
Bacillus Calmette–Guérin-Induced Human Mast Cell Activation Relies on IL-33 Priming

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
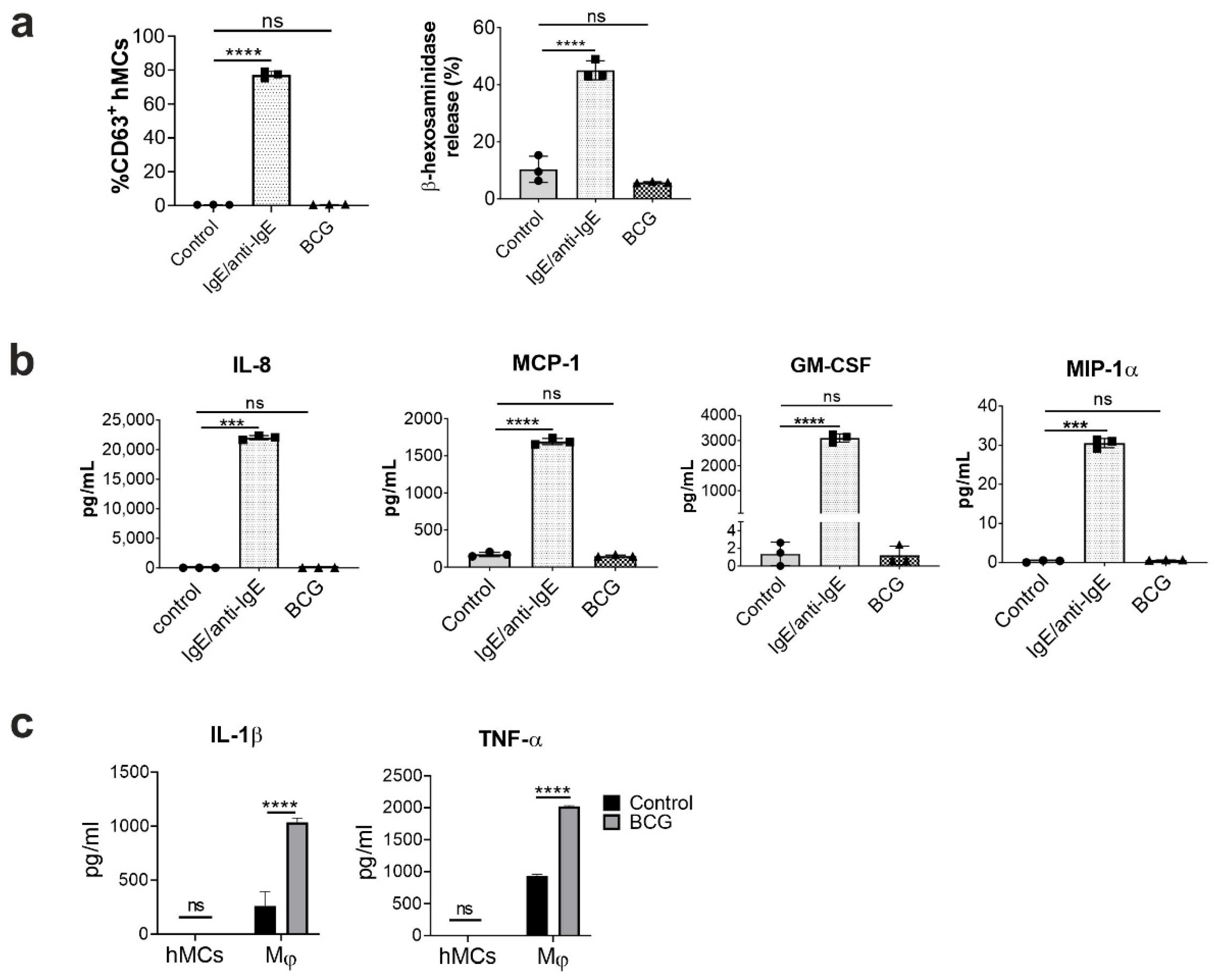
2.1. Human Mast Cells Are Resistant to BCG-Induced Activation
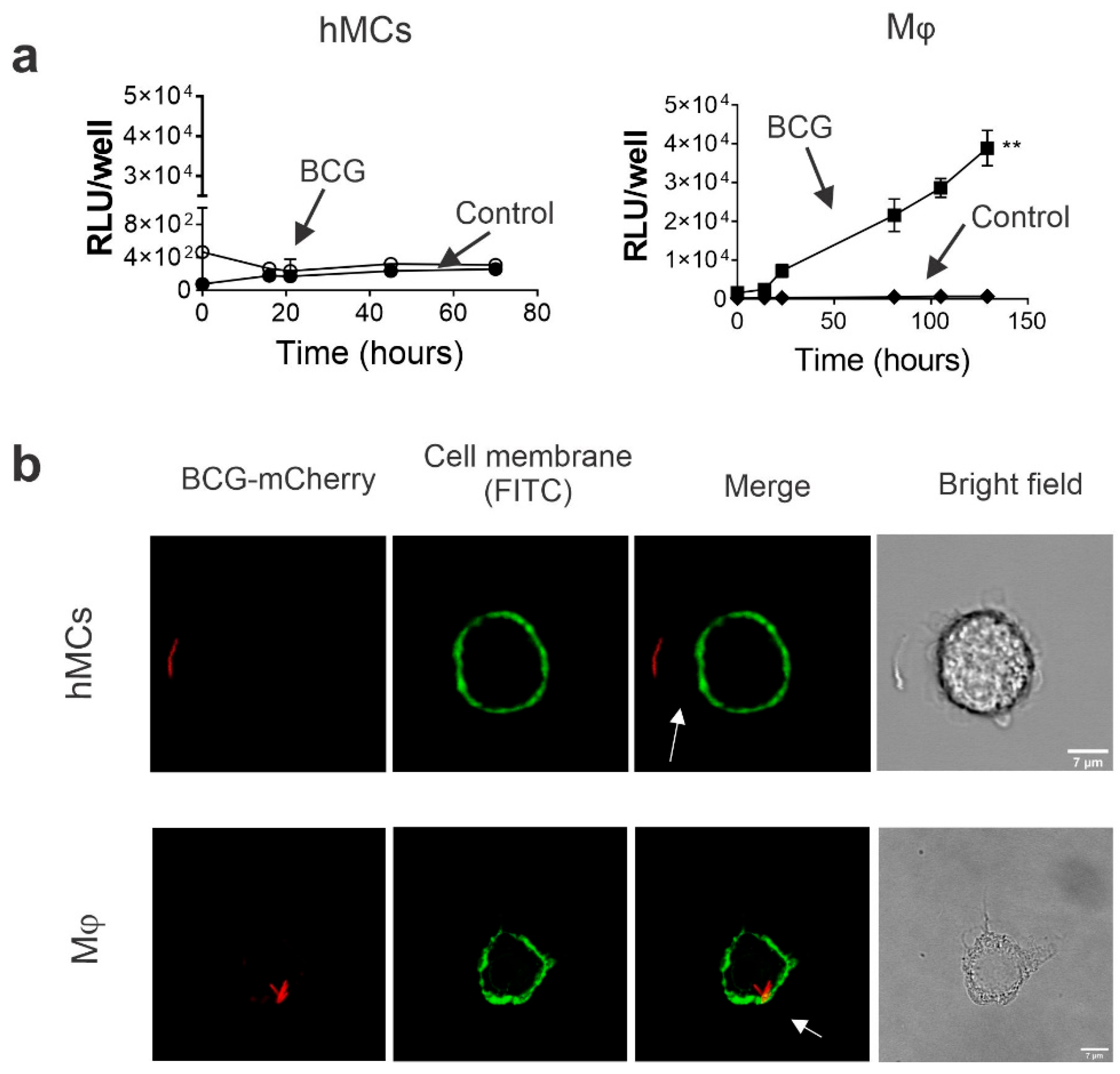
2.2. Human MCs Do Not Internalize and Promote BCG Intracellular Growth
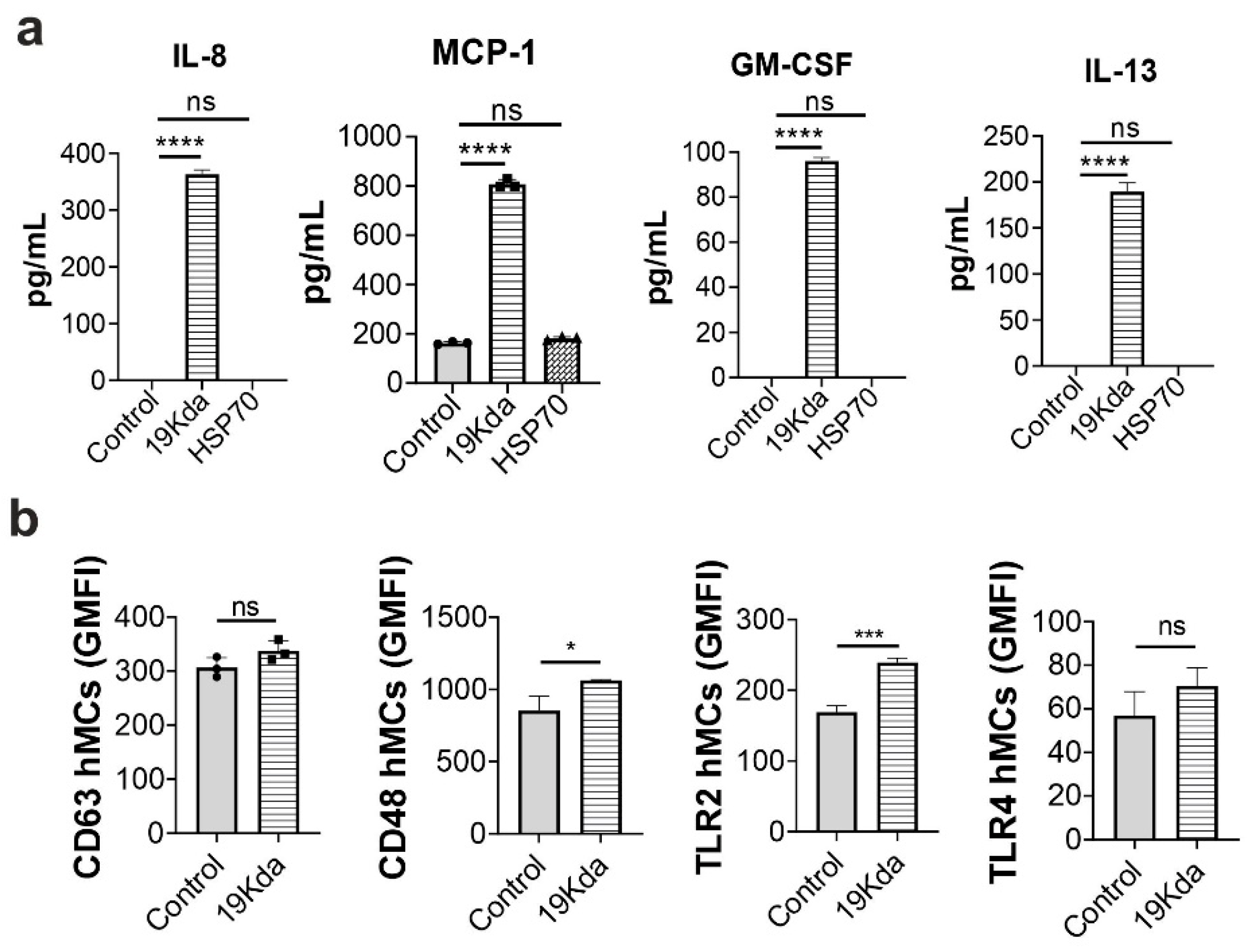
2.3. The Mtb 19-KDa Lipoprotein Induces CD48 Expression and Cytokines Secretion in hMCs
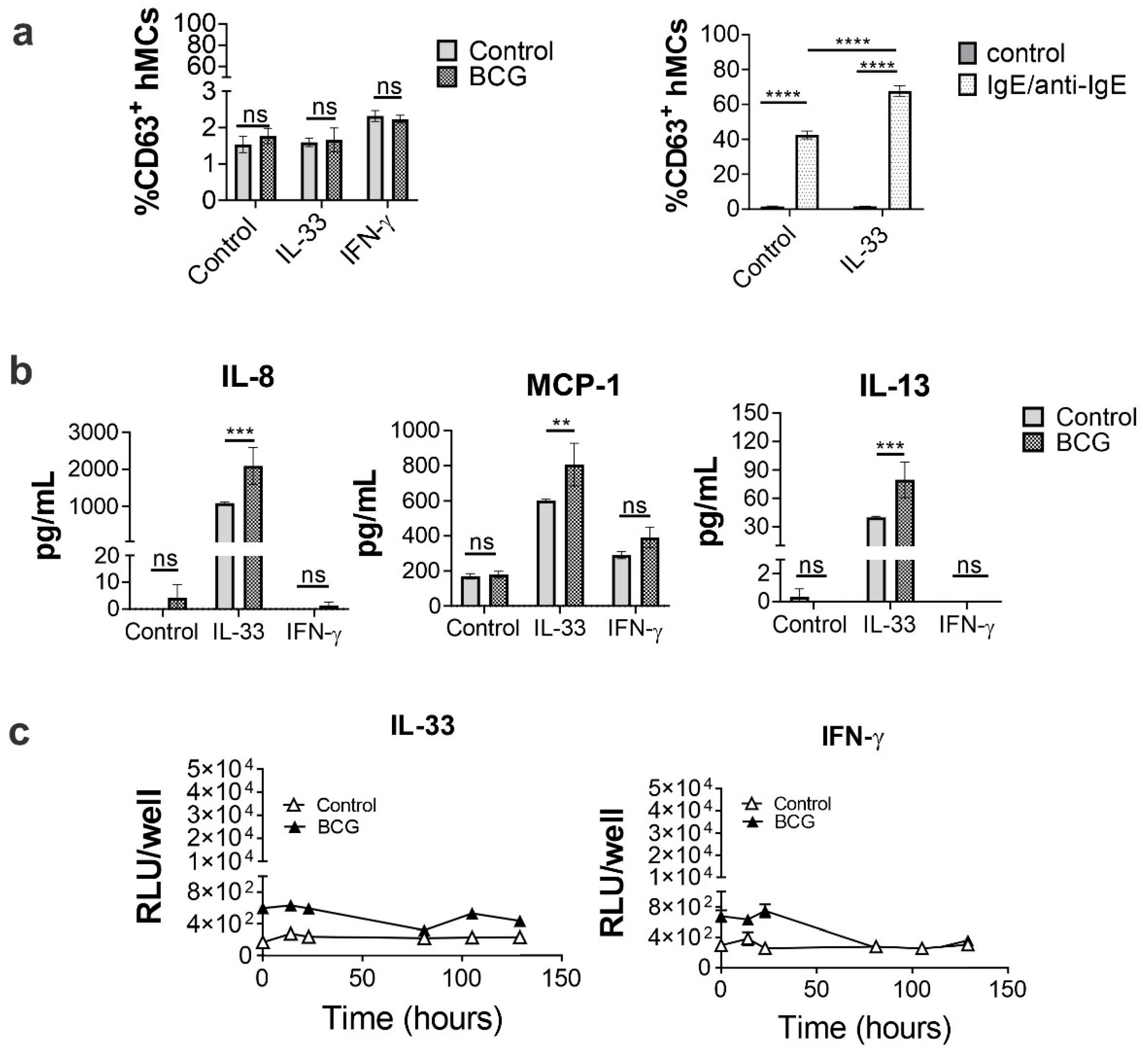
2.4. IL-33 Priming Alters hMC Responses upon BCG Exposure
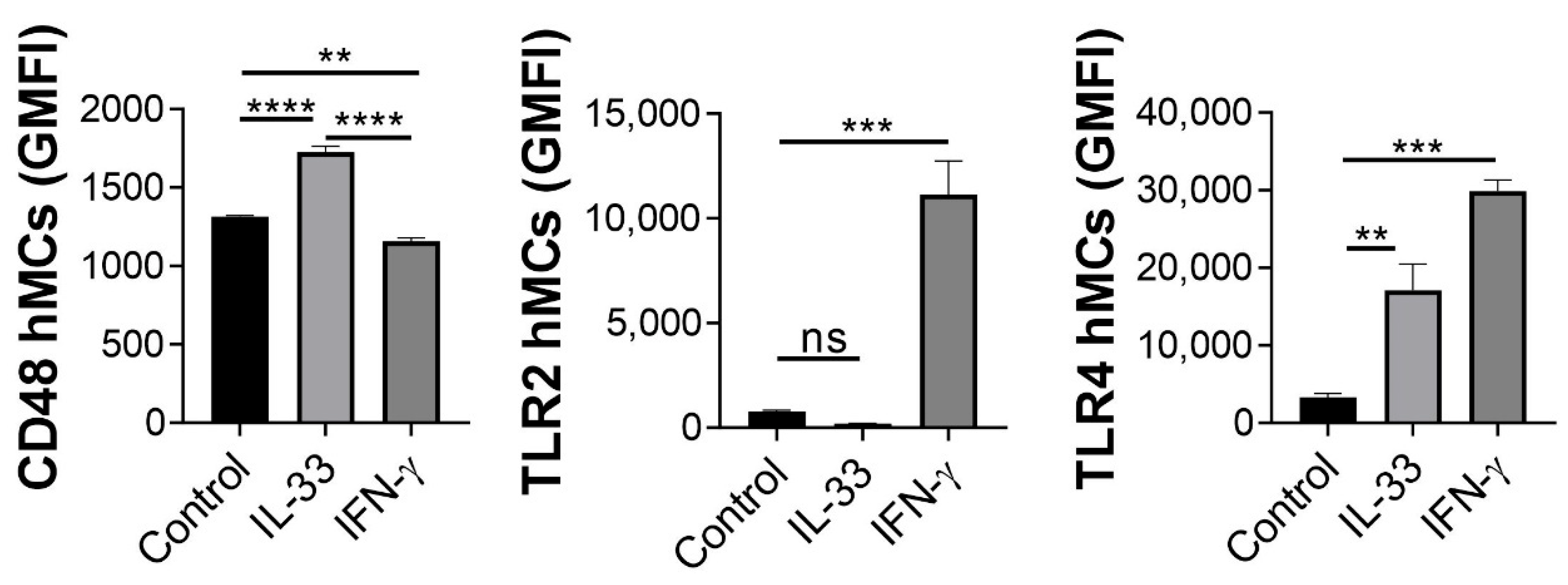
2.5. IL-33 Enhances hMC CD48 Expression
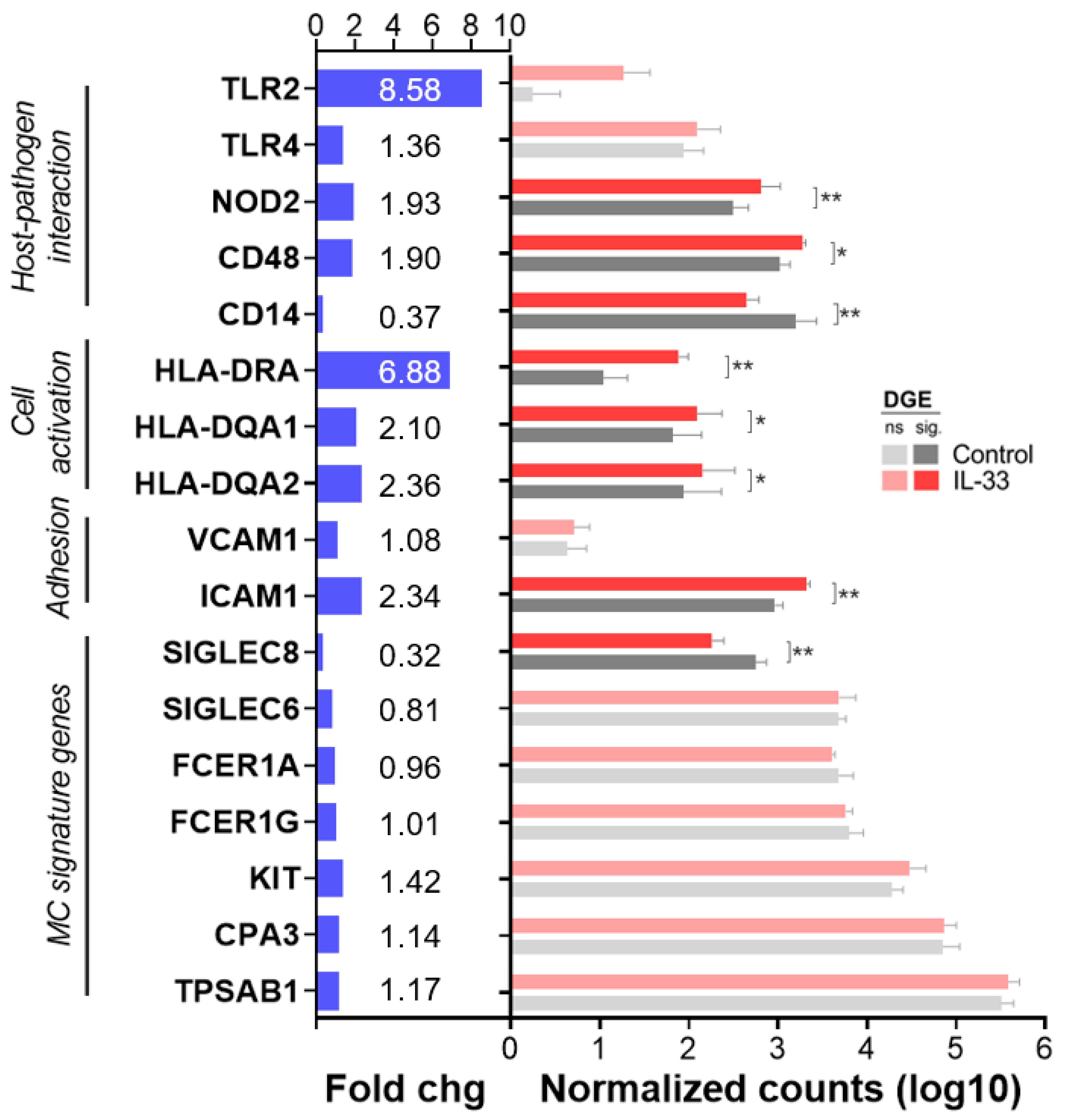
2.6. IL-33 Increases CD48, NOD2, ICAM-1 and MHC Class II mRNA Transcription in hMCs
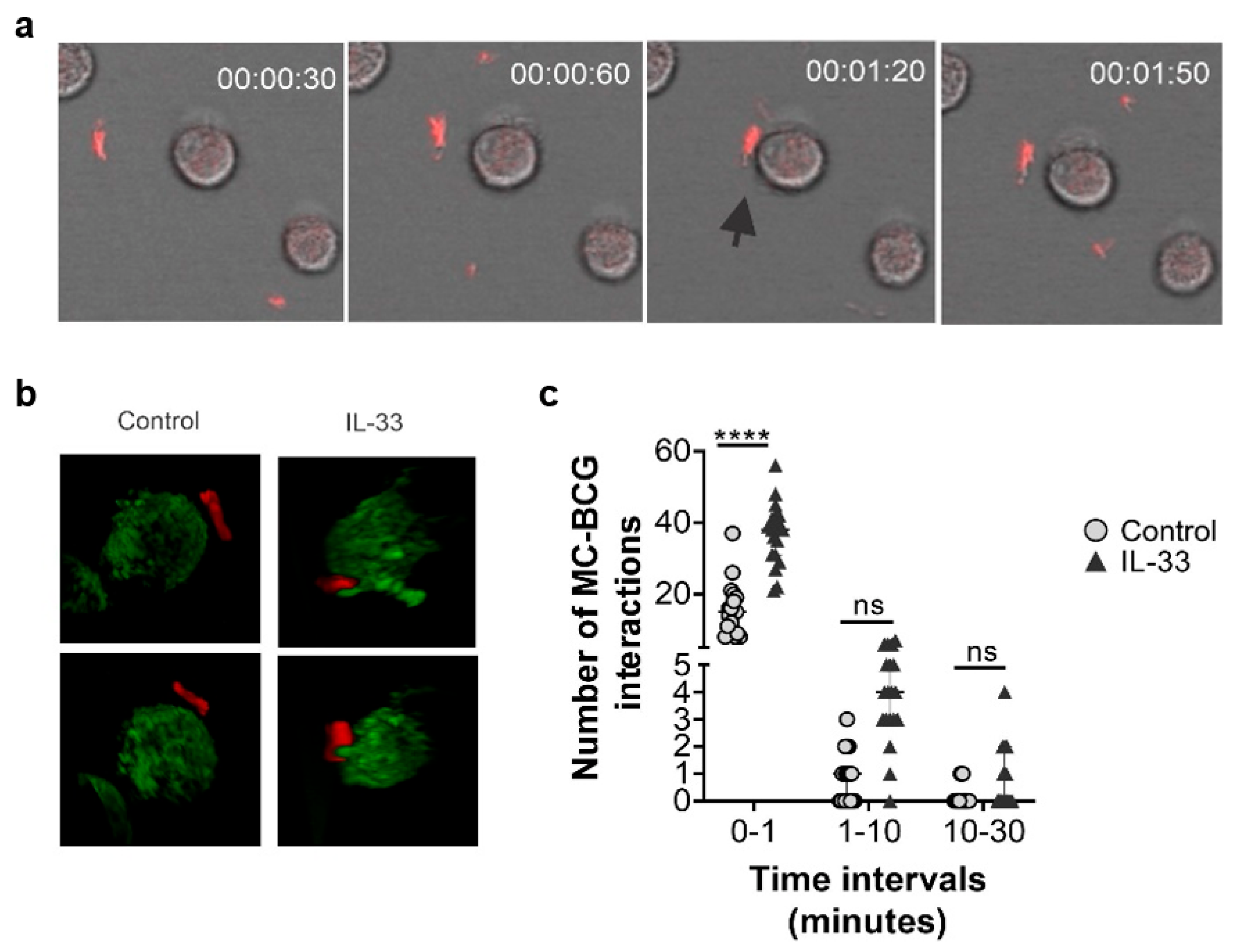
2.7. Human MC Primed with IL-33 Establish Higher Numbers of Rapid Interactions with BCG
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Cellular Culture
4.3. Bacterial Cultures
4.4. Cellular Priming, Stimulation and Infection
4.5. Flow Cytometry
4.6. Cytokine and Chemokine Secretion
4.7. β-Hexosaminidase Assay
4.8. BCG Intracellular Growth and Internalization
4.9. Confocal Microscopy
4.10. mRNA Extraction, Sequencing and Differential Gene Expression Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Dockrell, H.M.; Smith, S.G. What Have We Learnt about BCG Vaccination in the Last 20 Years? Front. Immunol. 2017, 8, 1134. [Google Scholar] [CrossRef]
- Davenne, T.; McShane, H. Why don’t we have an effective tuberculosis vaccine yet? Expert Rev. Vaccines 2016, 15, 1009–1013. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki-Nakashimada, M.A.; Unzueta, A.; Berenise Gamez-Gonzalez, L.; González-Saldaña, N.; Sorensen, R. BCG: A vaccine with multiple faces. Hum. Vaccines Immunother. 2020, 16, 1841–1850. [Google Scholar] [CrossRef] [PubMed]
