; Geschwind, D.H.; Kaur, M.; Lee, Y.-S.; Li, W.; Lowe, J.K.; et al. Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Mol. Psychiatry 2012, 17, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, D.; Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Croen, L.A.; Pessah, I.N.; Van De Water, J. Autism: Maternally derived antibodies specific for fetal brain proteins. Neurotoxicology 2008, 29, 226–231. [Google Scholar] [CrossRef] [Green Version]
- Braunschweig, D.; Duncanson, P.; Boyce, R.; Hansen, R.; Ashwood, P.; Pessah, I.N.; Hertz-Picciotto, I.; Van De Water, J. Behavioral correlates of maternal antibody status among children with autism. J. Autism Dev. Disord. 2012, 42, 1435–1445. [Google Scholar] [CrossRef] [Green Version]
- Dalton, P.; Deacon, R.; Blamire, A.; Pike, M.; McKinlay, I.; Stein, J.; Styles, P.; Vincent, A. Maternal neuronal antibodies associated with autism and a language disorder. Ann. Neurol. 2003, 53, 533–537. [Google Scholar] [CrossRef]
- Martínez-Cerdeño, V.; Camacho, J.; Fox, E.; Miller, E.; Ariza, J.; Kienzle, D.; Plank, K.; Noctor, S.C.; Van De Water, J. Prenatal exposure to autism-specific maternal autoantibodies alters proliferation of cortical neural precursor cells, enlarges brain, and increases neuronal size in adult animals. Cereb. Cortex 2016, 26, 374–383. [Google Scholar] [CrossRef]
- Reed, M.D.; Yim, Y.S.; Wimmer, R.D.; Kim, H.; Ryu, C.; Welch, G.M.; Andina, M.; King, H.O.; Waisman, A.; Halassa, M.M.; et al. IL-17a promotes sociability in mouse models of neurodevelopmental disorders. Nature 2020, 577, 249–253. [Google Scholar] [CrossRef]
- Ramirez-Celis, A.; Becker, M.; Nuño, M.; Schauer, J.; Aghaeepour, N.; Van de Water, J. Risk assessment analysis for maternal autoantibody-related autism (MAR-ASD): A subtype of autism. Mol. Psychiatry 2021, 26, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, A.; Van de Water, J. The role of the immune system in autism spectrum disorder. Neuropsychopharmacology 2017, 42, 284–298. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.A.; Swedo, S.E. Post-infectious autoimmune disorders: Sydenham’s chorea, PANDAS and beyond. Brain Res. 2015, 1617, 144–154. [Google Scholar] [CrossRef] [PubMed]
- MCPAP News. Available online: https://www.mcpap.com/pdf/Vol19Feb19MCPAPNews.pdf (accessed on 19 February 2019).
- Autism Speaks. Available online: https://www.autismspeaks.org/expert-opinion/what-pandas-how-it-different-autism (accessed on 4 April 2014).
- Goncalves, M.V.M.; Harger, R.; Braatz, V.; Parolin, L.F.; Eboni, A.C.B.; Fontana, M.A.N.; Anacleto, A.; Fragoso, Y.D. Pediatric acute-onset neuropsychiatric syndrome (PANS) misdiagnosed as autism spectrum disorder. Immunol. Lett. 2018, 203, 52–53. [Google Scholar] [CrossRef]
- Swedo, S.E.; Leonard, H.L.; Mittleman, B.B.; Allen, A.J.; Rapoport, J.L.; Dow, S.P.; Kanter, M.E.; Chapman, F.; Zabriskie, J. Identification of children with pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections by a marker associated with rheumatic fever. Am. J. Psychiatry. 1997, 154, 110–112. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.W. Streptococcus and rheumatic fever. Curr. Opin. Rheumatol. 2012, 24, 408–416. [Google Scholar] [CrossRef]
- Cunningham, M.W. Rheumatic fever, autoimmunity, and molecular mimicry: The streptococcal connection. Int. Rev. Immunol. 2014, 33, 314–329. [Google Scholar] [CrossRef] [Green Version]
