
Digitoxin Affects Metabolism, ROS Production and Proliferation in Pancreatic Cancer Cells Differently Depending on the Cell Phenotype

Abstract
:1. Introduction
2. Results
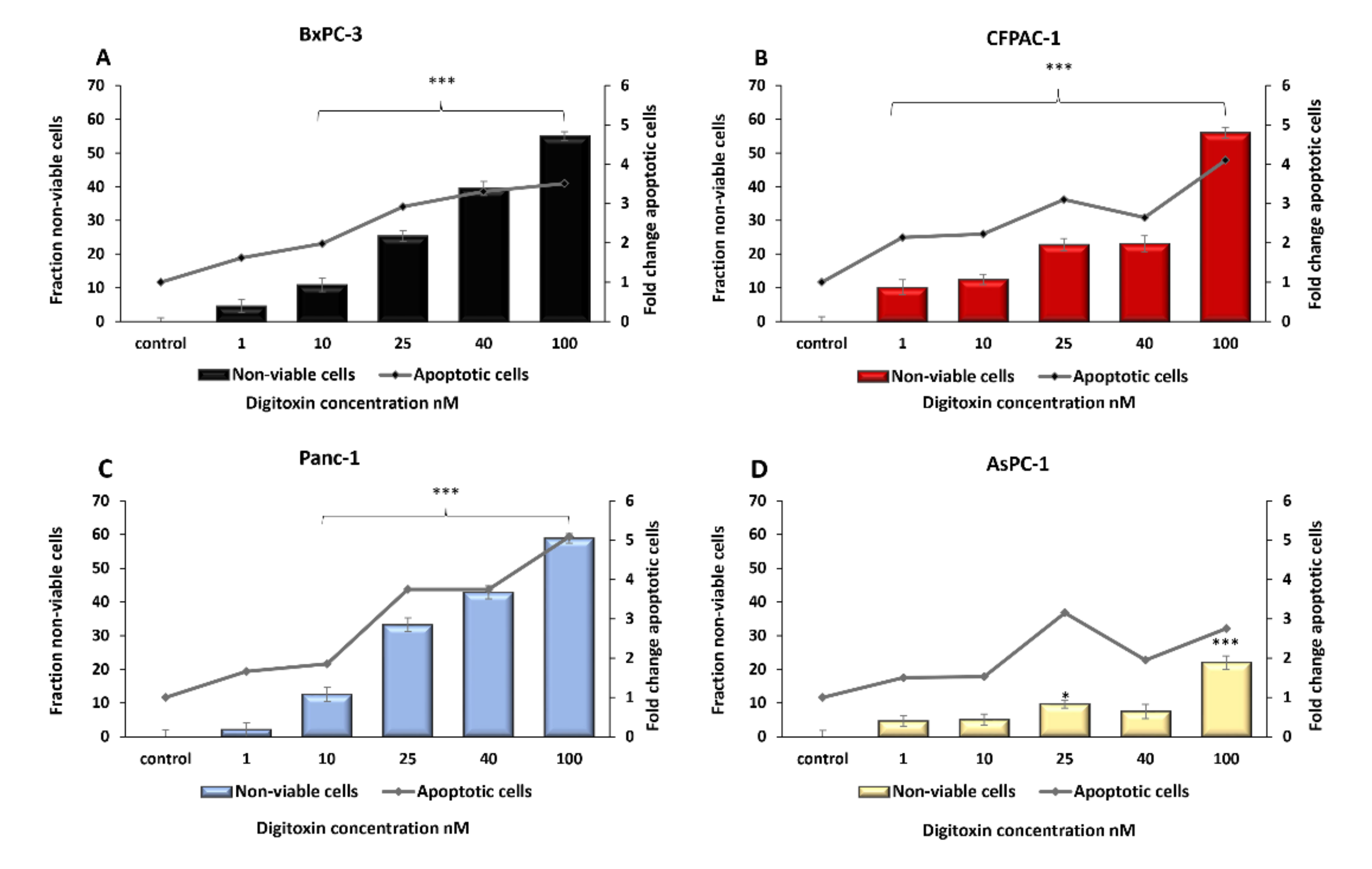
2.1. Digitoxin Treatment Decreased Pancreatic Cancer Cell Viability and Induced Apoptosis
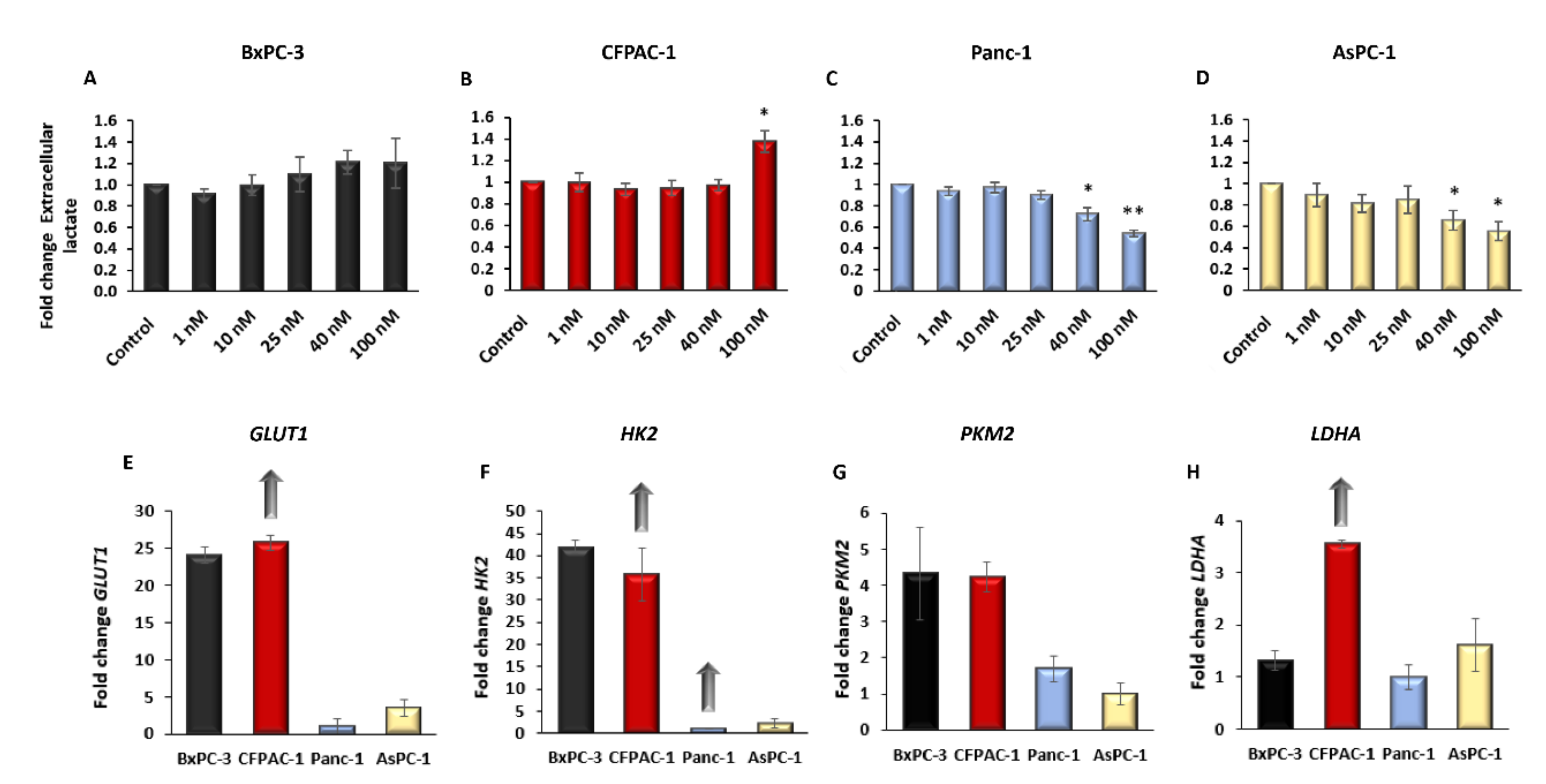
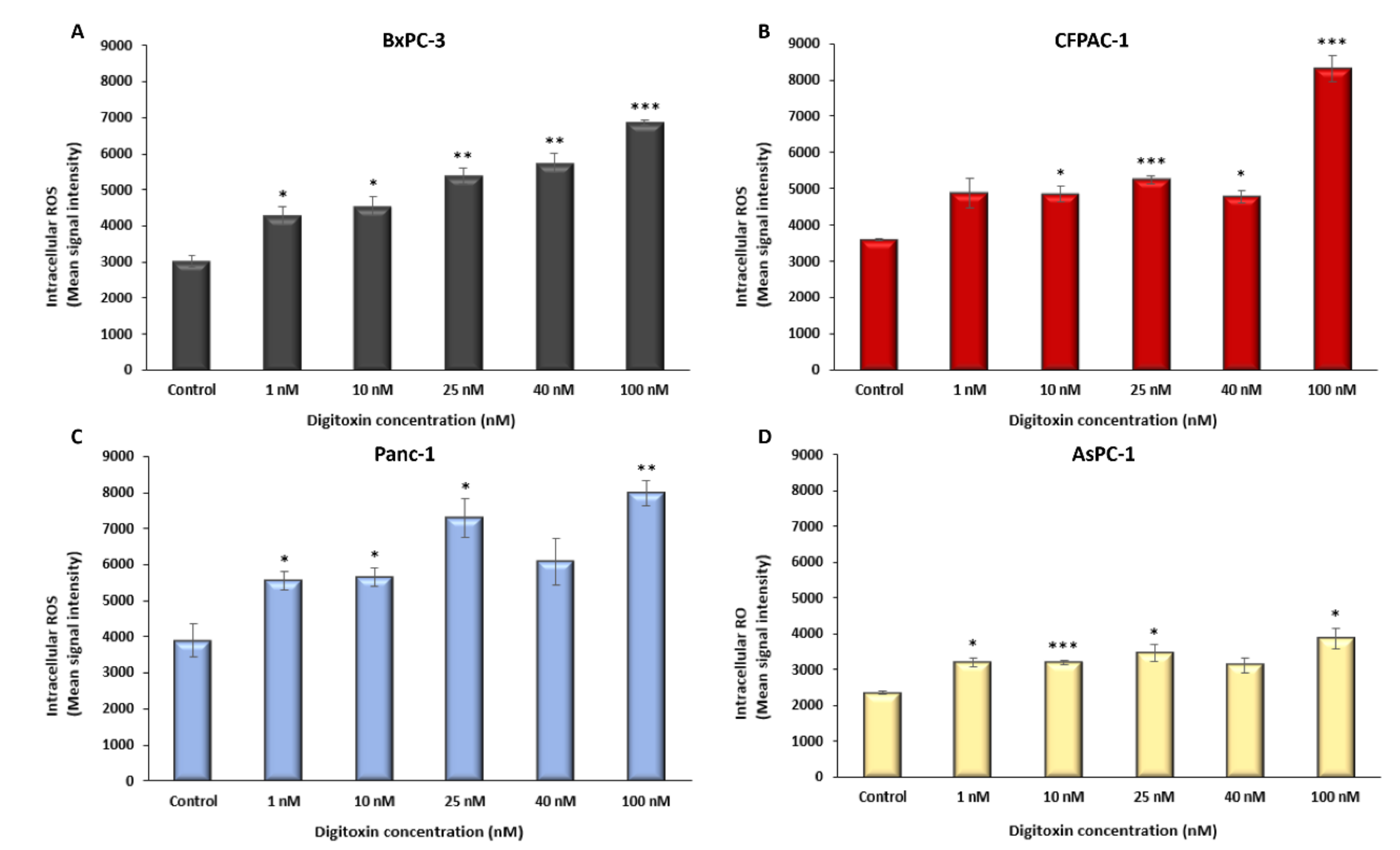
2.2. Digitoxin Affects the Rate of Aerobic Glycolysis and Increase Intracellular ROS
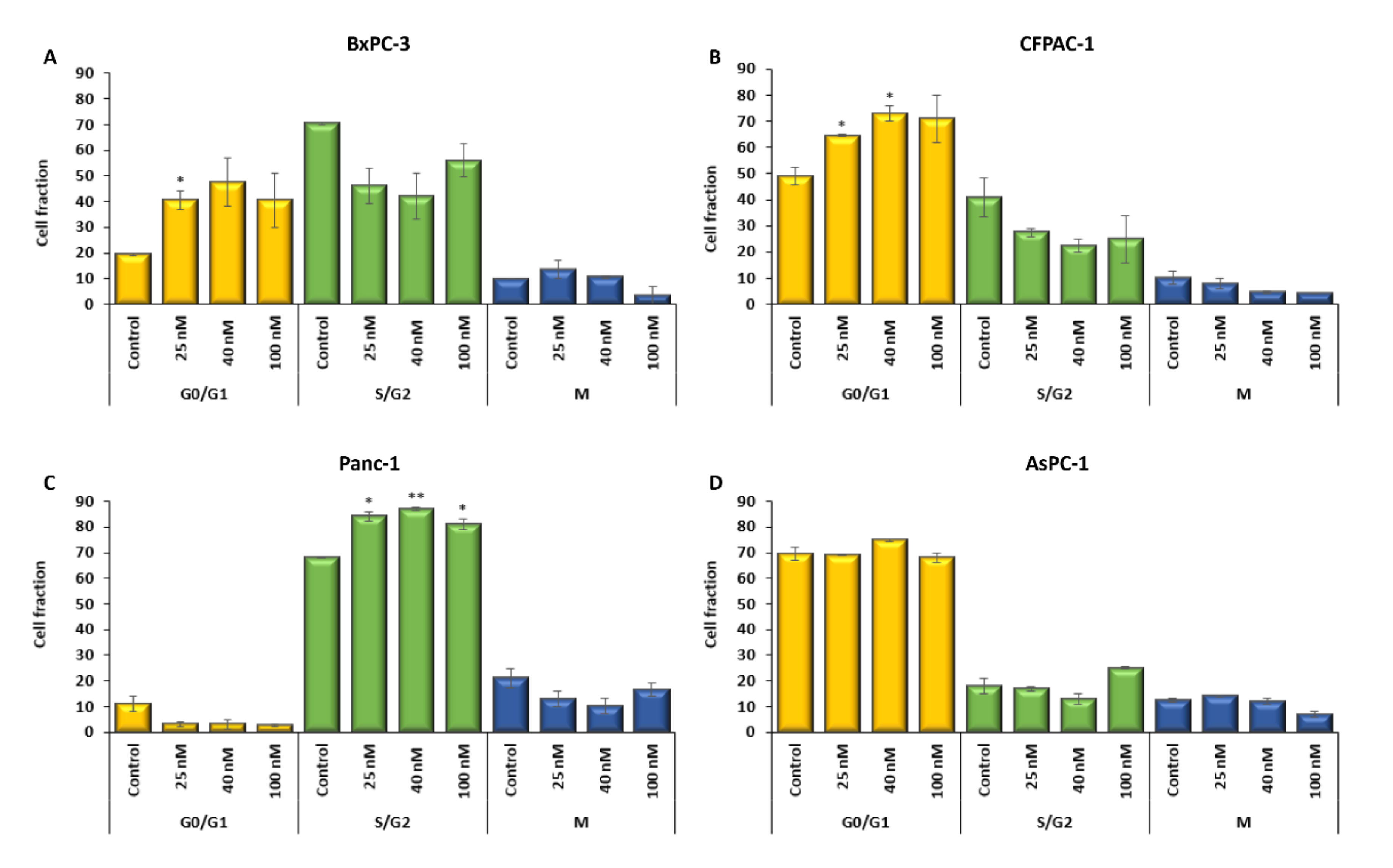
2.3. The Cell Cycle Is Affected by Digitoxin Treatment
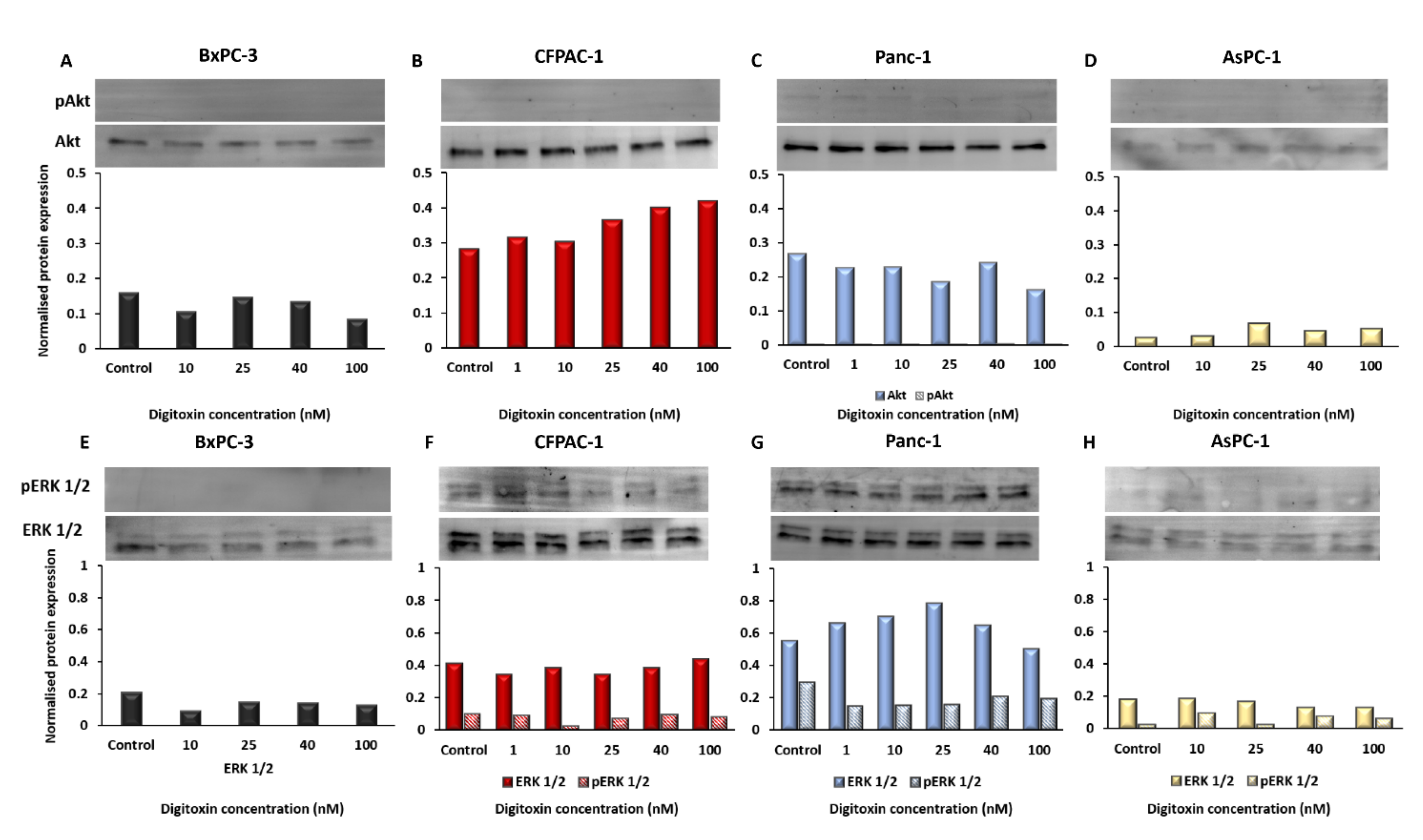
2.4. Oncogenic Signaling in The PDAC Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Culturing
4.2. Seeding and Treatment with Digitoxin
4.3. RNA-Extraction, cDNA Synthesis, and Real-Time PCR
4.4. Protein Extraction and Western Blot (WB)
4.5. Cell Viability Assay
4.6. Caspase 3/7 Assay
4.7. Extracellular Lactate- Aerobic Glycolysis
4.8. Intracellular ROS
4.9. Cell Cycle Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Becker, A.E.; Hernandez, Y.G.; Frucht, H.; Lucas, A.L. Pancreatic ductal adenocarcinoma: Risk factors, screening, and early detection. World J. Gastroenterol. 2014, 20, 11182–11198. [Google Scholar] [CrossRef] [PubMed]
- Ansari, D.; Gustafsson, A.; Andersson, R. Update on the management of pancreatic cancer: Surgery is not enough. World J. Gastroenterol. 2015, 21, 3157–3165. [Google Scholar] [CrossRef] [PubMed]
- Pereira, N.P.; Corrêa, J.R. Pancreatic cancer: Treatment approaches and trends. J. cancer metastatis treat. 2018, 4, 30. [Google Scholar] [CrossRef] [Green Version]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Duronio, R.J.; Xiong, Y. Signaling pathways that control cell proliferation. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Moralli, S.; Tarrado-Castellarnau, M.; Miranda, A.; Cascante, M. Targeting cell cycle regulation in cancer therapy. Pharmacol. Ther. 2013, 138, 255–271. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; G, M.M.; G, S.