- Kowalewicz-Kulbat, M.; Locht, C. BCG and protection against inflammatory and auto-immune diseases. Expert Rev. Vaccines 2017, 16, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wang, Y.; Zhao, H.; Yao, W.; Yu, X. Anti-allergic action of bacillus Calmette-Guerin extract in experimental mast cell-mediated anaphylactic models. Mol. Med. Rep. 2017, 16, 6248–6254. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oseph, M.; Enting, D. Immune Responses in Bladder Cancer-Role of Immune Cell Populations, Prognostic Factors and Therapeutic Implications. Front. Oncol. 2019, 9, 1270. [Google Scholar]
- Yan, S.; Liu, R.; Mao, M.; Liu, Z.; Zhang, W.; Zhang, Y.; Li, J.; Peng, C.; Chen, X. Therapeutic effect of Bacillus Calmette-Guerin polysaccharide nucleic acid on mast cell at the transcriptional level. PeerJ 2019, 7, e7404. [Google Scholar] [CrossRef]
- Kremenovic, M.; Schenk, M.; Lee, D.J. Clinical and molecular insights into BCG immunotherapy for melanoma. J. Intern. Med. 2020, 288, 625–640. [Google Scholar] [CrossRef]
- Arts, R.J.W.; Moorlag, S.J.; Novakovic, B.; Li, Y.; Wang, S.Y.; Oosting, M.; Kumar, V.; Xavier, R.J.; Wijmenga, C.; Joosten, L.A.B.; et al. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe 2018, 23, 89–100.e5. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Netea, M.G. BCG-induced trained immunity: Can it offer protection against COVID-19? Nat. Rev. Immunol. 2020, 20, 335–337. [Google Scholar] [CrossRef]