- Shimasaki, C.; Frye, R.E.; Trifiletti, R.; Cooperstock, M.; Kaplan, G.; Melamed, I.; Greenberg, R.; Katz, A.; Fier, E.; Kem, D.; et al. Evaluation of the Cunningham Panel™ in pediatric autoimmune neuropsychiatric disorder associated with streptococcal infection (PANDAS) and pediatric acute-onset neuropsychiatric syndrome (PANS): Changes in antineuronal antibody titers parallel changes in patient symptoms. J. Neuroimmunol. 2020, 339, 577138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammad, S.S.; Sinclair, K.; Pillai, S.; Merheb, V.; Aumann, T.D.; Gill, D.; Dale, R.C.; Brilot, F. Herpes simplex encephalitis relapse with chorea is associated with autoantibodies to N-Methyl-D-aspartate receptor or dopamine-2 receptor. Mov. Disord. 2014, 29, 117–122. [Google Scholar] [CrossRef]
- Dalmau, J.; Lancaster, E.; Martinez-Hernandez, E.; Rosenfeld, M.R.; Balice-Gordon, R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011, 10, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Creten, C.; van der Zwaan, S.; Blankespoor, R.J.; Maatkamp, A.; Nicolai, J.; van Os, J.; Schieveld, J.N. Late onset autism and anti-NMDA-receptor encephalitis. Lancet 2011, 378, 98. [Google Scholar] [CrossRef]
- Hacohen, Y.; Wright, S.; Gadian, J.; Vincent, A.; Lim, M.; Wassmer, E.; Lin, J.P. N-methyl-d-aspartate (NMDA) receptor antibodies encephalitis mimicking an autistic regression. Dev. Med. Child Neurol. 2016, 58, 1092–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, S.; Walsh, C.; Hacohen, Y.; Muscal, E.; Jankovic, J.; Stocco, A.; Dale, R.C.; Vincent, A.; Lim, M.; King, M. Earlier treatment of NMDAR antibody encephalitis in children results in a better outcome. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e130. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.; Saha, B.; Riley, J.L. The Battle over mTOR: An Emerging Theatre in Host–Pathogen Immunity. PLoS Pathog. 2012, 8, e1002894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuluunbaatar, U.; Roller, R.; Feldman, M.E.; Brown, S.; Shokat, K.M.; Mohr, I. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev. 2010, 24, 2627–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Zou, X.; Feng, P.; Zhan, H.; Xiong, D.; Lang, J. Inhibition of the PI3K/AKT Signaling Pathway or Overexpression of Beclin1 Blocks Reinfection of Streptococcus pneumoniae After Infection of Influenza A Virus in Severe Community-Acquired Pneumonia. Inflammation 2019, 42, 1741–1753. [Google Scholar] [CrossRef]
- Cheng, Y.-L.; Kuo, C.-F.; Lu, S.-L.; Hiroko, O.; Wu, Y.-N.; Hsieh, C.-L.; Noda, T.; Wu, S.-R.; Anderson, R.; Lin, C.-F.; et al. Group A Streptococcus Induces LAPosomes via SLO/β1 Integrin/NOX2/ROS Pathway in Endothelial Cells That Are Ineffective in Bacterial Killing and Suppress Xenophagy. mBio 2019, 10, e02148-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Meng, M.; Li, M.; Guan, X.; Liu, J.; Gao, X.; Sun, Q.; Li, J.; Ma, C.; Wei, L. Integrin α5β1, as a Receptor of Fibronectin, Binds the FbaA Protein of Group A Streptococcus To Initiate Autophagy during Infection. mBio 2020, 11, e00771-20. [Google Scholar] [CrossRef]
- Toh, H.; Nozawa, T.; Minowa-Nozawa, A.; Hikichi, M.; Nakajima, S.; Aikawa, C.; Nakagawa, I. Group A Streptococcus modulates RAB1- and PIK3C3 complex-dependent autophagy. Autophagy 2020, 2, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, S.; Huston, W.M.; Johnson, M.; Tiberti, N.; Saunders, B.; O’Brien, B.; Burke, C.; Labbate, M.; Combes, V. Targeting the master regulator mTOR: A new approach to prevent the neurological of consequences of parasitic infections? Parasites Vectors 2017, 10, 581. [Google Scholar] [CrossRef] [Green Version]
- Gordon, E.B.; Hart, G.T.; Tran, T.; Waisberg, M.; Akkaya, M.; Skinner, J.; Zinöcker, S.; Pena, M.; Yazew, T.; Qi, C.-F.; et al. Inhibiting the Mammalian target of rapamycin blocks the development of experimental cerebral malaria. mBio 2015, 6, e00725. [Google Scholar] [CrossRef] [Green Version]
