
Functional Versatility of the Human 2-Oxoadipate Dehydrogenase in the L-Lysine Degradation Pathway toward Its Non-Cognate Substrate 2-Oxopimelic Acid

 ,
,  , and
, and
Abstract
1. Introduction
2. Results and Discussion
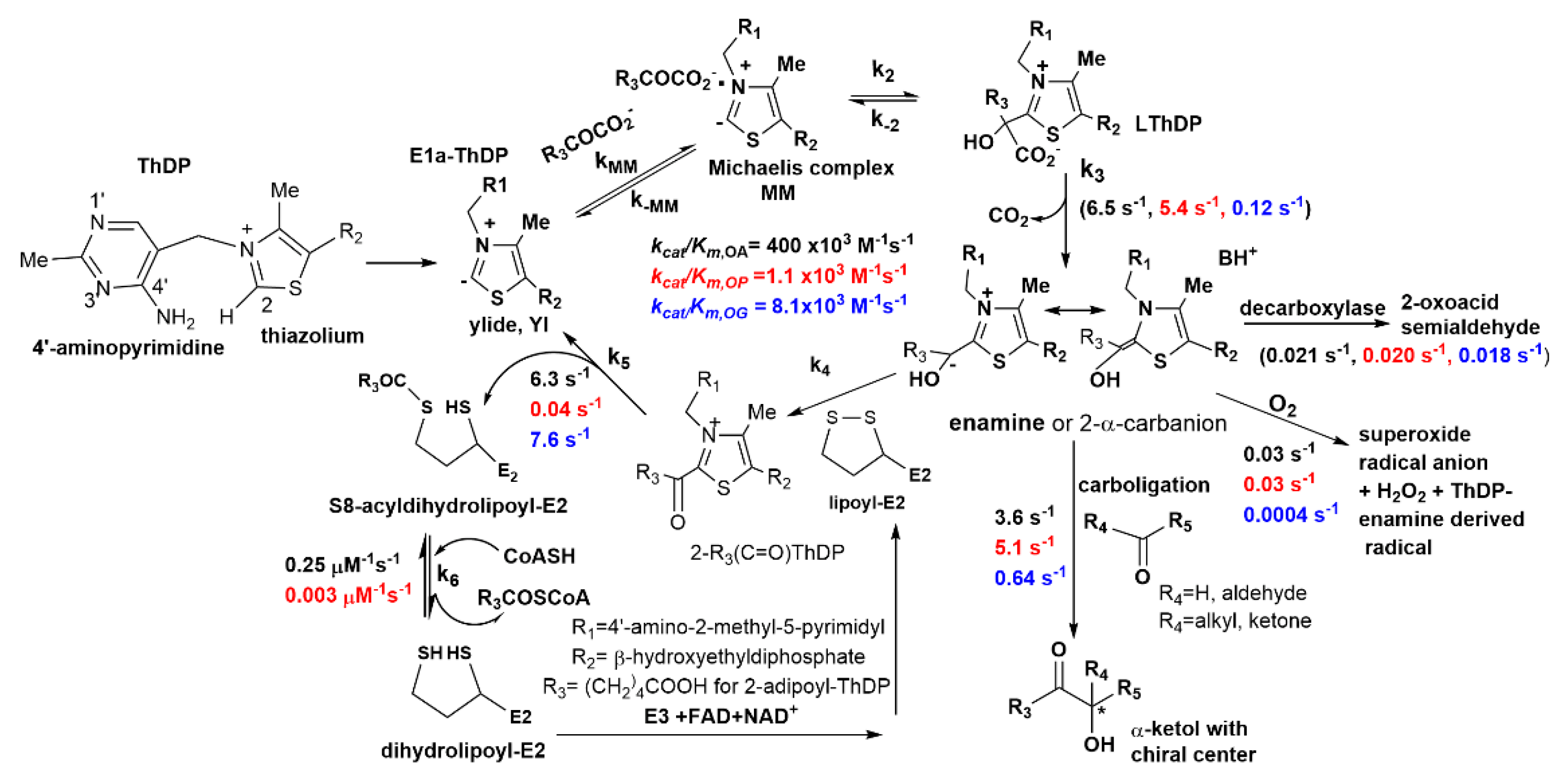
2.1. The E1a Oxidizes 2-Oxopimelic Acid at Rates Similar to the Rates with Its Substrate 2-Oxoadipate through Decarboxylation and Enamine Intermediate Formation
(E1a-Thdp-enamine intermediate) + CO2
S8-glutaryldihydrolipoyl-E2o (reductive glutarylation)
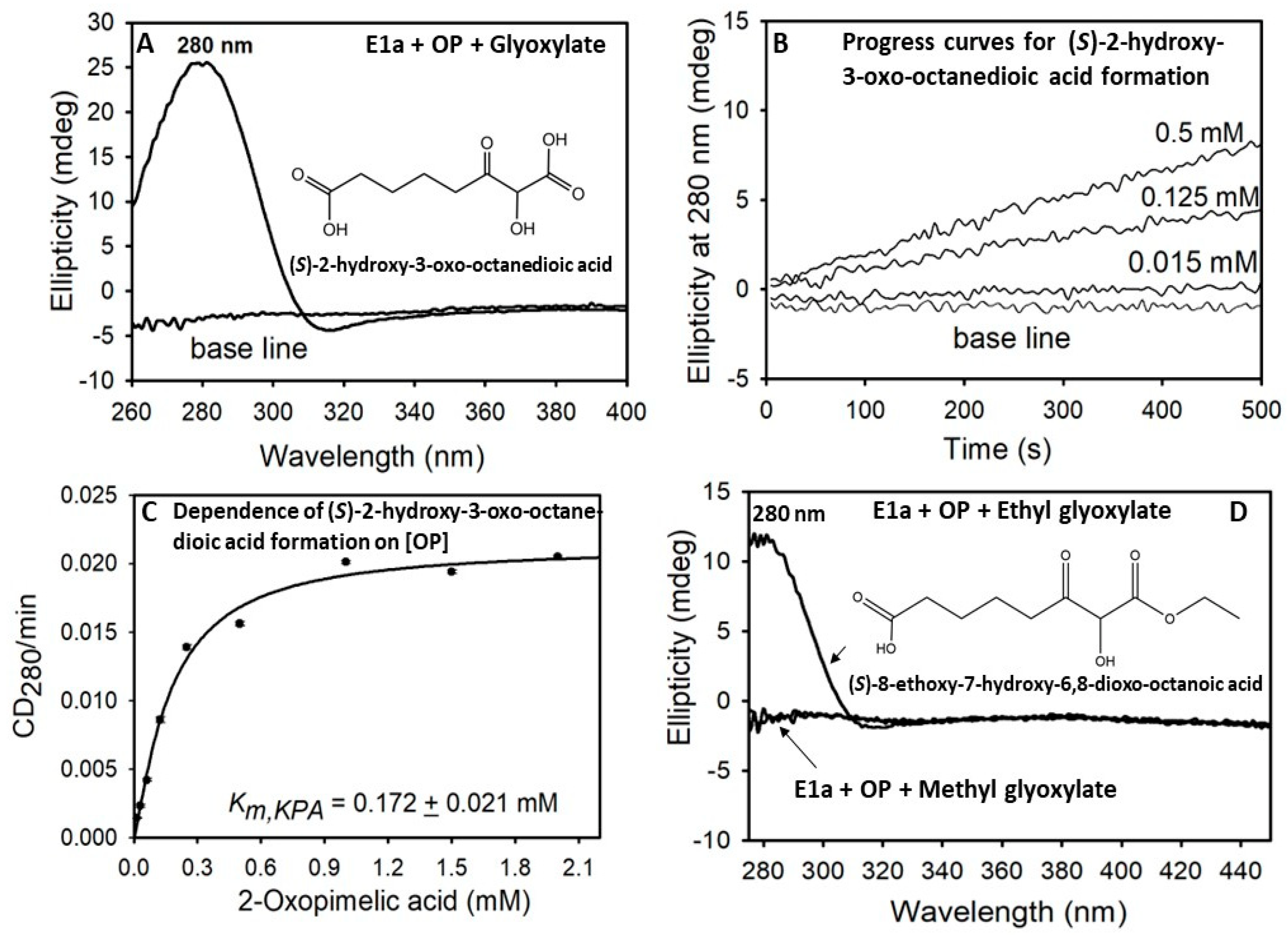
2.2. The E1a-ThDP-Enamine Intermediate Formed on Decarboxylation of OP Is Catalytically Competent in the E1a-Side Reactions and Could Be an Efficient Source of H2O2 Production
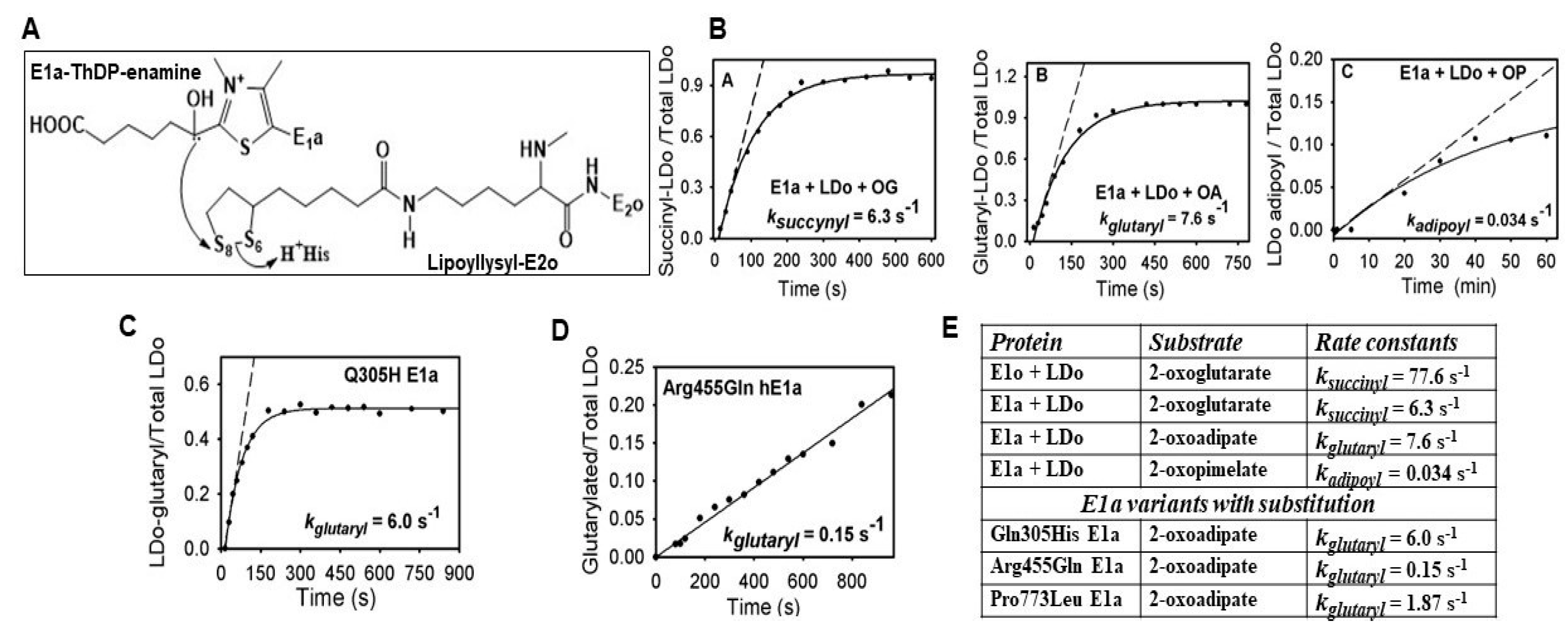
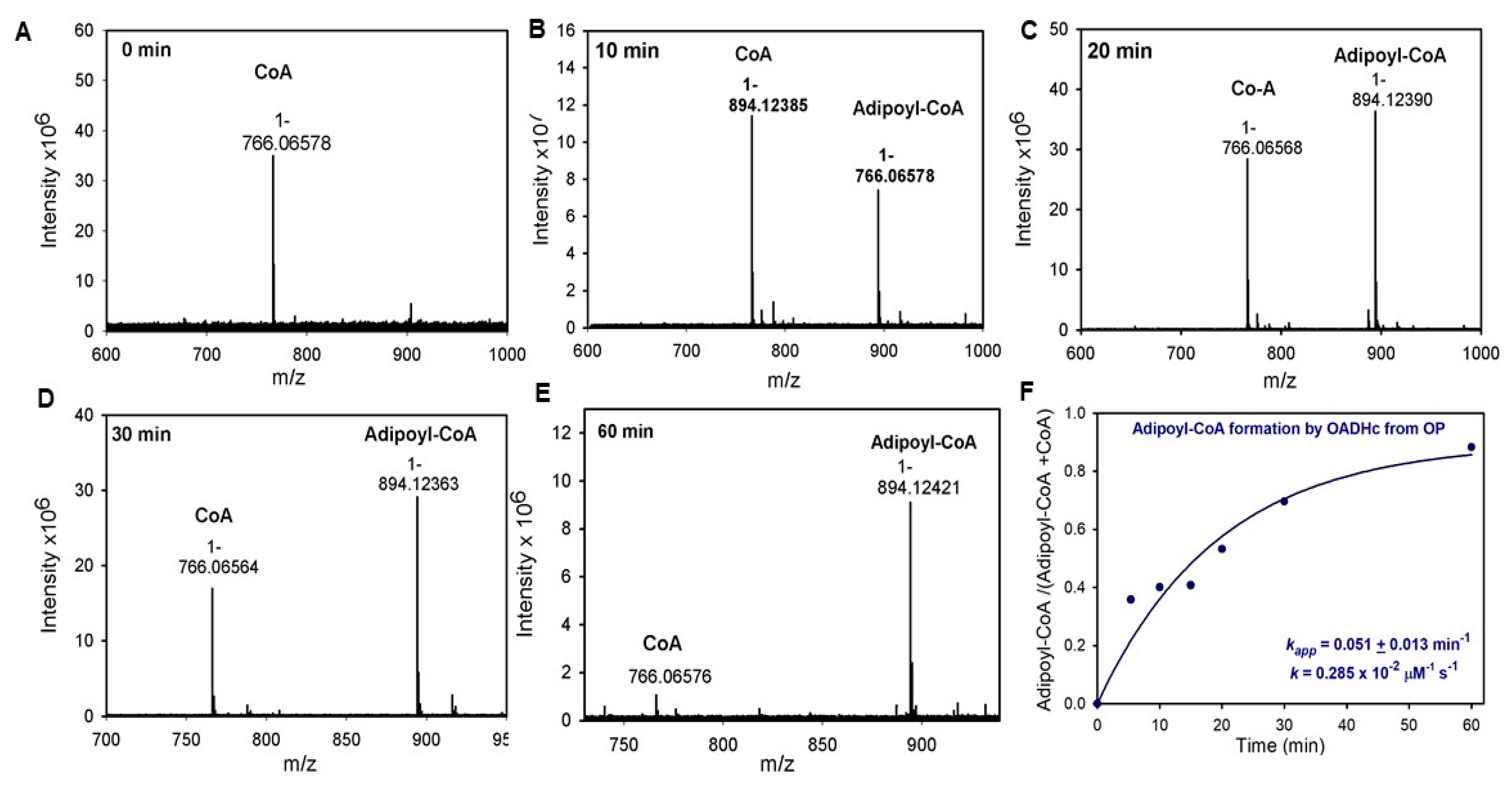
