
Distinct Longitudinal Changes in Immunoglobulin G N-Glycosylation Associate with Therapy Response in Chronic Inflammatory Diseases

 , , ,
, , ,
Abstract
1. Introduction
2. Results
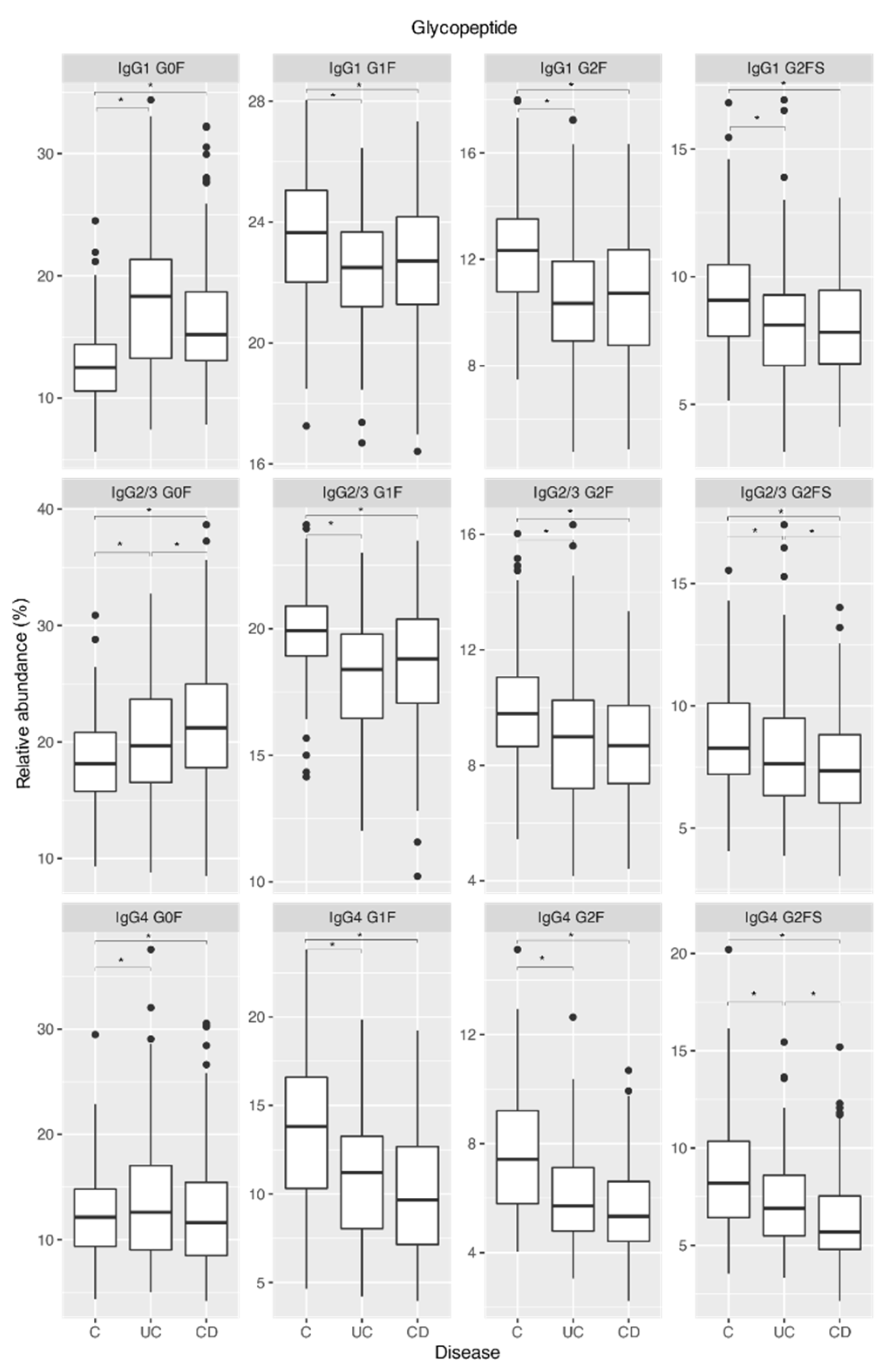
2.1. IgG Fc Galactosylation and Sialylation Are Lower in Chronic Inflammatory Diseases Patients Compared to Controls
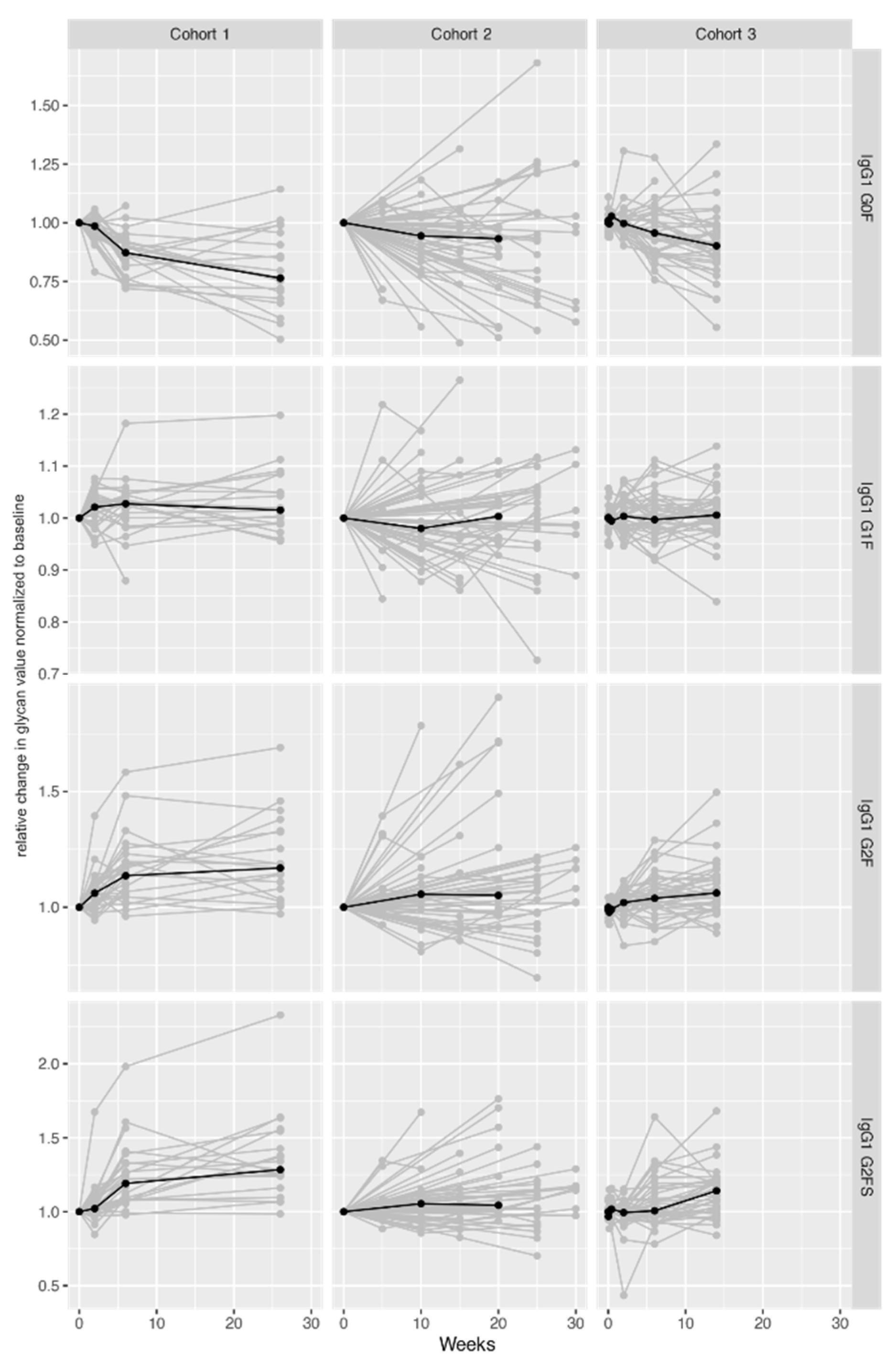
2.2. IgG Fc Galactosylation and Sialylation Levels Increase during Therapy

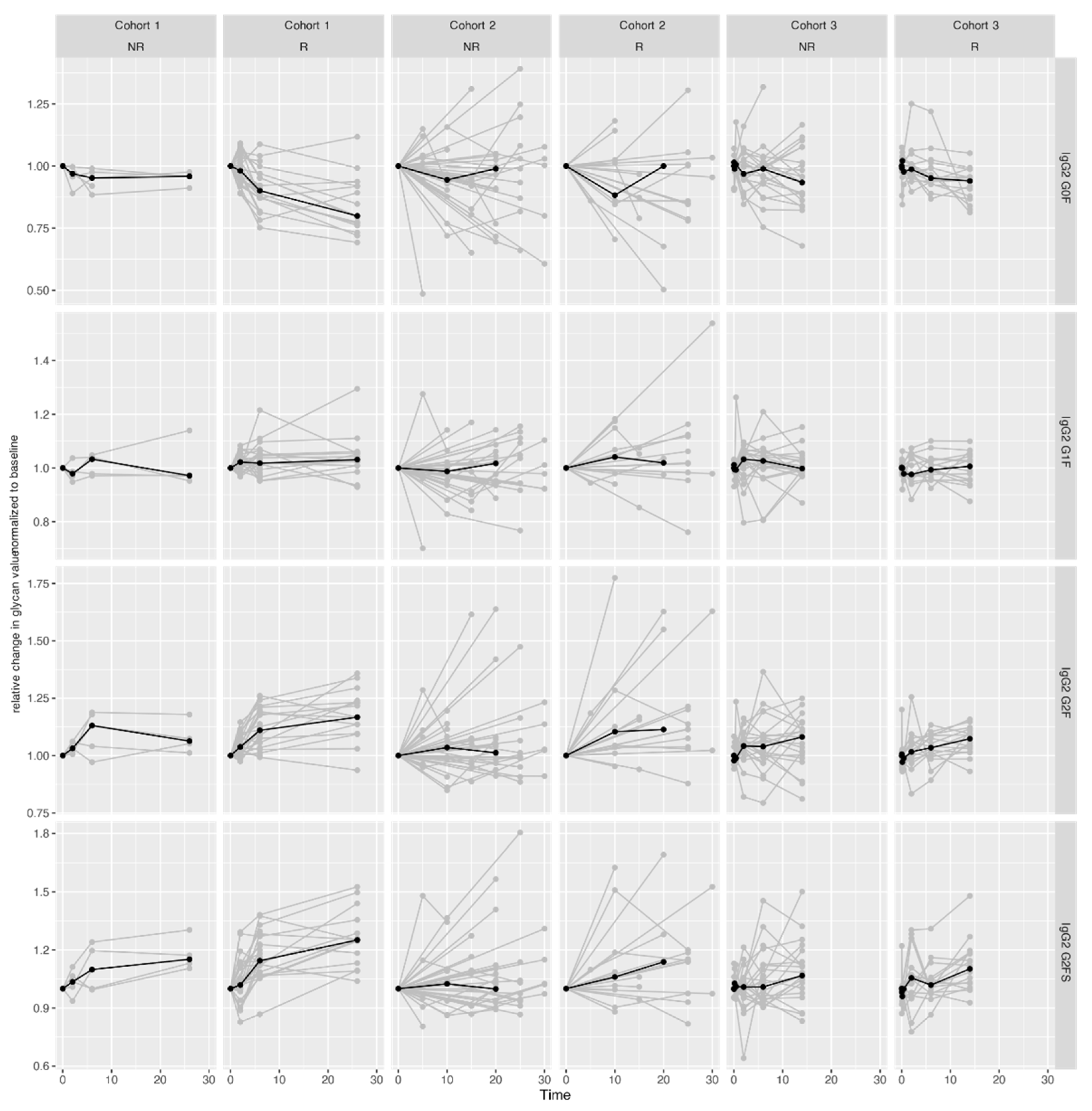
2.3. Pronounced IgG Fc Galactosylation Associates with Clinical Remission in CID Therapy

3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Immunoglobulin G Isolation
4.3. IgG Trypsin Digestion and Solid-Phase Extraction of Glycopeptides
4.4. Liquid Chromatography-Mass Spectrometry Analysis of IgG Fc Glycopeptides
4.5. Data Processing
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADCC | antibody-dependent cellular cytotoxicity |
| AS | ankylosing spondylitis |
| CD | Crohn’s disease |
| CDC | complement-dependent cytotoxicity |
| CID | chronic inflammatory diseases |
| Fc | fragment crystallizable |
| GlcNAc | N-acetylglucosamine |
| IBD | inflammatory bowel disease |
| IgG | immunoglobulin G |
| G | galactose |
| F | fucose |
| S | N-acetylneuraminic acid (sialic acid) |
| IL-6 | interleukin 6 |
| LC-MS | liquid chromatography-mass spectrometry |
| PBS | phosphate buffered saline |
| PsA | psoriatic arthritis |
| RA | rheumatoid arthritis |
| SLE | systemic lupus erythematosus |
| S/N | signal-to-noise |
| TNF | tumor necrosis factor |
| UC | ulcerative colitis |
References
- Lucafò, M.; Franca, R.; Selvestrel, D.; Curci, D.; Pugnetti, L.; Decorti, G.; Stocco, G. Pharmacogenetics of Treatments for Inflammatory Bowel Disease. Expert Opin. Drug Metab. Toxicol. 2018, 14, 1209–1223. [Google Scholar] [CrossRef]
- Sands, B.E.; Peyrin-Biroulet, L.; Loftus, E.V.; Danese, S.; Colombel, J.-F.; Törüner, M.; Jonaitis, L.; Abhyankar, B.; Chen, J.; Rogers, R.; et al. Vedolizumab versus Adalimumab for Moderate-to-Severe Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1215–1226. [Google Scholar] [CrossRef]
- Effenberger, M.; Reider, S.; Waschina, S.; Bronowski, C.; Enrich, B.; Adolph, T.E.; Koch, R.; Moschen, A.R.; Rosenstiel, P.; Aden, K.; et al. Microbial Butyrate Synthesis Indicates Therapeutic Efficacy of Azathioprine in IBD Patients. J. Crohns. Colitis 2021, 15, 88–98. [Google Scholar] [CrossRef]