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.S.; Xu, Z.W.; Yi, T.L.; Xu, R.C.; Li, J.; Zhang, W.B.; Zhang, S.; Sun, H.T.; Yu, Z.Q.; Xu, H.X.; et al. Ouabain suppresses the growth and migration abilities of glioma U-87MG cells through inhibiting the Akt/mTOR signaling pathway and downregulating the expression of HIF-1α. Mol. Med. Rep. 2018, 17, 5595–5600. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Qian, D.Z.; Tan, Y.S.; Lee, K.; Gao, P.; Ren, Y.R.; Rey, S.; Hammers, H.; Chang, D.; Pili, R.; et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 19579–19586. [Google Scholar] [CrossRef] [Green Version]
- Haux, J. Digitoxin is a potential anticancer agent for several types of cancer. Med. Hypotheses. 1999, 53, 543–548. [Google Scholar] [CrossRef]
- Haux, J.; Klepp, O.; Spigset, O.; Tretli, S. Digitoxin medication and cancer; case control and internal dose-response studies. BMC Cancer 2001, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, W.; Taubert, K.; Narahara, K. Nonradioactive Serum Digoxin and Digitoxin Levels. Arch. Intern. Med. 1972, 130, 31–36. [Google Scholar] [CrossRef]
- Kepp, O.; Menger, L.; Vacchelli, E.; Adjemian, S.; Martins, I.; Ma, Y.; Sukkurwala, A.Q.; Michaud, M.; Galluzzi, L.; Zitvogel, L.; et al. Anticancer activity of cardiac glycosides: At the frontier between cell-autonomous and immunological effects. Oncoimmunology 2012, 1, 1640–1642. [Google Scholar] [CrossRef] [Green Version]
- Frankel, A.E.; Eskiocak, U.; Gill, J.G.; Yuan, S.; Ramesh, V.; Froehlich, T.W.; Ahn, C.; Morrison, S.J. Digoxin Plus Trametinib Therapy Achieves Disease Control in BRAF Wild-Type Metastatic Melanoma Patients. Neoplasia 2017, 19, 255–260. [Google Scholar] [CrossRef]
- Li, Q.; Pogwizd, S.M.; Prabhu, S.D.; Zhou, L. Inhibiting Na+/K+ ATPase can impair mitochondrial energetics and induce abnormal Ca2+ cycling and automaticity in guinea pig cardiomyocytes. PLoS One 2014, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kembro, J.M.; Aon, M.A.; Winslow, R.L.; O′Rourke, B.; Cortassa, S. Integrating mitochondrial energetics, redox and ROS metabolic networks: A two-compartment model. Biophys. J. 2013, 104, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [Green Version]