2.3. Generation of NADH from OP Is Strongly Diminished Due to the Diminished Rate of Reductive Transfer of the Adipoyl Moiety from the E1a to the E2o Component Affected by One-Carbon Substrate Elongation
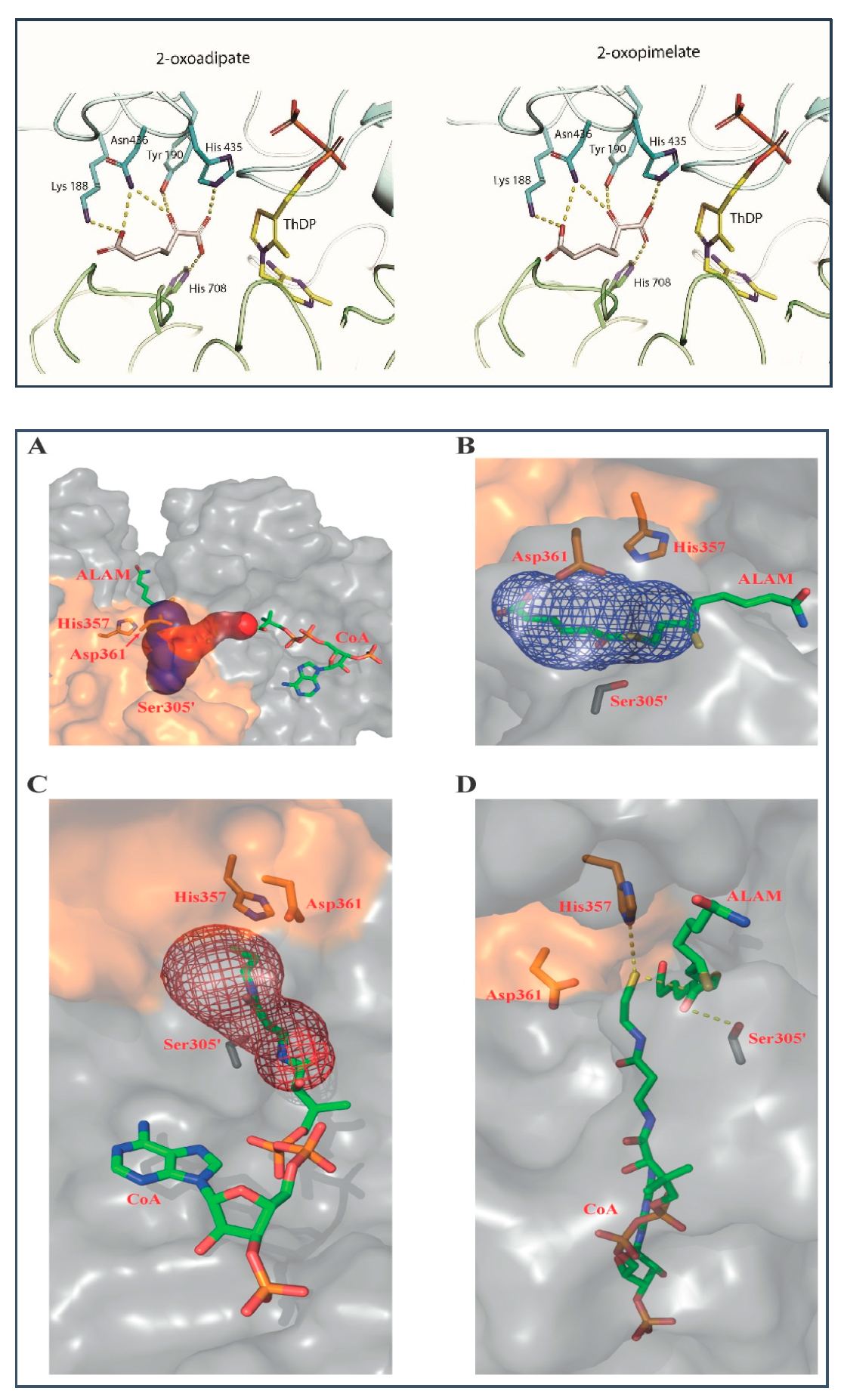
2.4. Binding Models of E1a with OP and of E2o with S8-Adipoyldihydrolipoamide in the Active Centers
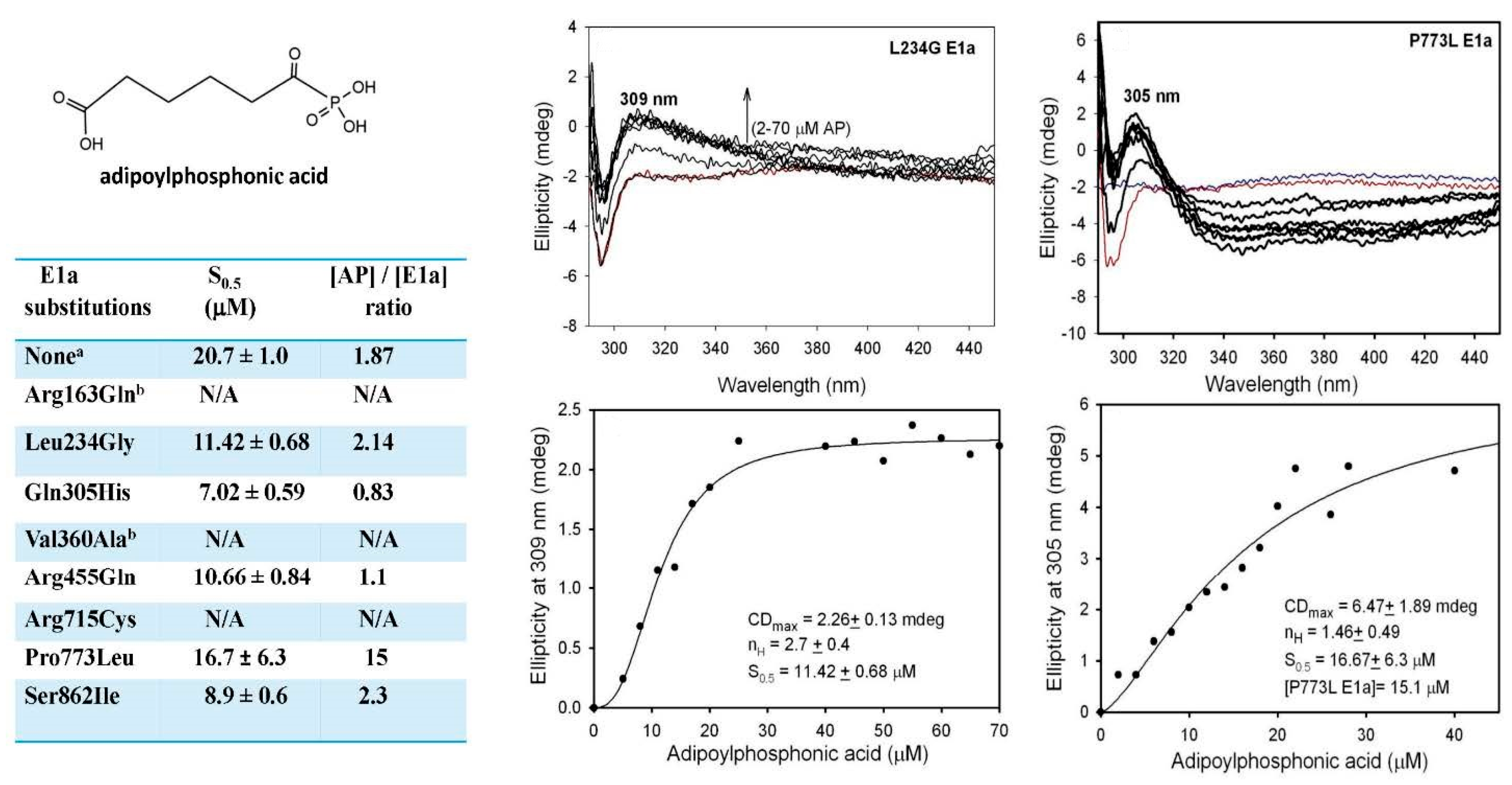
2.5. The E1a-Specific Activity and the E1a Side-Reactions with OP Are Affected by E1a Substitutions Associated with AMOXAD and EoE and Showed a Great Similarity to the Behavior with OA
2.5.1. Functional Characterization of the E1a Variants with Disease-Causing Mutations
2.5.2. Studies on E1a Variants Encoded by DHTKD1 with Genetic Mutations Linked to AMOXAD and EoE
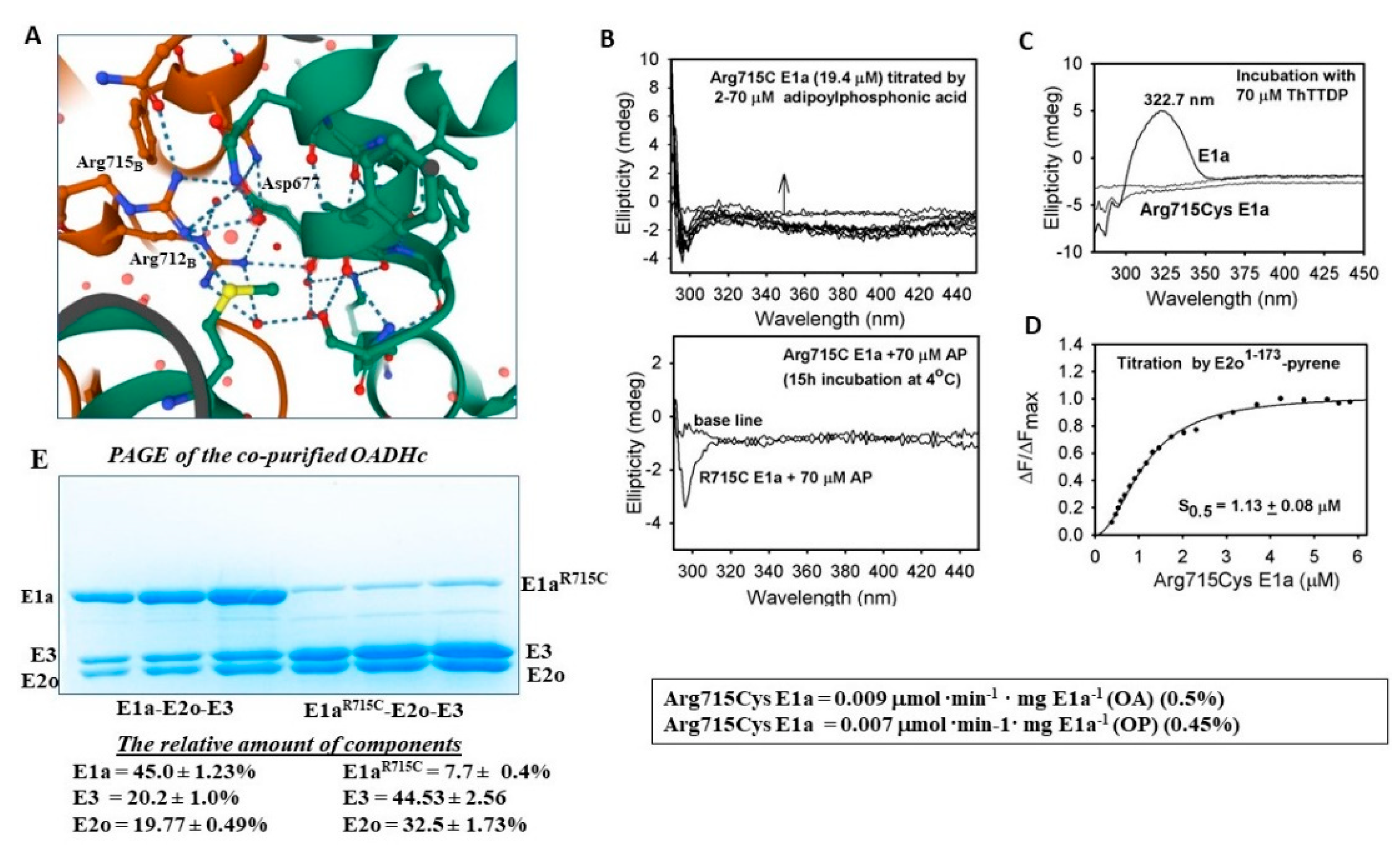