- Aden, K.; Rehman, A.; Waschina, S.; Pan, W.H.; Walker, A.; Lucio, M.; Nunez, A.M.; Bharti, R.; Zimmerman, J.; Bethge, J.; et al. Metabolic Functions of Gut Microbes Associate with Efficacy of Tumor Necrosis Factor Antagonists in Patients with Inflammatory Bowel Diseases. Gastroenterology 2019, 157, 1279–1292.e11. [Google Scholar] [CrossRef]
- Derer, S.; Till, A.; Haesler, R.; Sina, C.; Grabe, N.; Jung, S.; Nikolaus, S.; Kuehbacher, T.; Groetzinger, J.; Rose-John, S.; et al. MTNF Reverse Signalling Induced by TNFα Antagonists Involves a GDF-1 Dependent Pathway: Implications for Crohn’s Disease. Gut 2013, 62, 376–386. [Google Scholar] [CrossRef][Green Version]
- Kennedy, N.A.; Heap, G.A.; Green, H.D.; Hamilton, B.; Bewshea, C.; Walker, G.J.; Thomas, A.; Nice, R.; Perry, M.H.; Bouri, S.; et al. Predictors of Anti-TNF Treatment Failure in Anti-TNF-Naive Patients with Active Luminal Crohn’s Disease: A Prospective, Multicentre, Cohort Study. Lancet Gastroenterol. Hepatol. 2019, 4, 341–353. [Google Scholar] [CrossRef]
- Syversen, S.W.; Goll, G.L.; Jørgensen, K.K.; Sandanger, Ø.; Sexton, J.; Olsen, I.C.; Gehin, J.E.; Warren, D.J.; Brun, M.K.; Klaasen, R.A.; et al. Effect of Therapeutic Drug Monitoring vs Standard Therapy During Infliximab Induction on Disease Remission in Patients with Chronic Immune-Mediated Inflammatory Diseases: A Randomized Clinical Trial. JAMA 2021, 325, 1744–1754. [Google Scholar] [CrossRef]
- Zeissig, S.; Rosati, E.; Dowds, C.M.; Aden, K.; Bethge, J.; Schulte, B.; Pan, W.H.; Mishra, N.; Zuhayra, M.; Marx, M.; et al. Vedolizumab Is Associated with Changes in Innate Rather than Adaptive Immunity in Patients with Inflammatory Bowel Disease. Gut 2019, 68, 25–39. [Google Scholar] [CrossRef]
- Marek, K.W.; Vijay, I.K.; Marth, J.D. A Recessive Deletion in the GlcNAc-1-Phosphotransferase Gene Results in Peri-Implantation Embryonic Lethality. Glycobiology 1999, 9, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Schachter, H. Complex N-Glycans: The Story of the Yellow Brick Road. Glycoconj. J. 2014, 31, 1–5. [Google Scholar] [CrossRef]
- Varki, A.; Gagneux, P. Biological Functions of Glycans. In Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2015; ISBN 9781621824213. [Google Scholar]
- Hanić, M.; Trbojević-Akmačić, I.; Lauc, G. Inflammatory Bowel Disease—Glycomics Perspective. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in Health and Disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed]