- Parra, J.; Marcoux, J.; Poncin, I.; Canaan, S.; Herrmann, J.L.; Nigou, J.; Burlet-Schiltz, O.; Rivière, M. Scrutiny of Mycobacterium tuberculosis 19 kDa antigen proteoforms provides new insights in the lipoglycoprotein biogenesis paradigm. Sci. Rep. 2017, 7, 43682. [Google Scholar] [CrossRef]
- Rao, V.; Dhar, N.; Shakila, H.; Singh, R.; Khera, A.; Jain, R.; Naseema, M.; Paramasivan, C.N.; Narayanan, P.R.; Ramanathan, V.D.; et al. Increased Expression of Mycobacterium tuberculosis 19 kDa Lipoprotein Obliterates the Protective Efficacy of BCG by Polarizing Host Immune Responses to the Th2 Subtype. Scand. J. Immunol. 2005, 61, 410–417. [Google Scholar] [CrossRef]
- Kim, W.S.; Kim, J.-S.; Kim, H.M.; Kwon, K.W.; Eum, S.-Y.; Shin, S.J. Comparison of immunogenicity and vaccine efficacy between heat-shock proteins, HSP70 and GrpE, in the DnaK operon of Mycobacterium tuberculosis. Sci. Rep. 2018, 8, 14411. [Google Scholar] [CrossRef]
- Moliva, J.I.; Turner, J.; Torrelles, J.B. Immune Responses to Bacillus Calmette-Guérin Vaccination: Why Do They Fail to Protect against Mycobacterium tuberculosis? Front. Immunol. 2017, 8, 407. [Google Scholar] [CrossRef]
- Piliponsky, A.M.; Romani, L. The contribution of mast cells to bacterial and fungal infection immunity. Immunol. Rev. 2018, 282, 188–197. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, K.M.; Goenka, A.; Alonso-Rasgado, M.T.; Hernández-Pando, R.; Bulfone-Paus, S. The Role of Mast Cells in Tuberculosis: Orchestrating Innate Immune Crosstalk? Front. Immunol. 2017, 8, 1290. [Google Scholar] [CrossRef]
- Johnson-Weaver, B.; Choi, H.W.; Abraham, S.N.; Staats, H.F. Mast cell activators as novel immune regulators. Curr. Opin. Pharmacol. 2018, 41, 89–95. [Google Scholar] [CrossRef]
- Chan, B.C.L.; Lam, C.W.K.; Tam, L.-S.; Wong, C.K. IL33: Roles in Allergic Inflammation and Therapeutic Perspectives. Front. Immunol. 2019, 10, 364. [Google Scholar] [CrossRef]