2.5.3. Evidence for the Formation of a Pre-Decarboxylation Intermediate from OP by E1a Variants with Mutations
3. Materials and Methods
3.1. Protein Expression and Purification

3.2. Overall Activity of NADH Production upon Assembly of E1a with E2o and E3
3.3. E1a-Specific Activity
3.4. Measurement for Formation of Adipoyl Semialdehyde by E1a from OP
3.5. Measurement of the Rate of H2O2 Production by Fluorescence Spectroscopy
3.6. Circular Dichroism Spectroscopy for Detection of Carboligase Products, (S)-2-Hydroxy-3-Oxo-Octanedioic and (S)-8-Ethoxy-7-Hydroxy-6,8-Dioxo-Octanoic Acid
3.7. Circular Dichroism Titration of E1a and E1a Variants by Adipoylphosphonic Acid and by Thiamin 2-Thiothiazolone Diphosphate
3.8. Reductive Acylation of LDo by E1a and OA or OP In Vitro as Detected by Fourier Transform Mass Spectrometry
3.9. Enzymatic Synthesis of Adipoyl-CoA
3.10. Fluorescence Titration of Pyrene-Labeled E2o1–173 Di-Domain by Arg715Cys E1a
3.11. Ligand Docking
4. Conclusions
- The functional promiscuity of the human E1a component has been demonstrated and adds to our understanding of the OADHc function in the L-lysine catabolism pathway. According to biochemical and structural findings, the E1a active centers accept OP with a one-carbon longer chain compared to the natural substrate OA, with no significant effect on the rate of decarboxylation and on the formation of the E1a-ThDP-enamine intermediate, as well as on all three side-reactions derived from the E1a-enamine intermediate. Importantly, the findings indicate that E1a is a better source of H2O2 production from OA and OP than E1o with its natural substrate OG, a finding that may potentially affect mitochondrial structure and function.