- Takai, T. Roles of Fc Receptors in Autoimmunity. Nat. Rev. Immunol. 2002, 2, 580–592. [Google Scholar] [CrossRef]
- Pincetic, A.; Bournazos, S.; Dilillo, D.J.; Maamary, J.; Wang, T.T.; Dahan, R.; Fiebiger, B.M.; Ravetch, J.V. Type i and Type II Fc Receptors Regulate Innate and Adaptive Immunity. Nat. Immunol. 2014, 15, 707–716. [Google Scholar] [CrossRef]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The Absence of Fucose but Not the Presence of Galactose or Bisecting N-Acetylglucosamine of Human IgG1 Complex-Type Oligosaccharides Shows the Critical Role of Enhancing Antibody-Dependent Cellular Cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef]
- Gornik, O.; Pavić, T.; Lauc, G. Alternative Glycosylation Modulates Function of IgG and Other Proteins—Implications on Evolution and Disease. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 1318–1326. [Google Scholar] [CrossRef]
- Novokmet, M.; Lukić, E.; Vučković, F.; Durić, Ž.; Keser, T.; Rajšl, K.; Remondini, D.; Castellani, G.; Gašparović, H.; Gornik, O.; et al. Changes in IgG and Total Plasma Protein Glycomes in Acute Systemic Inflammation. Sci. Rep. 2014, 4, 4347. [Google Scholar] [CrossRef]
- Gudelj, I.; Lauc, G.; Pezer, M. Immunoglobulin G Glycosylation in Aging and Diseases. Cell. Immunol. 2018, 333, 65–79. [Google Scholar] [CrossRef]
- Parekh, R.B.; Dwek, R.A.; Sutton, B.J.; Fernandes, D.L.; Leung, A.; Stanworth, D.; Rademacher, T.W.; Mizuochi, T.; Taniguchi, T.; Matsuta, K.; et al. Association of Rheumatoid Arthritis and Primary Osteoarthritis with Changes in the Glycosylation Pattern of Total Serum IgG. Nature 1985, 316, 452–457. [Google Scholar] [CrossRef]
- Vučkovïc, F.; Krištïc, J.; Gudelj, I.; Teruel, M.; Keser, T.; Pezer, M.; Pučïc-Bakovïc, M.; Štambuk, J.; Trbojevïc-Akmačïc, I.; Barrios, C.; et al. Association of Systemic Lupus Erythematosus with Decreased Immunosuppressive Potential of the IgG Glycome. Arthritis Rheumatol. 2015, 67, 2978–2989. [Google Scholar] [CrossRef]
- Miyoshi, E.; Shinzaki, S.; Fujii, H.; Iijima, H.; Kamada, Y.; Takehara, T. Role of Aberrant IgG Glycosylation in the Pathogenesis of Inflammatory Bowel Disease. Proteomics Clin. Appl. 2016, 10, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Šimurina, M.; de Haan, N.; Vučković, F.; Kennedy, N.A.; Štambuk, J.; Falck, D.; Trbojević-Akmačić, I.; Clerc, F.; Razdorov, G.; Khon, A.; et al. Glycosylation of Immunoglobulin G Associates with Clinical Features of Inflammatory Bowel Diseases. Gastroenterology 2018, 154, 1320–1333.e10. [Google Scholar] [CrossRef]