- Joulia, R.; L’Faqihi, F.-E.; Valitutti, S.; Espinosa, E. IL-33 fine tunes mast cell degranulation and chemokine production at the single-cell level. J. Allergy Clin. Immunol. 2016, 140, 497–509.e10. [Google Scholar] [CrossRef]
- Naqvi, N.; Srivastava, R.; Naskar, P.; Puri, N. Mast cells modulate early responses to Mycobacterium bovis Bacillus Calmette-Guerin by phagocytosis and formation of extracellular traps. Cell. Immunol. 2021, 365, 104380. [Google Scholar] [CrossRef]
- Muñoz, S.; Hernández-Pando, R.; Abraham, S.N.; Enciso, J.A. Mast Cell Activation by Mycobacterium tuberculosis: Mediator Release and Role of CD48. J. Immunol. 2003, 170, 5590–5596. [Google Scholar] [CrossRef] [PubMed]
- Dowell, A.C.; Cobby, E.; Wen, K.; Devall, A.J.; During, V.; Anderson, J.; James, N.D.; Cheng, K.K.; Zeegers, M.P.; Bryan, R.T.; et al. Interleukin-17-positive mast cells influence outcomes from BCG for patients with CIS: Data from a comprehensive characterisation of the immune microenvironment of urothelial bladder cancer. PLoS ONE 2017, 12, e0184841. [Google Scholar] [CrossRef] [PubMed]
- Stone, L. Mastering the immune microenvironment. Nat. Rev. Urol. 2017, 14, 639. [Google Scholar] [CrossRef] [PubMed]
- Johnzon, C.-F.; Rönnberg, E.; Pejler, G. The Role of Mast Cells in Bacterial Infection. Am. J. Pathol. 2015, 186, 4–14. [Google Scholar] [CrossRef]
- Motta, A.; Schmitz, C.; Rodrigues, L.; Ribeiro, F.; Teixeira, C.; Detanico, T.; Bonan, C.; Zwickey, H.; Bonorino, C. Mycobacterium tuberculosis heat-shock protein 70 impairs maturation of dendritic cells from bone marrow precursors, induces interleukin-10 production and inhibits T-cell proliferation in vitro. Immunology 2007, 121, 462–472. [Google Scholar] [CrossRef]