- Shuid, A.N.; Jayusman, P.A.; Shuid, N.; Ismail, J.; Kamal Nor, N.; Naina Mohamed, I. Update on Atypicalities of Central Nervous System in Autism Spectrum Disorder. Brain Sci. 2020, 10, 309. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, D.T.; Liu, X.G. mTOR signaling in T cell immunity and autoimmunity. Int. Rev. Immunol. 2015, 34, 50–66. [Google Scholar] [CrossRef]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 andmTORC2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Strauss, L.; Whiteside, T.L.; Knights, A.; Bergmann, C.; Knuth, A.; Zippelius, A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J. Immunol. 2007, 178, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zhu, M.; Che, X.; Wang, H.; Liang, X.J.; Wu, C.; Xue, X.; Yang, J. Lipopolysaccharide induces neuroinflammation in microglia by activating the MTOR pathway and downregulating Vps34 to inhibit autophagosome formation. J. Neuroinflamm. 2020, 17, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi-Fakhari, D.; Saffari, A.; Wahlster, L.; Sahin, M. Using tuberous sclerosis complex to understand the impact of MTORC1 signaling on mitochondrial dynamics and mitophagy in neurons. Autophagy 2017, 13, 754–756. [Google Scholar] [CrossRef]
- Lenzi, P.; Ferese, R.; Biagioni, F.; Fulceri, F.; Busceti, C.; Falleni, A.; Gambardella, S.; Frati, A.; Fornai, F. Rapamycin Ameliorates Defects in Mitochondrial Fission and Mitophagy in Glioblastoma Cells. Int. J. Mol. Sci. 2021, 22, 5379. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Wang, L.; Li, X.; Niu, X.; Xu, G.; Lv, P. Rapamycin alleviates cognitive impairment in murine vascular dementia: The enhancement of mitophagy by PI3K/AKT/mTOR axis. Tissue Cell 2021, 69, 101481. [Google Scholar] [CrossRef]
- Sharon, G.; Cruz, N.J.; Kang, D.W.; Gandal, M.J.; Wang, B.; Kim, Y.M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J.; et al. Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 2019, 177, 1600–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Lahham, S.H.; Peppelenbosch, M.P.; Roelofsen, H.; Vonk, R.J.; Venema, K. Biological effects of propionic acid in humans; metabolism, potential applications and underlying mechanisms. Biochim. Biophys. Acta 2010, 1801, 1175–1183. [Google Scholar] [CrossRef]
- Abdelli, L.S.; Samsam, A.; Naser, S.A. Propionic Acid Induces Gliosis and Neuro-inflammation through Modulation of PTEN/AKT Pathway in Autism Spectrum Disorder. Sci. Rep. 2019, 9, 8824. [Google Scholar] [CrossRef] [Green Version]
- Rose, S.; Bennuri, S.C.; Davis, J.E.; Wynne, R.; Slattery, J.C.; Tippett, M.; Delhey, L.; Melnyk, S.; Kahler, S.G.; MacFabe, D.F.; et al. Butyrate enhances mitochondrial function during oxidative stress in cell lines from boys with autism. Transl. Psychiatry 2018, 8, 42. [Google Scholar] [CrossRef]
- MacFabe, D.F. Enteric short-chain fatty acids: Microbial messengers of metabolism, mitochondria, and mind: Implications in autism spectrum disorders. Microb. Ecol. Health Dis. 2015, 26, 28177. [Google Scholar] [CrossRef] [Green Version]
- Trifonova, E.A.; Klimenko, A.I.; Mustafin, Z.S.; Lashin, S.A.; Kochetov, A.V. The mTOR Signaling Pathway Activity and Vitamin D Availability Control the Expression of Most Autism Predisposition Genes. Int. J. Mol. Sci. 2019, 20, 6332. [Google Scholar] [CrossRef] [Green Version]
- Lisse, T.S.; Liu, T.; Irmler, M.; Beckers, J.; Chen, H.; Adams, J.S.; Hewison, M. Gene targeting by the vitamin D response element binding protein reveals a role for vitamin D in osteoblast mTOR signaling. FASEB J. 2011, 25, 937–947. [Google Scholar] [CrossRef] [Green Version]