- Banda, N.K.; Wood, A.K.; Takahashi, K.; Levitt, B.; Rudd, P.M.; Royle, L.; Abrahams, J.L.; Stahl, G.L.; Holers, V.M.; Arend, W.P. Initiation of the Alternative Pathway of Murine Complement by Immune Complexes Is Dependent on N-Glycans in IgG Antibodies. Arthritis Rheum. 2008, 58, 3081–3089. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Wormald, M.R.; Rudd, P.M.; Fischer, P.B.; Dwek, R.A.; Sim, R.B. Glycosylation Changes of IgG Associated with Rheumatooid Arthritis Can Activate Complement via the Mannose-Binding Protein. Nat. Med. 1995, 1, 237–243. [Google Scholar] [CrossRef]
- Van de Geijn, F.E.; de Man, Y.A.; Wuhrer, M.; Willemsen, S.P.; Deelder, A.M.; Hazes, J.M.W.; Dolhain, R.J.E.M. Mannose-Binding Lectin Does Not Explain the Course and Outcome of Pregnancy in Rheumatoid Arthritis. Arthritis Res. Ther. 2011, 13, R10. [Google Scholar] [CrossRef]
- Karsten, C.M.C.; Pandey, M.M.K.; Figge, J.; Kilchenstein, R.; Taylor, P.P.R.P.; Rosas, M.; McDonald, J.U.J.; Orr, S.S.J.S.; Berger, M.; Petzold, D.; et al. Anti-Inflammatory Activity of IgG1 Mediated by Fc Galactosylation and Association of FcγRIIB and Dectin-1. Nat. Med. 2012, 18, 1401–1406. [Google Scholar] [CrossRef]
- Mihai, S.; Nimmerjahn, F. The Role of Fc Receptors and Complement in Autoimmunity. Autoimmun. Rev. 2013, 12, 657–660. [Google Scholar] [CrossRef]
- Peschke, B.; Keller, C.W.; Weber, P.; Quast, I.; Lünemann, J.D. Fc-Galactosylation of Human Immunoglobulin Gamma Isotypes Improves C1q Binding and Enhances Complement-Dependent Cytotoxicity. Front. Immunol. 2017, 8, 646. [Google Scholar] [CrossRef]
- Houde, D.; Peng, Y.; Berkowitz, S.A.; Engen, J.R. Post-Translational Modifications Differentially Affect IgG1 Conformation and Receptor Binding. Mol. Cell. Proteomics 2010, 9, 1716–1728. [Google Scholar] [CrossRef]
- Trbojevic Akmacic, I.; Ventham, N.T.; Theodoratou, E.; Vučković, F.; Kennedy, N.A.; Krištić, J.; Nimmo, E.R.; Kalla, R.; Drummond, H.; Štambuk, J.; et al. Inflammatory Bowel Disease Associates with Proinflammatory Potential of the Immunoglobulin G Glycome. Inflamm. Bowel Dis. 2015, 21, 1237–1247. [Google Scholar] [CrossRef]
- Parekh, R.B.; Isenberg, D.A.; Ansell, B.M.; Roitt, I.M.; Dwek, R.A.; Rademacher, T.W. Galactosylation of IgG Associated Oligosaccharides: Reduction in Patients with Adult and Juvenile Onset Rheumatoid Arthritis and Relation to Disease Activity. Lancet 1988, 331, 966–969. [Google Scholar] [CrossRef]