- Iikura, M.; Suto, H.; Kajiwara, N.; Oboki, K.; Ohno, T.; Okayama, Y.; Saito, H.; Galli, S.J.; Nakae, S. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab. Investig. 2007, 87, 971–978. [Google Scholar] [CrossRef]
- Swindle, E.J.; Brown, J.M.; Rådinger, M.; DeLeo, F.R.; Metcalfe, D.D. Interferon-γ enhances both the anti-bacterial and the pro-inflammatory response of human mast cells to Staphylococcus aureus. Immunology 2015, 146, 470–485. [Google Scholar] [CrossRef]
- Abraham, S.N.; John, A.L.S. Mast cell-orchestrated immunity to pathogens. Nat. Rev. Immunol. 2010, 10, 440–452. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, K.M.; Bahri, R.; Sattentau, C.; Roberts, I.S.; Goenka, A.; Bulfone-Paus, S. Human mast cells exhibit an individualized pattern of antimicrobial responses. Immunity Inflamm. Dis. 2020, 8, 198–210. [Google Scholar] [CrossRef]
- Ronnberg, E.; Guss, B.; Pejler, G. Infection of Mast Cells with Live Streptococci Causes a Toll-Like Receptor 2- and Cell-Cell Contact-Dependent Cytokine and Chemokine Response. Infect. Immun. 2010, 78, 854–864. [Google Scholar] [CrossRef]
- Stewart, G.R.; Wilkinson, K.A.; Newton, S.M.; Sullivan, S.M.; Neyrolles, O.; Wain, J.R.; Patel, J.; Pool, K.-L.; Young, D.B.; Wilkinson, R.J. Effect of Deletion or Overexpression of the 19-Kilodalton Lipoprotein Rv3763 on the Innate Response to Mycobacterium tuberculosis. Infect. Immun. 2005, 73, 6831–6837. [Google Scholar] [CrossRef]
- Bulut, Y.; Michelsen, K.S.; Hayrapetian, L.; Naiki, Y.; Spallek, R.; Singh, M.; Arditi, M. Mycobacterium Tuberculosis Heat Shock Proteins Use Diverse Toll-like Receptor Pathways to Activate Pro-inflammatory Signals. J. Biol. Chem. 2005, 280, 20961–20967. [Google Scholar] [CrossRef]