- The E2o component, an important enzyme in the TCA cycle, is known as a gatekeeper in intermediate channeling in 2-oxo acid dehydrogenase complexes [29,30,46,47,48,49,50]. Our findings suggest involvement of the E2o in the rate-limiting step at two levels in the mechanism, specifically, by controlling the rate of transfer of the acyl group between the E1a and E2o components (so called reductive acylation) as generally accepted, and by controlling the rate of transfer of the acyl group from S8-acyldihydrolipoyl-E2o to CoA in its active centers. At the same time, the rate of recycling of dihydrolipoyl-E2o to lipoyl-E2o proceeds faster [26,40,41].
- Evidence is presented that the E2o component of the 2-oxoglutarate dehydrogenase complex in the TCA cycle can produce acyl-CoA thioesters of different lengths, such as succinyl-CoA (C4), glutaryl-CoA (C5) and, less efficiently, adipoyl-CoA (C6), indicating its promiscuity, not recognized before.
- The E1a variants with mutations associated with AMOXAD and EoE affected the E1a-specific and the E1a-enamine side reactions with OP and showed a great similarity to that observed with OA. The mechanism underlying diminished E1a activity due to the Arg715Cys substitution is suggested. The data point to local conformational changes in the C-terminal region of the E1a homodimer, with consequences for ThDP cofactor and substrate binding, as well as for its assembly with E2o and E3.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Bunik, V.I.; Degtyarev, D. Structure-function relationships in the 2-oxo acid dehydrogenase family: Structure-specific signatures and functional predictions for the 2-oxoglutarate dehydrogenase-like proteins. Proteins 2008, 71, 874–890. [Google Scholar] [CrossRef]
- Nemeria, N.S.; Gerfen, G.; Nareddy, P.R.; Yang, L.; Zhang, X.; Szostak, M.; Jordan, F. The mitochondrial 2-oxoadipate and 2-oxoglutarate dehydrogenase complexes share their E2 and E3 components for their function and both generate reactive oxygen species. Free Radic. Biol. Med. 2018, 115, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Nemeria, N.S.; Gerfen, G.; Yang, L.; Zhang, X.; Jordan, F. Evidence for functional and regulatory crosstalk between the tricarboxylic acid cycle 2-oxoglutarate dehydrogenase complex and 2-oxoadipate dehydrogenase on the L-lysine, L-hydroxylysine and L-tryptophan degradation pathways from studies in vitro. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Leandro, J.; Dodatko, T.; Aten, J.; Nemeria, N.S.; Zhang, X.; Jordan, F.; Hendrickson, R.C.; Sanchez, R.; Yu, C.; DeVita, R.J.; et al. DHTKD1 and OGDHc display substrate overlap in cultured cells and form a hybrid 2-oxo acid dehydrogenase complex in vivo. Hum. Mol. Genet. 2020, 29, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhu, H.; Gu, M.; Luo, Q.; Ding, J.; Yao, Y.; Chen, F.; Wang, Z. DHTKD1 is essential for mitochondrial biogenesis and function maintenance. FEBS Lett. 2013, 587, 3587–3592. [Google Scholar] [CrossRef]
- Sherrill, J.D.; Kiran, K.C.; Wang, X.; Wen, T.; Chamberlin, A.; Stucke, E.M.; Collins, M.H.; Abonia, J.P.; Peng, Y.; Wu, Q.; et al. Whole-exome sequencing uncovers oxidoreductases DHTKD1 and OGDHL as linkers between mitochondrial dysfunction and eosinophilic esophagitis. JCI Insight 2018, 3, e99922. [Google Scholar] [CrossRef]
- Wang, C.; Calcutt, M.W.; Ferguson, J.F. Knock-out of DHTKD1 alters mitochondrial respiration and function and may represent a novel pathway in cardiometabolic disease risk. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef]
- Danhauser, K.; Sauer, S.W.; Haack, T.B.; Wieland, T.; Staufner, C.; Graf, E.; Zschocke, J.; Strom, T.M.; Traub, T.; Okun, J.G.; et al. DHTKD1 mutations cause 2-aminoadipic and 2-oxoadipic aciduria. Am. J. Hum. Gen. 2012, 91, 1082–1087. [Google Scholar] [CrossRef]
- Stiles, A.R.; Venturoni, L.; Mucci, G.; Elbalalesy, N.; Woontner, M.; Goodmann, S.; Abdenur, J.E. New cases of DHTKD1 mutations in patients with 2-ketoadipic aciduria. JIMD Rep. 2015, 25, 15–19. [Google Scholar]
- Hagen, J.; Brinke, H.; Wanders, R.J.A.; Knegt, A.C.; Oussoren, E.; Hoogeboom, A.J.M.; Ruijter, G.J.G.; Becker, D.; Schwab, K.O.; Franke, I.; et al. Genetic basis of alpha-aminoadipic and alpha-ketoadipic aciduria. J. Inherit. Metab. Dis. 2015, 38, 873–879. [Google Scholar] [CrossRef]
- Xu, W.Y.; Gu, M.M.; Sun, L.H.; Guo, W.T.; Zhu, H.B.; Ma, J.F.; Yuan, W.T.; Kuang, Y.; Ji, B.J.; Wu, X.L.; et al. A nonsense mutation in DHTKD1 causes Charcot-Marie-Tooth type 2 in a large Chinese pedigree. Am. J. Hum. Genet. 2012, 91, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.Y.; Zhu, H.; Shen, Y.; Wan, Y.H.; Tu, X.D.; Wu, W.T.; Tang, L.; Zhang, H.X.; Lu, S.Y.; Jin, X.L.; et al. DHTKD1 deficiency causes Charcot-Marie-Tooth disease in mice. Mol. Cell Biol. 2018, 38, e00085-18. [Google Scholar] [CrossRef] [PubMed]
- Karakaya, M.; Storbeck, M.; Strathmann, E.A.; Delle Vedove, A.; Hölker, I.; Altmueller, J.; Naghiyeva, L.; Schmitz-Steinkrüger, L.; Vezyroglou, K.; Motameny, S.; et al. Targeted sequencing with expanded genes profile enables high diagnostic yield in non-5q-spinal muscular atrophies. Hum. Mutat. 2018, 39, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Osmanovic, A.; Gogol, I.; Martens, H.; Widjaja, M.; Müller, K.; Schreiber-Katz, O.; Feuerhake, F.; Langhans, C.-D.; Schmidt, G.; Anderson, P.M.; et al. Heterozygous DHTKD1 Variants in Two European Cohorts of Amyotrophic Lateral Sclerosis patients. Genes 2022, 13, 84. [Google Scholar] [CrossRef]
- Biagosch, C.; Ediga, R.D.; Hensler, S.V.; Faerberboeck, M.; Kuehn, R.; Wurst, W.; Meitinger, T.; Kölker, S.; Sauer, S.; Prokisch, H. Elevated glutaric acid levels in Dhtkd1-/Gcdh—Double knockout mice challenge our current understanding of lysine metabolism. Biochim. Biophys. Acta Mol. Basis. Dis. 2017, 1863, 2220–2228. [Google Scholar] [CrossRef]