- Murdaca, G.; Tonacci, A.; Negrini, S.; Greco, M.; Borro, M.; Puppo, F.; Gangemi, S. Emerging role of vitamin D in autoimmune diseases: An update on evidence and therapeutic implications. Autoimmun. Rev. 2019, 18, 102350. [Google Scholar] [CrossRef]
- Trifonova, E.A.; Klimenko, A.I.; Mustafin, Z.S.; Lashin, S.A.; Kochetov, A.V. Do Autism Spectrum and Autoimmune Disorders Share Predisposition Gene Signature Due to mTOR Signaling Pathway Controlling Expression? Int. J. Mol. Sci. 2021, 22, 5248. [Google Scholar] [CrossRef]
- Caron, E.; Ghosh, S.; Matsuoka, Y.; Ashton-Beaucage, D.; Therrien, M.; Lemieux, S.; Perreault, C.; Roux, P.P.; Kitano, H. A comprehensive map of the mTOR signaling network. Mol. Syst. Biol. 2010, 6, 453. [Google Scholar] [CrossRef] [PubMed]
- Gandin, V.; Masvidal, L.; Hulea, L.; Gravel, S.-P.; Cargnello, M.; McLaughlan, S.; Cai, Y.; Balanathan, P.; Morita, M.; Rajakumar, A.; et al. nanoCAGE reveals 50 UTR features that define specific modes of translation of functionally related MTOR-sensitive mRNAs. Genome Res. 2016, 26, 636–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazón-Cabrera, R.; Vandormael, P.; Somers, V. Antigenic Targets of Patient and Maternal Autoantibodies in Autism Spectrum Disorder. Front. Immunol. 2019, 10, 1474. [Google Scholar] [CrossRef] [Green Version]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The regulation and function of lactate dehydrogenase a: Therapeutic potential in brain tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Young, J.I.; Hong, E.P.; Castle, J.C.; Crespo-Barreto, J.; Bowman, A.B.; Rose, M.F.; Kang, D.; Richman, R.; Johnson, J.M.; Berget, S.; et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 2005, 102, 17551–17558. [Google Scholar] [CrossRef] [Green Version]
- Nagai, J.; Baba, R.; Ohshima, T. CRMPs function in neurons and glial cells: Potential therapeutic targets for neurodegenerative diseases and CNS injury. Mol. Neurobiol. 2017, 54, 4243–4256. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Yoon, S.O.; Kubota, K.; Mendoza, M.C.; Gygi, S.P.; Blenis, J. p90 ribosomal S6 kinase and p70 ribosomal S6 kinase link phosphorylation of the eukaryotic chaperonin containing TCP-1 to growth factor, insulin, and nutrient signaling. J. Biol. Chem. 2009, 284, 14939–14948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pape, K.; Tamouza, R.; Leboyer, M.; Zipp, F. Immunoneuropsychiatry—Novel perspectives on brain disorders. Nat. Rev. Neurol. 2019, 15, 317–328. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trifonova, E.A.; Mustafin, Z.S.; Lashin, S.A.; Kochetov, A.V. Abnormal mTOR Activity in Pediatric Autoimmune Neuropsychiatric and MIA-Associated Autism Spectrum Disorders. Int. J. Mol. Sci. 2022, 23, 967. https://doi.org/10.3390/ijms23020967
Trifonova EA, Mustafin ZS, Lashin SA, Kochetov AV. Abnormal mTOR Activity in Pediatric Autoimmune Neuropsychiatric and MIA-Associated Autism Spectrum Disorders. International Journal of Molecular Sciences. 2022; 23(2):967. https://doi.org/10.3390/ijms23020967
Chicago/Turabian StyleTrifonova, Ekaterina A., Zakhar S. Mustafin, Sergey A. Lashin, and Alex V. Kochetov. 2022. "Abnormal mTOR Activity in Pediatric Autoimmune Neuropsychiatric and MIA-Associated Autism Spectrum Disorders" International Journal of Molecular Sciences 23, no. 2: 967. https://doi.org/10.3390/ijms23020967
APA StyleTrifonova, E. A., Mustafin, Z. S., Lashin, S. A., & Kochetov, A. V. (2022). Abnormal mTOR Activity in Pediatric Autoimmune Neuropsychiatric and MIA-Associated Autism Spectrum Disorders. International Journal of Molecular Sciences, 23(2), 967. https://doi.org/10.3390/ijms23020967