- Liu, L.; Liu, J.; Niu, G.; Xu, Q.; Chen, Q. Mycobacterium tuberculosis 19-kDa lipoprotein induces Toll-like receptor 2-dependent peroxisome proliferator-activated receptor γ expression and promotes inflammatory responses in human macrophages. Mol. Med. Rep. 2015, 11, 2921–2926. [Google Scholar] [CrossRef]
- Becker, K.; Sander, P. Mycobacterium tuberculosis lipoproteins in virulence and immunity-fighting with a double-edged sword. FEBS Lett. 2016, 590, 3800–3819. [Google Scholar] [CrossRef]
- Pai, R.K.; Pennini, M.E.; Tobian, A.A.R.; Canaday, D.H.; Boom, W.H.; Harding, C.V. Prolonged Toll-Like Receptor Signaling by Mycobacterium tuberculosis and Its 19-Kilodalton Lipoprotein Inhibits Gamma Interferon-Induced Regulation of Selected Genes in Macrophages. Infect. Immun. 2004, 72, 6603–6614. [Google Scholar] [CrossRef]
- Pennini, M.E.; Pai, R.K.; Schultz, D.C.; Boom, W.H.; Harding, C.V. Mycobacterium tuberculosis 19-kDa Lipoprotein Inhibits IFN-γ-Induced Chromatin Remodeling of MHC2TA by TLR2 and MAPK Signaling. J. Immunol. 2006, 176, 4323–4330. [Google Scholar] [CrossRef]
- Bischoff, S.C. Role of mast cells in allergic and non-allergic immune responses: Comparison of human and murine data. Nat. Rev. Immunol. 2007, 7, 93–104. [Google Scholar] [CrossRef]
- McLachlan, J.B.; Shelburne, C.P.; Hart, J.P.; Pizzo, S.V.; Goyal, R.; Brooking-Dixon, R.; Staats, H.; Abraham, S.N. Mast cell activators: A new class of highly effective vaccine adjuvants. Nat. Med. 2008, 14, 536–541. [Google Scholar] [CrossRef]
- Kayamuro, H.; Yoshioka, Y.; Abe, Y.; Arita, S.; Katayama, K.; Nomura, T.; Yoshikawa, T.; Kubota-Koketsu, R.; Ikuta, K.; Okamoto, S.; et al. Interleukin-1 Family Cytokines as Mucosal Vaccine Adjuvants for Induction of Protective Immunity against Influenza Virus. J. Virol. 2010, 84, 12703–12712. [Google Scholar] [CrossRef]