- Maverakis, E.; Kim, K.; Shimoda, M.; Gershwin, M.E.; Patel, F.; Wilken, R.; Raychaudhuri, S.; Ruhaak, L.R.; Lebrilla, C.B. Glycans in the Immune System and The Altered Glycan Theory of Autoimmunity: A Critical Review. J. Autoimmun. 2015, 57, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Seeling, M.; Brückner, C.; Nimmerjahn, F. Differential Antibody Glycosylation in Autoimmunity: Sweet Biomarker or Modulator of Disease Activity? Nat. Rev. Rheumatol. 2017, 13, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Hess, C.; Winkler, A.; Lorenz, A.K.; Holecska, V.; Blanchard, V.; Eiglmeier, S.; Schoen, A.L.; Bitterling, J.; Stoehr, A.D.; Petzold, D.; et al. T Cell-Independent B Cell Activation Induces Immunosuppressive Sialylated IgG Antibodies. J. Clin. Investig. 2013, 123, 3788–3796. [Google Scholar] [CrossRef]
- Kavur, M.M.; Lauc, G.; Pezer, M. Systems Glycobiology: Immunoglobulin G Glycans as Biomarkers and Functional Effectors in Aging and Diseases. Compr. Glycosci. Second Ed. 2021, 12, 439–478. [Google Scholar] [CrossRef]
- Pasek, M.; Duk, M.; Podbielska, M.; Sokolik, R.; Szechiński, J.; Lisowska, E.; Krotkiewski, H. Galactosylation of IgG from Rheumatoid Arthritis (RA) Patients—Changes during Therapy. Glycoconj. J. 2006, 23, 463–471. [Google Scholar] [CrossRef]
- Croce, A.; Firuzi, O.; Altieri, F.; Eufemi, M.; Agostino, R.; Priori, R.; Bombardieri, M.; Alessandri, C.; Valesini, G.; Saso, L. Effect of Infliximab on the Glycosylation of IgG of Patients with Rheumatoid Arthritis. J. Clin. Lab. Anal. 2007, 21, 303–314. [Google Scholar] [CrossRef]
- Van Beneden, K.; Coppieters, K.; Laroy, W.; De Keyser, F.; Hoffman, I.E.; Van Den Bosch, F.; Vander Cruyssen, B.; Drennan, M.; Jacques, P.; Rottiers, P.; et al. Reversible Changes in Serum Immunoglobulin Galactosylation during the Immune Response and Treatment of Inflammatory Autoimmune Arthritis. Ann. Rheum. Dis. 2009, 68, 1360–1365. [Google Scholar] [CrossRef]
- Collins, E.S.; Galligan, M.C.; Saldova, R.; Adamczyk, B.; Abrahams, J.L.; Campbell, M.P.; Ng, C.T.; Veale, D.J.; Murphy, T.B.; Rudd, P.M.; et al. Glycosylation Status of Serum in Inflammatory Arthritis in Response to Anti-TNF Treatment. Rheumatology 2013, 52, 1572–1582. [Google Scholar] [CrossRef]