- Leandro, J.; Khamrui, S.; Wang, H.; Suebsuwong, C.; Nemeria, N.S.; Huynh, K.; Moustakim, M.; Secor, C.; Wang, M.; Dodatko, T.; et al. Inhibition and crystal structure of the human DHTKD1-thiamin diphosphate complex. ACS Chem. Biol. 2020, 15, 2041–2047. [Google Scholar] [CrossRef] [PubMed]
- Blickling, S.; Renner, C.; Laber, B.; Pohlenz, H.-D.; Holak, T.A.; Hubner, R. Reaction mechanism of Escherichia coli dihydrodipicolinate synthase investigated by X-ray crystallography and NMR Spectroscopy. Biochemistry 1997, 36, 24–33. [Google Scholar] [CrossRef]
- Shrivastava, P.; Navratna, V.; Silla, Y.; Dewangan, R.P.; Pramanik, A.; Chaudhary, S.; Rayasam, G.; Kumar, A.; Gopal, B.; Ramachandran, S. Inhibition of Mycobacterium tuberculosis dihydro-dipicolinate synthase by alpha-ketopimelic acid and its other structural analogues. Sci. Rep. 2016, 6, 30827. [Google Scholar] [CrossRef]
- Manandhar, M.; Cronan, J.E. Pimelic acid, the first precursor of the Bacillus subtilis biotin synthesis pathway, exists as the free acid and is assembled by fatty acid synthesis. Mol. Microbiol. 2017, 104, 595–607. [Google Scholar] [CrossRef]
- Hu, Y.; Cronan, J.E. α-Proteobacteria synthesize biotin precursor pimeloyl-CoA using BioZ 3-ketoacyl-ACP synthase and lysine catabolism. Nat. Commun. 2020, 11, 5598. [Google Scholar] [CrossRef]
- Cronan, J.E. Advances in synthesis of biotin and assembly of lipoic acid. Curr. Opin. Chem. Biol. 2018, 47, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Sirithanakorn, C.; Cronan, J.E. Biotin, a universal and essential cofactor: Synthesis, ligation and regulation. FEMS Microbiol. Rev. 2021, 45, fuab003. [Google Scholar] [CrossRef] [PubMed]
- Westin, M.A.K.; Hunt, M.C.; Alexson, S.E.H. The identification of a succinyl-CoA thioesterase suggests a novel pathway for succinate production in peroxisomes. J. Biol. Chem. 2005, 280, 38125–38132. [Google Scholar] [CrossRef] [PubMed]
- Bouatra, S.; Aziat, F.; Mandal, R.; Guo, A.C.; Wilson, M.R.; Knox, C.; Bjorndahl, T.C.; Krishnamurthy, R.; Saleem, F.; Liu, P.; et al. The Human Urine Metabolome. PLoS ONE 2013, 8, e73076. [Google Scholar]
- Fiermonte, G.; Dolce, V.; Palmieri, L.; Ventura, M.; Runswick, M.J.; Palmieri, F.; Walker, J.E. Identification of the human mitochondrial oxodicarboxylate carrier: Bacterial expression, reconstitution, functional characterization, tissue distribution, and chromosomal location. J. Biol. Chem. 2001, 276, 8225–8230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Nemeria, N.S.; Leandro, J.; Houten, S.M.; Lazarus, M.; Gerfen, G.; Ozohanics, O.; Ambrus, A.; Nagy, B.; Brukh, R.; et al. Structure-function analyses of the G729R 2-oxoadipate dehydrogenase genetic variant associated with disorder of L-lysine metabolism. J. Biol. Chem. 2020, 295, 8078–8095. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Nemeria, N.S.; Chandrasekhar, K.; Kumaran, S.; Arjunan, P.; Reynolds, S.; Calero, G.; Brukh, R.; Kakalis, L.; Jordan, F. Structure and function of the catalytic domain of the dihydrolipoyl acetyltransferase component in Escherichia coli pyruvate dehydrogenase complex. J. Biol. Chem. 2014, 289, 15215–15230. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, G.; Foster, W.R.; Bailey, H.J.; Hicks, K.G.; Sauer, S.W.; Dimitrow, B.; McCorvie, T.J.; Okum, J.G.; Rutter, J.; Kolker, S.; et al. Crystal structure and interaction studies of human DHTKD1 provide insight into a mitochondrial megacomplex in lysine catabolism. Int. Union Cryst. J. 2020, 7, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Nagy, B.; Polak, M.; Ozohanics, O.; Zambo, Z.; Szabo, E.; Hubert, A.; Jordan, F.J.; Novaček, J.; Adam-Vizi, V.; Ambrus, A. Structure of the dihydrolipoamide succinyltransferase (E2) component of the human alpha-ketoglutarate dehydrogenase complex (hKGDHc) revealed by cryo-EM and cross-linking mass spectrometry: Implications for the overall hKGDHc structure. Biochim. Biophys. Acta-Gen. Subj. 2021, 1865, 129889. [Google Scholar] [CrossRef]
- Mattevi, A.; Obmolova, G.; Kalk, K.H.; Teplyakov, A.; Hol, W.G. Crystallographic analysis of substrate binding and catalysis in dihydrolipoyl transacetylase (E2p). Biochemistry 1993, 32, 3887–3901. [Google Scholar] [CrossRef]
- Schrödinger LLC. The PyMOL Molecular Graphics System; Version 2; Schrödinger LLC: New York, NY, USA, 2015; Available online: https://pymol.org/ (accessed on 1 January 2020).
- Zhou, J.; Yang, L.; Ozohanics, O.; Zhang, X.; Wang, J.; Ambrus, A.; Arjunan, P.; Brukh, R.; Nemeria, N.S.; Furey, W.; et al. A multipronged approach unravels unprecedented protein-protein interactions in the human 2-oxoglutarate dehydrogenase multienzyme complex. J. Biol. Chem. 2018, 293, 19213–19227. [Google Scholar] [CrossRef]
- Nemeria, N.; Yan, Y.; Zhang, Z.; Brown, A.M.; Arjunan, P.; Furey, W.; Guest, J.R.; Jordan, F. Inhibition of the Escherichia coli pyruvate dehydrogenase complex E1 subunit and its tyrosine 177 variants by thiamin 2-thiazolone and thiamin 2-thiothiazolon diphosphates. Evidence for reversible tight-binding inhibition. J. Biol. Chem. 2001, 276, 45969–45978. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Syobodová, R.; Berka, K.; Bazgier, V.; Valankar, S.; Burley, S.K.; Koća, J.; Rose, A.S. Mol* Viewer; modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]