- Tu, B.P.; Kudlicki, A.; Rowicka, M.; McKnight, S.L. Logic of the yeast metabolic cycle: Temporal compartmentalization of cellular processes. Science 2005, 310, 1152–1158. [Google Scholar] [CrossRef] [Green Version]
- Kaludercic, N.; Deshwal, S.; Lisa, F.D. Reactive oxygen species and redox compartmentalization. Front. Physiol. 2014, 5, 285. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.G.; Goswami, P.C. A redox cycle within the cell cycle: Ring in the old with the new. Oncogene 2007, 26, 1101–1109. [Google Scholar] [CrossRef] [Green Version]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Liu, T.; O′Rourke, B. Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. J. Bioenerg. Biomembr. 2009, 41, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Cortassa, S.; Aon, M.A.; Marbán, E.; Winslow, R.L.; O′Rourke, B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys. J. 2003, 84, 2734–2755. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.-C.; Senese, S.; France, B.; Gholkar, A.A.; Damoiseaux, R.; Torres, J.Z. Computational Cell Cycle Profiling of Cancer Cells for Prioritizing FDA-Approved Drugs with Repurposing Potential. Sci. Rep. 2017, 7, 11261. [Google Scholar] [CrossRef] [Green Version]
- Burhans, W.C.; Heintz, N.H. The cell cycle is a redox cycle: Linking phase-specific targets to cell fate. Free Radic. Biol. Med. 2009, 47, 1282–1293. [Google Scholar] [CrossRef]
- Yano, S.; Miwa, S.; Mii, S.; Hiroshima, Y.; Uehara, F.; Yamamoto, M.; Kishimoto, H.; Tazawa, H.; Bouvet, M.; Fujiwara, T.; et al. Invading cancer cells are predominantly in G0/G1 resulting in chemoresistance demonstrated by real-time FUCCI imaging. Cell Cycle 2014, 13, 953–960. [Google Scholar] [CrossRef] [Green Version]