- Villarreal, D.; Siefert, R.J.; Weiner, D.B. Alarmin IL-33 elicits potent TB-specific cell-mediated responses. Hum. Vaccines Immunother. 2015, 11, 1954–1960. [Google Scholar] [CrossRef]
- Enoksson, M.; Möller-Westerberg, C.; Wicher, G.; Fallon, P.G.; Forsberg-Nilsson, K.; Lunderius-Andersson, C.; Nilsson, G. Intraperitoneal influx of neutrophils in response to IL-33 is mast cell–dependent. Blood 2013, 121, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Heitmann, L.; Dar, M.A.; Schreiber, T.; Erdmann, H.; Behrends, J.; Mckenzie, A.N.J.; Brombacher, F.; Ehlers, S.; Hölscher, C. The IL-13/IL-4Rα axis is involved in tuberculosis-associated pathology. J. Pathol. 2014, 234, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rodriguez, K.M.; Bini, E.I.; Gamboa-Domínguez, A.; Espitia-Pinzón, C.I.; Huerta-Yepez, S.; Bulfone-Paus, S.; Hernández-Pando, R. Differential mast cell numbers and characteristics in human tuberculosis pulmonary lesions. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Malaviya, R.; Twesten, N.J.; A Ross, E.; Abraham, S.N.; Pfeifer, J.D. Mast cells process bacterial Ags through a phagocytic route for class I MHC presentation to T cells. J. Immunol. 1996, 156, 1490–1496. [Google Scholar]
- Féger, F.; Varadaradjalou, S.; Gao, Z.; Abraham, S.N.; Arock, M. The role of mast cells in host defense and their subversion by bacterial pathogens. Trends Immunol. 2002, 23, 151–158. [Google Scholar] [CrossRef]
- Pinke, K.H.; de Lima, H.G.; Cunha, F.Q.; Lara, V.S. Mast cells phagocyte Candida albicans and produce nitric oxide by mechanisms involving TLR2 and Dectin-1. Immunobiology 2016, 221, 220–227. [Google Scholar] [CrossRef]