- Clerc, F.; Novokmet, M.; Dotz, V.; Reiding, K.R.; de Haan, N.; Kammeijer, G.S.M.; Dalebout, H.; Bladergroen, M.R.; Vukovic, F.; Rapp, E.; et al. Plasma N-Glycan Signatures Are Associated with Features of Inflammatory Bowel Diseases. Gastroenterology 2018, 155, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Gorski, A.; Korczak-Kowalska, G.; Nowaczyk, M.; Paczek, L.; Gaciong, Z. The Effect of Azathioprine on Terminal Differentiation of Human B Lymphocytes. Immunopharmacology 1983, 6, 259–266. [Google Scholar] [CrossRef]
- Winkelstein, A. The Effects of Azathioprine and 6 MP on Immunity. J. Immunopharmacol. 1979, 1, 429–454. [Google Scholar] [CrossRef]
- Pala, O.; Diaz, A.; Blomberg, B.B.; Frasca, D. B Lymphocytes in Rheumatoid Arthritis and Effects of Anti-TNF-α Agents on B Lymphocytes: Review of the Literature. Clin. Ther. 2018, 40, 1034. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lehotay, M.; Anastassiades, T.; Harrison, M.; Brockhausen, I. The Effect of TNF-α on Glycosylation Pathways in Bovine Synoviocytes. Biochem. Cell Biol. 2004, 82, 559–568. [Google Scholar] [CrossRef]
- Veny, M.; Garrido-Trigo, A.; Corraliza, A.M.; Masamunt, M.C.; Bassolas-Molina, H.; Esteller, M.; Arroyes, M.; Tristán, E.; Fernández-Clotet, A.; Ordás, I.; et al. Dissecting Common and Unique Effects of Anti-A4β7 and Anti-Tumor Necrosis Factor Treatment in Ulcerative Colitis. J. Crohns. Colitis 2021, 15, 441. [Google Scholar] [CrossRef]
- Reiding, K.R.; Vreeker, G.C.M.; Bondt, A.; Bladergroen, M.R.; Hazes, J.M.W.; van der Burgt, Y.E.M.; Wuhrer, M.; Dolhain, R.J.E.M. Serum Protein N-Glycosylation Changes with Rheumatoid Arthritis Disease Activity during and after Pregnancy. Front. Med. 2017, 4, 241. [Google Scholar] [CrossRef]
- Van de Geijn, F.E.; Wuhrer, M.; Selman, M.H.J.; Willemsen, S.P.; de Man, Y.A.; Deelder, A.M.; Hazes, J.M.W.; Dolhain, R.J.E.M. Immunoglobulin G Galactosylation and Sialylation Are Associated with Pregnancy-Induced Improvement of Rheumatoid Arthritis and the Postpartum Flare: Results from a Large Prospective Cohort Study. Arthritis Res. Ther. 2009, 11, R193. [Google Scholar] [CrossRef]
- Stümer, J.; Biermann, M.H.C.; Knopf, J.; Magorivska, I.; Kastbom, A.; Svärd, A.; Janko, C.; Bilyy, R.; Schett, G.; Sjöwall, C.; et al. Altered Glycan Accessibility on Native Immunoglobulin G Complexes in Early Rheumatoid Arthritis and Its Changes during Therapy. Clin. Exp. Immunol. 2017, 189, 372–382. [Google Scholar] [CrossRef]