- da Veiga Moreira, J.; Peres, S.; Steyaert, J.-M.; Bigan, E.; Paulevé, L.; Nogueira, M.L.; Schwartz, L. Cell cycle progression is regulated by intertwined redox oscillators. Theor. Biol. Med. Model. 2015, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Mukai, K.; Hishiki, T.; Kubo, A.; Ohmura, M.; Sugiura, Y.; Matsuura, T.; Nagahata, Y.; Hayakawa, N.; Yamamoto, T.; et al. Energy management by enhanced glycolysis in G1-phase in human colon cancer cells in vitro and in vivo. Mol. Cancer Res. 2013, 11, 973–985. [Google Scholar] [CrossRef] [Green Version]
- Beresford, M.J.; Wilson, G.D.; Makris, A. Measuring proliferation in breast cancer: Practicalities and applications. Breast Cancer Res. 2006, 8, 216. [Google Scholar] [CrossRef] [Green Version]
- Chiu, J.; Dawes, I.W. Redox control of cell proliferation. Trends Cell Biol. 2012, 22, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Gable, M.E.; Abdallah, S.L.; Najjar, S.M.; Liu, L.; Askari, A. Digitalis-induced cell signaling by the sodium pump: On the relation of Src to Na(+)/K(+)-ATPase. Biochem. Biophys. Res. Commun. 2014, 446, 1151–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Cai, T.; Yuan, Z.; Wang, H.; Liu, L.; Haas, M.; Maksimova, E.; Huang, X.Y.; Xie, Z.J. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol. Cell 2006, 17, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, M.; Tian, J.; Liu, L.; Pierre, S.; Liu, J.; Shapiro, J.; Xie, Z.-J. Identification of a Pool of Non-pumping Na/K-ATPase. J. Biol. Chem. 2007, 282, 10585–10593. [Google Scholar] [CrossRef] [Green Version]
- Souza e Souza, K.F.C.; Moraes, B.P.T.; Paixao, I.C.N.D.P.; Burth, P.; Silva, A.R.; Gonçalves-de-Albuquerque, C.F. Na+/K+-ATPase as a Target of Cardiac Glycosides for the Treatment of SARS-CoV-2 Infection. Front. pharmacol. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Lemire, M.; Zhang, A.; Chan-Seng-Yue, M.; Wilson, G.; Grant, R.C.; Merico, D.; Lungu, I.; et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 2019, 35, 267–282. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Yang, D.; Zou, J.; Ma, W.; Wu, R.; Zhao, W.; Zhang, Y.; Xiao, H.; Gong, X.; Zhang, M.; et al. Systematic Interpretation of Comutated Genes in Large-Scale Cancer Mutation Profiles. Mol. Cancer Ther. 2010, 9, 2186–2195. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Polosukhina, D.; Love, H.D.; Correa, H.; Su, Z.; Dahlman, K.B.; Pao, W.; Moses, H.L.; Arteaga, C.L.; Lovvorn, H.N.; Zent, R.; et al. Functional KRAS mutations and a potential role for PI3K/AKT activation in Wilms tumors. Mol. Oncol. 2017, 11, 405–421. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta. Pharm. Sin. B. 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. PKM2, cancer metabolism, and the road ahead. EMBO Reports 2016, 17, 1721–1730. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [Green Version]
- Graziano, F.; Ruzzo, A.; Giacomini, E.; Ricciardi, T.; Aprile, G.; Loupakis, F.; Lorenzini, P.; Ongaro, E.; Zoratto, F.; Catalano, V.; et al. Glycolysis gene expression analysis and selective metabolic advantage in the clinical progression of colorectal cancer. Pharmacogenomics J. 2017, 17, 258–264. [Google Scholar] [CrossRef]
- Anderson, M.; Marayati, R.; Moffitt, R.; Yeh, J.J. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget 2017, 8, 56081–56094. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Hanai, J.; Ren, J.G.; Kats, L.; Burgess, K.; Bhargava, P.; Signoretti, S.; Billiard, J.; Duffy, K.J.; Grant, A.; et al. Targeting lactate dehydrogenase--a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014, 19, 795–809. [Google Scholar] [CrossRef] [Green Version]
- Koukourakis, M.I.; Giatromanolaki, A.; Simopoulos, C.; Polychronidis, A.; Sivridis, E. Lactate dehydrogenase 5 (LDH5) relates to up-regulated hypoxia inducible factor pathway and metastasis in colorectal cancer. Clin. Exp. Metastasis 2005, 22, 25–30. [Google Scholar] [CrossRef]