- Nemeria, N.S.; Gerfen, G.; Guevara, E.; Nareddy, P.P.; Szostak, M.; Jordan, F. The human Krebs cycle 2-oxoglutarate dehydrogenase complex creates an additional source of superoxide/hydrogen peroxide from 2-oxoadipate as alternative substrate. Free Radic. Biol. Med. 2017, 108, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Nemeria, N.S.; Ambrus, A.; Patel, H.; Gerfen, G.; Adam-Vizi, V.; Tretter, L.; Zhou, J.; Wang, J.; Jordan, F. Human 2-oxoglutarate dehydrogenase complex E1 component forms a thiamin-derived radical by aerobic oxidation of the enamine intermediate. J. Biol. Chem. 2014, 289, 29859–29873. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Perevoschikova, I.V.; Goncalves, R.L.S.; Hey-Mogensen, M.; Brand, M.D. The determination and analysis of site-specific rates of mitochondria reactive oxygen species production. Methods Enzymol. 2013, 526, 189–217. [Google Scholar]
- Vinogradov, M.; Kaplun, A.; Vyazmensky, M.; Engel, S.; Golbik, R.; Tittmann, K.; Uhlemann, K.; Meshalkina, L.; Barak, Z.; Hübner, G.; et al. Monitoring the acetohydroxy acid synthase reaction and related carboligations by circular dichroism spectroscopy. Anal. Biochem. 2005, 342, 126–133. [Google Scholar] [CrossRef]
- Nemeria, N.S.; Shome, B.; DeColli, A.A.; Heflin, K.; Begley, T.P.; Meyers Freel, C.; Jordan, F. Competence of thiamin diphosphate-dependent enzymes with 2′-methoxythiamin diphosphate derived from bacimethrin, a naturally occurring thiamin anti-vitamin. Biochemistry 2016, 55, 1135–1148. [Google Scholar] [CrossRef]
- Chakraborty, J.; Nemeria, N.S.; Zhang, X.; Nareddy, P.R.; Szostak, M.; Farinas, E.; Jordan, F. Engineering 2-oxoglutarate dehydrogenase to a 2-oxo aliphatic dehydrogenase complex by optimizing consecutive components. AIChE J. 2019, 66, e16769. [Google Scholar] [CrossRef]
- Chakraborty, J.; Nemeria, N.S.; Farinas, E.; Jordan, F. Catalysis of transthiolacylation in the active centers of dihydrolipoamide acyltransacetylase components of 2-oxo acid dehydrogenase complexes. FEBS Open Bio 2018, 8, 880–896. [Google Scholar] [CrossRef]
- Parthasarathy, A.; Pierik, A.J.; Kahnt, J.; Zelder, O.; Buckel, W. Substrate specificity of 2-hydroxyglutaryl-CoA dehydratase from Clostridium symbiosum: Toward a bio-based production of adipic acid. Biochemistry 2011, 50, 3540–3550. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, G.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Chovancova, E.; Pavelka, A.; Benes, P.; Stmad, O.; Brezovsky, J.; Kozlikova, B.; Gora, A.; Sustr, V.; Klvana, M.; Medek, P.; et al. CAVER 3: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures. PLoS Comput. Biol. 2012, 8, e1002708. [Google Scholar] [CrossRef]
- Jordan, F.; Nemeria, N.S.; Gerfen, G. Human 2-oxoglutarate dehydrogenase and 2- oxoadipate dehydrogenase both generate superoxide/H2O2 in a side reaction and each could contribute to oxidative stress in mitochondria. Neurochem. Res. 2019, 44, 2325–2335. [Google Scholar] [CrossRef]
- Fries, M.; Stott, K.M.; Reynolds, S.; Perham, R.N. Distinct modes of recognition of the lipoyl domain as substrate by the E1 and E3 components of the pyruvate dehydrogenase multienzyme complex. J. Mol. Biol. 2007, 366, 132–139. [Google Scholar] [CrossRef]
- Jones, D.D.; Perham, R.N. The role of loop and beta-turn residues as structural and functional determinants for the lipoyl domain from the Escherichia coli 2-oxoglutarate dehydrogenase complex. Biochem. J. 2008, 409, 357–366. [Google Scholar] [CrossRef]
- Stott, K.M.; Yusof, A.M.; Perham, R.H.; Jones, D.D. A surface loop directs conformational switching of a lipoyl domain between a folded and a novel misfolded structure. Structure 2009, 17, 1117–1127. [Google Scholar] [CrossRef][Green Version]
- Kato, M.; Wynn, R.M.; Chuang, J.L.; Brautigam, C.A.; Custorio, M.; Chuang, D.T. A synchronized substrate-gating mechanism revealed by cubic-core structure of the bovine branched-chain α-ketoacid dehydrogenase complex. EMBO J. 2006, 25, 5983–5994. [Google Scholar] [CrossRef]
- Škerlová, J.; Berndtsson, J.; Nolte, H.; Ott, M.; Stenmark, P. Structure of the native pyruvate dehydrogenase complex reveals the mechanism of substrate insertion. Nat. Commun. 2021, 12, 5277. [Google Scholar] [CrossRef]
- Jordan, F. Current mechanistic understanding of thiamin diphosphate-dependent enzymatic reactions. Nat. Prod. Rep. 2003, 20, 184–201. [Google Scholar] [CrossRef]
- Baykal, A.; Chakraborty, S.; Dodoo, A.; Jordan, F. Synthesis with good enantiomeric excess of both enantiomers of α-ketols and acetolactates by two thiamin diphosphate-dependent decarboxylases. Bioorg. Chem. 2006, 34, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Brandt, G.S.; Kneen, M.M.; Petsko, G.A.; Ringe, D.; McLeish, M.J. Active -site engineering of benzaldehyde lyase shows that point mutation can confer both new reactivity and susceptibility to mechanism-based inhibition. J. Am. Chem. Soc. 2010, 132, 438–439. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.; Walter, L.; Kolter, G.; Pohl, M.; Müller, M.; Tittmann, K. Conversion of pyruvate decarboxylase into enantioselective carboligase with biosynthetic potential. J. Am. Chem. Soc. 2011, 133, 3609–3616. [Google Scholar] [CrossRef]
- Hailes, H.C.; Rother, D.; Müller, M.; Westphal, R.; Ward, J.M.; Pleiss, J.; Vogel, C.; Pohl, M. Engineering stereoselectivity of ThDP-dependent enzymes. FEBS J. 2013, 280, 6374–6394. [Google Scholar] [CrossRef]
- Müller, M.; Sprenger, G.A.; Pohl, M. C-C bond formation using ThDP-dependent lyases. Curr. Opin. Chem. Biol. 2013, 17, 261–270. [Google Scholar] [CrossRef]
- Beigi, M.; Waltzer, S.; Zarei, A.; Eggeling, L.; Sprenger, G.A.; Müller, M. TCA cycle involved enzymes SucA and Kgd, as well as MenD: Efficient biocatalysts for asymmetric C-C bond formation. Org. Lett. 2013, 15, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Kasparyan, E.; Richter, M.; Dresen, C.; Walter, L.S.; Fuchs, G.; Leeper, F.J.; Wacker, T.; Andrade, S.L.A.; Kolter, G.; Pohl, M.; et al. Asymmetric Stetter reactions catalyzed by thiamine diphosphate-dependent enzymes. Appl. Microbiol. Biotechnol. 2014, 98, 9681–9690. [Google Scholar] [CrossRef] [PubMed]