- Váradi, C.; Holló, Z.; Póliska, S.; Nagy, L.; Szekanecz, Z.; Váncsa, A.; Palatka, K.; Guttman, A. Combination of IgG N-Glycomics and Corresponding Transcriptomics Data to Identify Anti-TNF-α Treatment Responders in Inflammatory Diseases. Electrophoresis 2015, 36, 1330–1335. [Google Scholar] [CrossRef]
- Pučić, M.; Knežević, A.; Vidič, J.; Adamczyk, B.; Novokmet, M.; Polašek, O.; Gornik, O.; Šupraha-Goreta, S.; Wormald, M.R.; Redžić, I.; et al. High Throughput Isolation and Glycosylation Analysis of IgG–Variability and Heritability of the IgG Glycome in Three Isolated Human Populations. Mol. Cell. Proteomics 2011, 10, M111.010090. [Google Scholar] [CrossRef] [PubMed]
- Keser, T.; Vučković, F.; Barrios, C.; Zierer, J.; Wahl, A.; Akinkuolie, A.O.; Štambuk, J.; Nakić, N.; Pavić, T.; Periša, J.; et al. Effects of Statins on the Immunoglobulin G Glycome. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1152–1158. [Google Scholar] [CrossRef]
- Jansen, B.C.; Falck, D.; De Haan, N.; Hipgrave Ederveen, A.L.; Razdorov, G.; Lauc, G.; Wuhrer, M. LaCyTools: A Targeted Liquid Chromatography-Mass Spectrometry Data Processing Package for Relative Quantitation of Glycopeptides. J. Proteome Res. 2016, 15, 2198–2210. [Google Scholar] [CrossRef] [PubMed]
- Dard, P.; Lefranc, M.P.; Osipova, L.; Sanchez-Mazas, A. DNA Sequence Variability of IGHG3 Alleles Associated to the Main G3m Haplotypes in Human Populations. Eur. J. Hum. Genet. 2001, 9, 765–772. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| IgG Subclass | Glycan | Effect | Standard Error | p-Value | Adjusted p-Value |
|---|---|---|---|---|---|
| IgG1 | G0F | −2.93 × 10−2 | 4.00 × 10−3 | 2.41 × 10−13 | 7.24 × 10−13 |
| G1F | 7.98 × 10−4 | 3.41 × 10−3 | 8.15 × 10−1 | 8.15 × 10−1 | |
| G2F | 2.25 × 10−2 | 3.12 × 10−3 | 5.64 × 10−13 | 1.35 × 10−12 | |
| G2FS1 | 2.89 × 10−2 | 3.23 × 10−3 | 3.62 × 10−19 | 4.34 × 10−18 | |
| IgG2/3 | G0F | −2.10 × 10−2 | 3.06 × 10−3 | 7.74 × 10−12 | 1.55 × 10−11 |
| G1F | 4.25 × 10−3 | 3.33 × 10−3 | 2.02 × 10−1 | 2.20 × 10−1 | |
| G2F | 2.09 × 10−2 | 2.56 × 10−3 | 3.25 × 10−16 | 1.30 × 10−15 | |
| G2FS1 | 2.16 × 10−2 | 2.48 × 10−3 | 2.76 × 10−18 | 1.66 × 10−17 | |
| IgG4 | G0F | −1.95 × 10−2 | 3.13 × 10−3 | 5.03 × 10−10 | 8.62 × 10−10 |
| G1F | −6.43 × 10−3 | 2.68 × 10−3 | 1.65 × 10−2 | 1.98 × 10−2 | |
| G2F | 1.19 × 10−2 | 3.59 × 10−3 | 9.35 × 10−4 | 1.25 × 10−3 | |
| G2FS1 | 1.48 × 10−2 | 3.12 × 10−3 | 2.19 × 10−6 | 3.28 × 10−6 |
| IgG Subclass | Glycan | Effect | Standard Error | p-Value | Adjusted p-Value |