- Mi, C.; Cao, X.; Ma, K.; Wei, M.; Xu, W.; Lin, Y.; Zhang, J.; Wang, T.-y. Digitoxin promotes apoptosis and inhibits proliferation and migration by reducing HIF-1α and STAT3 in KRAS mutant human colon cancer cells. Chem. Biol. Interact. 2022, 351, 109729. [Google Scholar] [CrossRef]
- Lee, D.H.; Cheul, O.S.; Giles, A.J.; Jung, J.; Gilbert, M.R.; Park, D.M. Cardiac glycosides suppress the maintenance of stemness and malignancy via inhibiting HIF-1α in human glioma stem cells. Oncotarget 2017, 8, 40233–40245. [Google Scholar] [CrossRef] [Green Version]
- Lindholm, H.; Ejeskär, K.; Szekeres, F. The Na+/K+-ATPase subunit α3 expression correlates to digitoxin treatment efficiency in pancreatic cancer cells. Med. Int. 2022. manuscript in preparation; to be submitted. [Google Scholar]
- James, A.D.; Richardson, D.A.; Oh, I.W.; Sritangos, P.; Attard, T.; Barrett, L.; Bruce, J.I.E. Cutting off the fuel supply to calcium pumps in pancreatic cancer cells: Role of pyruvate kinase-M2 (PKM2). Br. J. Cancer 2020, 122, 266–278. [Google Scholar] [CrossRef]
- Dejos, C.; Gkika, D.; Cantelmo, A.R. The Two-Way Relationship Between Calcium and Metabolism in Cancer. Front. Cell Dev. Biol. 2020, 8, 1–14. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Daemen, A.; Peterson, D.; Sahu, N.; McCord, R.; Du, X.; Liu, B.; Kowanetz, K.; Hong, R.; Moffat, J.; Gao, M.; et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, E4410–E4417. [Google Scholar] [CrossRef] [Green Version]
- Riquelme, E.; Behrens, C.; Lin, H.Y.; Simon, G.; Papadimitrakopoulou, V.; Izzo, J.; Moran, C.; Kalhor, N.; Lee, J.J.; Minna, J.D.; et al. Modulation of EZH2 Expression by MEK-ERK or PI3K-AKT Signaling in Lung Cancer Is Dictated by Different KRAS Oncogene Mutations. Cancer Research 2016, 76, 675–685. [Google Scholar] [CrossRef] [Green Version]
- Céspedes, M.V.; Sancho, F.J.; Guerrero, S.; Parreño, M.; Casanova, I.; Pavón, M.A.; Marcuello, E.; Trias, M.; Cascante, M.; Capellà, G.; et al. K-ras Asp12 mutant neither interacts with Raf, nor signals through Erk and is less tumorigenic than K-ras Val12. Carcinogenesis 2006, 27, 2190–2200. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Fritsche-Guenther, R.; Zasada, C.; Mastrobuoni, G.; Royla, N.; Rainer, R.; Roßner, F.; Pietzke, M.; Klipp, E.; Sers, C.; Kempa, S. Alterations of mTOR signaling impact metabolic stress resistance in colorectal carcinomas with BRAF and KRAS mutations. Sci. Rep. 2018, 8, 9204. [Google Scholar] [CrossRef]
- James, A.D.; Patel, W.; Butt, Z.; Adiamah, M.; Dakhel, R.; Latif, A.; Uggenti, C.; Swanton, E.; Imamura, H.; Siriwardena, A.K.; et al. The Plasma Membrane Calcium Pump in Pancreatic Cancer Cells Exhibiting the Warburg Effect Relies on Glycolytic ATP. J. Biol. Chem. 2015, 290, 24760–24771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta. Mol. Cell. Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 2019, 54, 1155–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuto, R.; Pinton, P.; Ferrari, D.; Chami, M.; Szabadkai, G.; Magalhães, P.J.; Virgilio, F.D.; Pozzan, T. Calcium and apoptosis: Facts and hypotheses. Oncogene 2003, 22, 8619–8627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Icard, P.; Fournel, L.; Wu, Z.; Alifano, M.; Lincet, H. Interconnection between Metabolism and Cell Cycle in Cancer. Trends Biochem. Sci. 2019, 44, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.; Qi, M.; Chan, C.; Leung, P.; Ye, G.; Lei, Y.; Liu, A.; Xue, F.; Liu, D.; Ye, W.; et al. Digitoxin inhibits HeLa cell growth through the induction of G2/M cell cycle arrest and apoptosis in vitro and in vivo. Int. J. Oncol. 2020, 57, 562–573. [Google Scholar] [CrossRef]
- Kaushik, V.; Yakisich, J.S.; Azad, N.; Kulkarni, Y.; Venkatadri, R.; Wright, C.; Rojanasakul, Y.; Iyer, A.K.V. Anti-Tumor Effects of Cardiac Glycosides on Human Lung Cancer Cells and Lung Tumorspheres. J. Cell. Physiol. 2017, 232, 2497–2507. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, Y.; Hou, L.; Wang, W.; Li, K.; Zhang, Z.; Du, B.; Kong, D. Digoxin exerts anticancer activity on human nonsmall cell lung cancer cells by blocking PI3K/Akt pathway. Biosci. Rep. 2021, 41, 1–13. [Google Scholar] [CrossRef]