- Espinosa-Cueto, P.; Magallanes-Puebla, A.; Mancilla, R. Phosphate starvation enhances phagocytosis of Mycobacterium bovis/BCG by macrophages. BMC Immunol. 2020, 21, 34. [Google Scholar] [CrossRef]
- Ito, T.; Egusa, C.; Maeda, T.; Numata, T.; Nakano, N.; Nishiyama, C.; Tsuboi, R. IL-33 promotes MHC class II expression in murine mast cells. Immunity Inflamm. Dis. 2015, 3, 196–208. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Rodriguez, K.M.; Goenka, A.; Thomson, D.D.; Bahri, R.; Tontini, C.; Salcman, B.; Hernandez-Pando, R.; Bulfone-Paus, S. Bacillus Calmette–Guérin-Induced Human Mast Cell Activation Relies on IL-33 Priming. Int. J. Mol. Sci. 2022, 23, 7549. https://doi.org/10.3390/ijms23147549
Garcia-Rodriguez KM, Goenka A, Thomson DD, Bahri R, Tontini C, Salcman B, Hernandez-Pando R, Bulfone-Paus S. Bacillus Calmette–Guérin-Induced Human Mast Cell Activation Relies on IL-33 Priming. International Journal of Molecular Sciences. 2022; 23(14):7549. https://doi.org/10.3390/ijms23147549
Chicago/Turabian StyleGarcia-Rodriguez, Karen M., Anu Goenka, Darren D. Thomson, Rajia Bahri, Chiara Tontini, Barbora Salcman, Rogelio Hernandez-Pando, and Silvia Bulfone-Paus. 2022. "Bacillus Calmette–Guérin-Induced Human Mast Cell Activation Relies on IL-33 Priming" International Journal of Molecular Sciences 23, no. 14: 7549. https://doi.org/10.3390/ijms23147549
APA StyleGarcia-Rodriguez, K. M., Goenka, A., Thomson, D. D., Bahri, R., Tontini, C., Salcman, B., Hernandez-Pando, R., & Bulfone-Paus, S. (2022). Bacillus Calmette–Guérin-Induced Human Mast Cell Activation Relies on IL-33 Priming. International Journal of Molecular Sciences, 23(14), 7549. https://doi.org/10.3390/ijms23147549