|---|---|---|---|---|---|
| IgG1 | G0F | −1.45 × 10−2 | 7.61 × 10−3 | 5.61 × 10−2 | 1.35 × 10−1 |
| G1F | −3.86 × 10−3 | 6.50 × 10−3 | 5.52 × 10−1 | 6.02 × 10−1 | |
| G2F | 8.27 × 10−3 | 6.25 × 10−3 | 1.85 × 10−1 | 3.18 × 10−1 | |
| G2FS1 | 7.89 × 10−3 | 6.38 × 10−3 | 2.16 × 10−1 | 3.24 × 10−1 | |
| IgG2/3 | G0F | −1.36 × 10−2 | 5.58 × 10−3 | 1.50 × 10−2 | 6.00 × 10−2 |
| G1F | 1.24 × 10−2 | 5.75 × 10−3 | 3.08 × 10−2 | 9.23 × 10−2 | |
| G2F | 1.81 × 10−2 | 5.16 × 10−3 | 4.54 × 10−4 | 5.44 × 10−3 | |
| G2FS1 | 8.14 × 10−3 | 5.34 × 10−3 | 1.28 × 10−1 | 2.55 × 10−1 | |
| IgG4 | G0F | −1.69 × 10−2 | 5.85 × 10−3 | 3.75 × 10−3 | 2.25 × 10−2 |
| G1F | −5.56 × 10−3 | 5.75 × 10−3 | 3.34 × 10−1 | 4.00 × 10−1 | |
| G2F | 6.82 × 10−4 | 7.43 × 10−3 | 9.27 × 10−1 | 9.27 × 10−1 | |
| G2FS1 | 7.06 × 10−3 | 6.61 × 10−3 | 2.85 × 10−1 | 3.80 × 10−1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Štambuk, J.; Vučković, F.; Habazin, S.; Hanić, M.; Novokmet, M.; Nikolaus, S.; Tran, F.; Schreiber, S.; Franke, A.; Rosenstiel, P.; et al. Distinct Longitudinal Changes in Immunoglobulin G N-Glycosylation Associate with Therapy Response in Chronic Inflammatory Diseases. Int. J. Mol. Sci. 2022, 23, 8473. https://doi.org/10.3390/ijms23158473
Štambuk J, Vučković F, Habazin S, Hanić M, Novokmet M, Nikolaus S, Tran F, Schreiber S, Franke A, Rosenstiel P, et al. Distinct Longitudinal Changes in Immunoglobulin G N-Glycosylation Associate with Therapy Response in Chronic Inflammatory Diseases. International Journal of Molecular Sciences. 2022; 23(15):8473. https://doi.org/10.3390/ijms23158473
Chicago/Turabian StyleŠtambuk, Jerko, Frano Vučković, Siniša Habazin, Maja Hanić, Mislav Novokmet, Susanna Nikolaus, Florian Tran, Stefan Schreiber, Andre Franke, Philip Rosenstiel, and et al. 2022. "Distinct Longitudinal Changes in Immunoglobulin G N-Glycosylation Associate with Therapy Response in Chronic Inflammatory Diseases" International Journal of Molecular Sciences 23, no. 15: 8473. https://doi.org/10.3390/ijms23158473
APA StyleŠtambuk, J., Vučković, F., Habazin, S., Hanić, M., Novokmet, M., Nikolaus, S., Tran, F., Schreiber, S., Franke, A., Rosenstiel, P., Lauc, G., Aden, K., & Pezer, M. (2022). Distinct Longitudinal Changes in Immunoglobulin G N-Glycosylation Associate with Therapy Response in Chronic Inflammatory Diseases. International Journal of Molecular Sciences, 23(15), 8473. https://doi.org/10.3390/ijms23158473