- Hoffman, A.; Spetner, L.M.; Burke, M. Ramifications of a redox switch within a normal cell: Its absence in a cancer cell. Free Radic. Biol. Med. 2008, 45, 265–268. [Google Scholar] [CrossRef]
- Park, S.-Y.; Nam, J.-S. The force awakens: Metastatic dormant cancer cells. Exp. Mol. Med. 2020, 52, 569–581. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ATCC® No. | Name | Histology | Tumour Source | Mutant Gene |
|---|---|---|---|---|
| CRL-1687™ | BxPC-3 | Adenocarcinoma | Primary | BRAF, CDKN2A, MAP2K4, SMAD4, TP53 |
| CRL-1918™ | CFPAC-1 | Ductal adenocarcinoma | Metastasis, liver | KRAS p. G12V, SMAD4, TP53 |
| CRL-1469™ | Panc-1 | Adenocarcinoma | Primary | KRAS p. G12D, TP53 |
| CRL-1682™ | AsPC-1 | Adenocarcinoma | Xenograft | KRAS p. G12D, CDKN2A, MAP2K4, FBXW7, TP53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindholm, H.; Ejeskär, K.; Szekeres, F. Digitoxin Affects Metabolism, ROS Production and Proliferation in Pancreatic Cancer Cells Differently Depending on the Cell Phenotype. Int. J. Mol. Sci. 2022, 23, 8237. https://doi.org/10.3390/ijms23158237
Lindholm H, Ejeskär K, Szekeres F. Digitoxin Affects Metabolism, ROS Production and Proliferation in Pancreatic Cancer Cells Differently Depending on the Cell Phenotype. International Journal of Molecular Sciences. 2022; 23(15):8237. https://doi.org/10.3390/ijms23158237
Chicago/Turabian StyleLindholm, Heléne, Katarina Ejeskär, and Ferenc Szekeres. 2022. "Digitoxin Affects Metabolism, ROS Production and Proliferation in Pancreatic Cancer Cells Differently Depending on the Cell Phenotype" International Journal of Molecular Sciences 23, no. 15: 8237. https://doi.org/10.3390/ijms23158237
APA StyleLindholm, H., Ejeskär, K., & Szekeres, F. (2022). Digitoxin Affects Metabolism, ROS Production and Proliferation in Pancreatic Cancer Cells Differently Depending on the Cell Phenotype. International Journal of Molecular Sciences, 23(15), 8237. https://doi.org/10.3390/ijms23158237