- Baraibar, A.G.; Lieres, E.; Wiechert, W.; Pohl, M.; Rother, D. Effective production of (S)-α-hydroxy ketones: A reaction engineering approach. Top Catal. 2014, 57, 401–411. [Google Scholar] [CrossRef]
- Schapfl, M.; Baier, S.; Fries, A.; Ferlaino, S.; Waltzer, S.; Müller, M.; Sprenger, G.A. Extended substrate range of thiamin diphosphate-dependent MenD enzyme by coupling of two C-C-bonding reactions. Appl. Microbiol. Biotechn. 2018, 102, 8359–8372. [Google Scholar] [CrossRef]
- Kulig, J.; Sehl, T.; Mackfeld, U.; Wiechert, W.; Pohl, M.; Rother, D. An Enzymatic 2-step cofactor and co-product recycling cascade towards a chiral 1,2-diol. Part I: Cascade design. Adv. Synth. Catal. 2019, 361, 2607–2615. [Google Scholar] [CrossRef]
- Shim, D.J.; Nemeria, N.S.; Balakrishnan, A.; Patel, H.; Song, J.; Jordan, F.; Farinas, E.T. Assignment of function to Histidines 260 and 298 by engineering the E1 component of the Escherichia coli 2-oxoglutarate dehydrogenase complex; substitutions that lead to acceptance of substrates lacking the 5-carboxyl group. Biochemistry 2011, 50, 7705–7709. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Overall NADH Activity a,e | E1a-Specific Activity b | ||
|---|---|---|---|
| μmol·min−1·mg E1a−1 | μmol·min−1·mg E1a−1 | ||
| OA | OP | OA | OP |
| 1.38 ± 0.25 | 0.012 ± 0.002 | 1.86 ± 0.09 | 1.56 ± 0.15 |
| kcat = 4.8 ± 0.9 s−1 | kcat = 0.04 ± 0.003 s−1 | kcat = 6.5 s−1 | kcat = 5.4 s−1 |
| kcat/Km = 400 × 103 M−1 s−1 | kcat/Km = 1.1 × 103 M−1 s−1 | kcat/Km = 1326 × 103 M−1 s−1 | kcat/Km = 146 × 103 M−1 s−1 |
| Glutarylation/Adipoylation c | Glutaryl-CoA formation d | Adipoyl-CoA formation d | |
| kglutaryl = 8.2 s−1 | kadipoyl= 0.028 s−1 | kglutaryl-CoA = 0.245 μM−1 s−1 | kadipoyl-CoA = 0.003 μM−1 s−1 |
| E1a carboligation activity f | H2O2 activity of E1a f | ||
| μmol ·min−1·mg E1a−1 | nmol min−1∙mg E1a−1 | ||
| OA | OP | OA | OP |
| 1.04 ± 0.17 | 1.46 ± 0.05 | 8.44 ± 1.69 | 8.36 ± 0.78 |
| kcat = 3.6 ± 0.41 s−1 | kcat = 5.1 ± 0.15 s−1 | kcat = 0.029 ± 0.006 s−1 | kcat = 0.029 ± 0.003 s−1 |
| kcat/Km = 736 × 103 M−1 s−1 | kcat/Km = 138 × 103 M−1 s−1 | kcat/Km = 5.9 × 103 M−1 s−1 | kcat/Km = 0.78 × 103 M−1 s−1 |
| Glutaric acid semialdehyde formation f | Adipic acid semialdehyde formation f | ||
| nmol·min−1·mg E1a−1 | nmol·min−1·mg E1a−1 | ||
| 6.05 ± 0.46 | 5.7 ± 1.1 | ||
| kcat = 0.021 ± 0.002 s−1 | kcat = 0.020 ± 0.003 s−1 | ||
| E1a Substitutions | E1a-Specific Activity c (μmol min−1·mg E1a−1) | H2O2 Activity (nmol·min−1·mg E1a−1) | E1a Carboligation Activity (μmol·min−1·mg E1a−1) | |||
|---|---|---|---|---|---|---|
| OA | OP | OA | OP | OA | OP | |
| None a | 1.86 ± 0.09 (100%) | 1.56 ± 0.15 (100%) | 8.44 ± 1.69 (100%) | 8.37 ± 0.78 (100%) | 1.04 ± 0.17 (100%) | 1.46 ± 0.05 (100%) |
| E1a variants associated with AMOXAD | ||||||
| Leu234Gly | 0.081 ± 0.007 (4.35%) | 0.091 ± 0.01 (5.8%) | 0.085 ± 0.024 (1.0%) | 0.070 ± 0.002 (0.84%) | 0.038 ± 0.006 (3.7%) | 0.041 ± 0.009 (2.8%) |
| Gln305His | 1.23 ± 0.03 (66%) | 1.00 ± 0.04 (64%) | 5.42 ± 0.89 (64%) | 8.37 ± 0.47 (100%) | 1.01 ± 0.11 (97%) | 1.34 ± 0.03 (92%) |
| Arg455Gln | 0.62 ± 0.09 (33%) | 0.352 ± 0.019 (23%) | 0.054 ± 0.004 (0.6%) | 0.32 ± 0.07 (3.8%) | 0.048 ± 0.012 (4.6%) | 0.043 ± 0.001 (3%) |
| Arg715Cys | 0.009 ± 0.001 (0.5%) | 0.007 ± 0.001 (0.45%) | 0.034 ± 0.002 (0.4%) | N/D | 0.02 ± 0.003 (1.9%) | 0.014 ± 0.002 (0.96%) |
| Pro773 Leu | 0.592 ± 0.031 (32%) | 0.543 ± 0.037 (35%) | 3.27 ± 0.11 (39%) | 4.88 ± 0.13 (58%) | 0.70 ± 0.02 (67%) | 0.902 ± 0.047 (62%) |
| E1a variants associated with EoE | ||||||
| None b | 0.54 ± 0.026 (100%) | 0.47 ± 0.01 (100%) | 1.41 ± 0.10 (100%) | 1.69 ± 0.08 (100%) | 0.268 ± 0.027 (100%) | 0.214 ± 0.013 (100%) |
| Arg163Gln | 0.037 ± 0.001 (6.9%) | 0.020 ± 0.004 (4.3%) | 0.113 ± 0.003 (8%) | 0.111 ± 0.004 (6.5%) | 0.021 ± 0.005 (7.8 %) | 0.012 ± 0.003 (5.6%) |
| Val360Ala | 0.195 ± 0.030 (36%) | 0.142 ± 0.007 (30%) | 0.29 ± 0.02 (21%) | 0.35 ± 0.030 (21%) | 0.085 ± 0.006 (32%) | 0.070 ± 0.02 (32.7%) |
| Ser862Ile | 0.571 ± 0.023 (100%) | 0.48 ± 0.01 (100%) | 1.08 ± 0.09 (77%) | 1.28 ± 0.05 (76%) | 0.296 ± 0.008 (100%) | 0.202 ± 0.029 (94%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemeria, N.S.; Nagy, B.; Sanchez, R.; Zhang, X.; Leandro, J.; Ambrus, A.; Houten, S.M.; Jordan, F. Functional Versatility of the Human 2-Oxoadipate Dehydrogenase in the L-Lysine Degradation Pathway toward Its Non-Cognate Substrate 2-Oxopimelic Acid. Int. J. Mol. Sci. 2022, 23, 8213. https://doi.org/10.3390/ijms23158213
Nemeria NS, Nagy B, Sanchez R, Zhang X, Leandro J, Ambrus A, Houten SM, Jordan F. Functional Versatility of the Human 2-Oxoadipate Dehydrogenase in the L-Lysine Degradation Pathway toward Its Non-Cognate Substrate 2-Oxopimelic Acid. International Journal of Molecular Sciences. 2022; 23(15):8213. https://doi.org/10.3390/ijms23158213
Chicago/Turabian StyleNemeria, Natalia S., Balint Nagy, Roberto Sanchez, Xu Zhang, João Leandro, Attila Ambrus, Sander M. Houten, and Frank Jordan. 2022. "Functional Versatility of the Human 2-Oxoadipate Dehydrogenase in the L-Lysine Degradation Pathway toward Its Non-Cognate Substrate 2-Oxopimelic Acid" International Journal of Molecular Sciences 23, no. 15: 8213. https://doi.org/10.3390/ijms23158213
APA StyleNemeria, N. S., Nagy, B., Sanchez, R., Zhang, X., Leandro, J., Ambrus, A., Houten, S. M., & Jordan, F. (2022). Functional Versatility of the Human 2-Oxoadipate Dehydrogenase in the L-Lysine Degradation Pathway toward Its Non-Cognate Substrate 2-Oxopimelic Acid. International Journal of Molecular Sciences, 23(15), 8213. https://doi.org/10.3390/ijms23158213