
Lymphangiogenesis and Lymphatic Barrier Dysfunction in Renal Fibrosis


Abstract
:1. Introduction
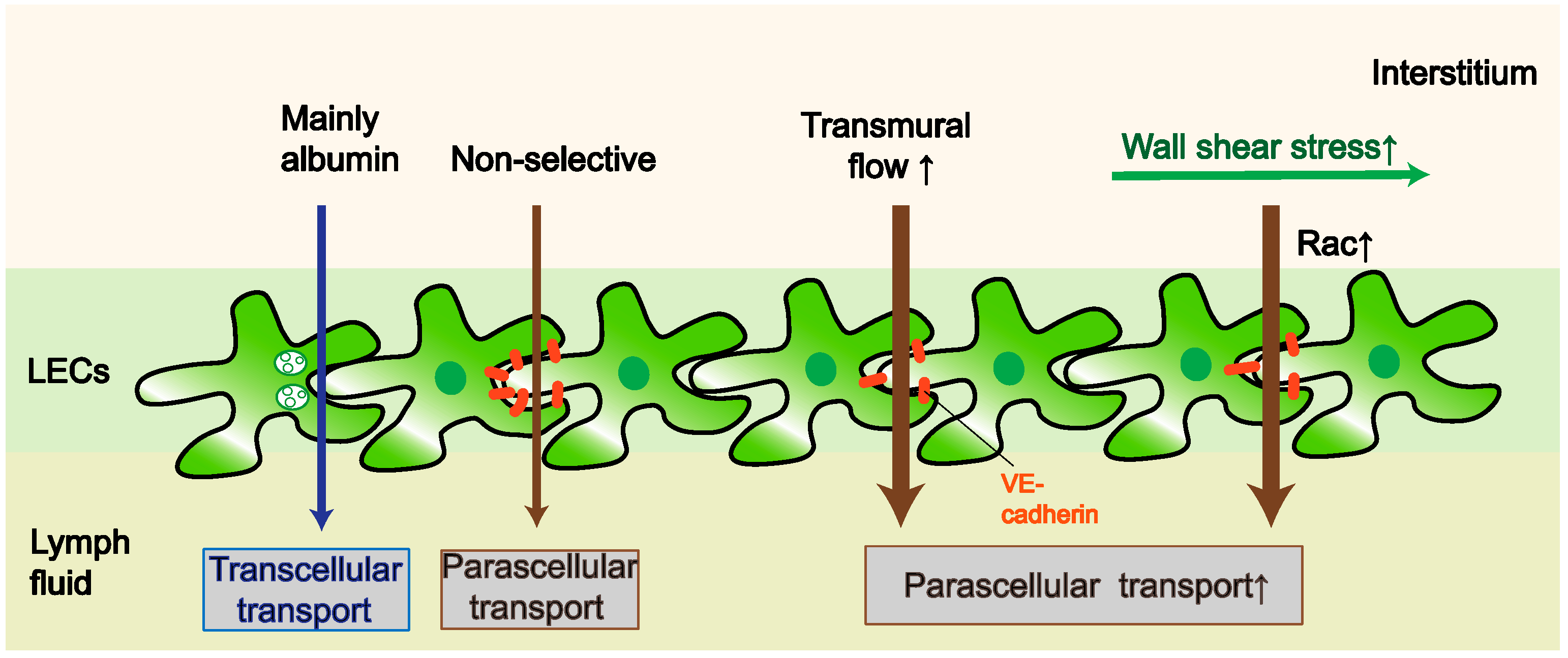
2. Permeability
2.1. Factors That Affect the Paracellular Pathway
2.2. Factors That Affect the Transcellular Pathway
2.3. Outstanding Questions Related to LV Permeability
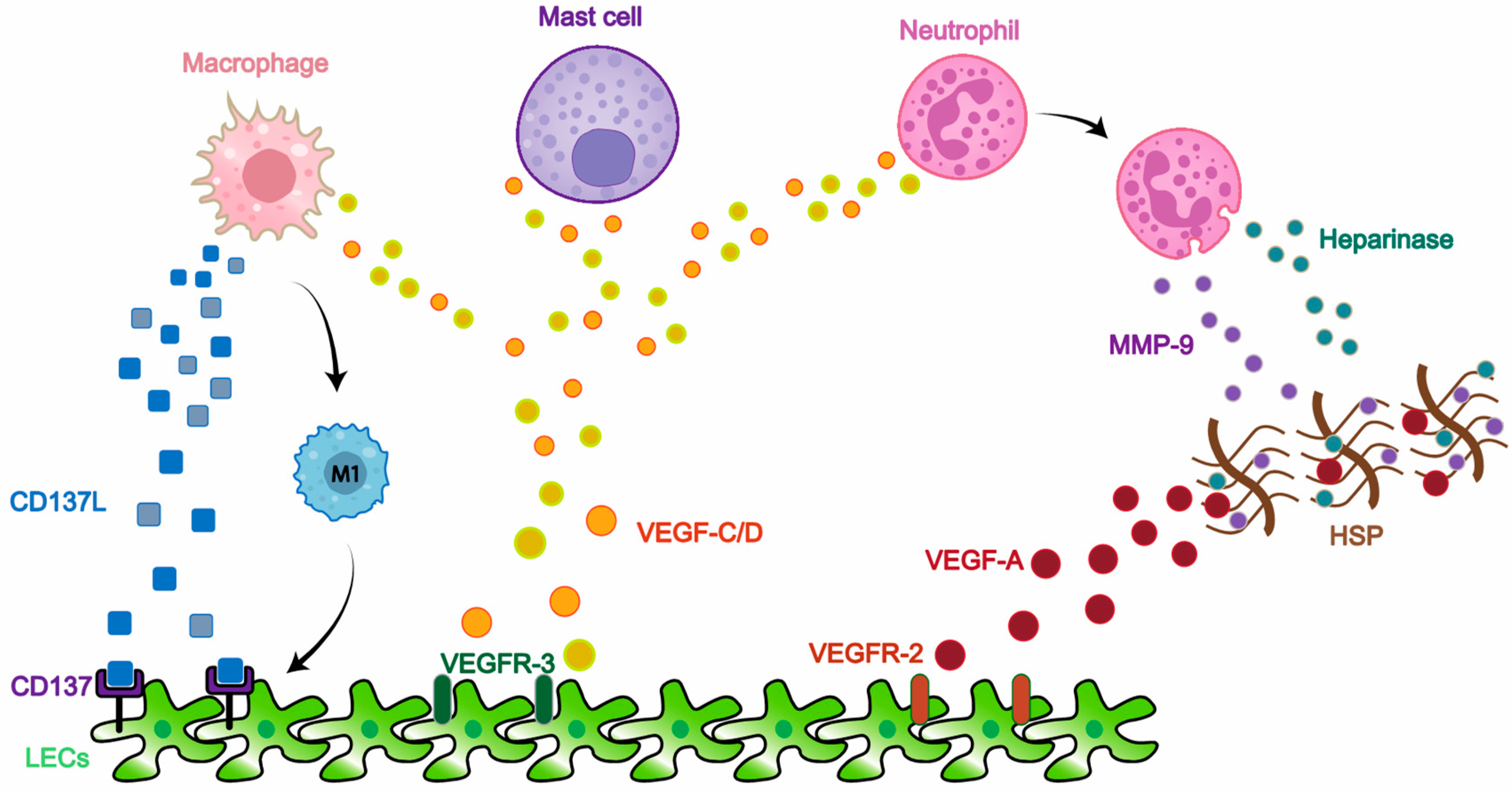
3. Proliferation
3.1. SS
3.2. Inflammation
4. Technologies of Lymphatic Research on Imaging and Function
5. LVs and Renal Fibrosis
5.1. The Renal Lymphatic System
5.2. Lymphatic Proliferation and Renal Fibrosis
5.3. Permeability and Renal Fibrosis
5.4. Lymphatic-Related Treatment of Fibrosis
5.5. The Lymphatic System: A New Pathway Mediating Crosstalk among Kidney and Other Organs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Petrova, T.V.; Koh, G.Y. Organ-specific lymphatic vasculature: From development to pathophysiology. J. Exp. Med. 2018, 215, 35–49. [Google Scholar] [CrossRef]
- Swartz, M.A. The physiology of the lymphatic system. Adv. Drug Deliv. Rev. 2001, 50, 3–20. [Google Scholar] [CrossRef]
- Mukherjee, A.; Hooks, J.; Dixon, J.B. Physiology: Lymph flow. In Lymphedema; Springer: Berlin/Heidelberg, Germany, 2018; pp. 91–111. [Google Scholar]
- Baluk, P.; Fuxe, J.; Hashizume, H.; Romano, T.; Lashnits, E.; Butz, S.; Vestweber, D.; Corada, M.; Molendini, C.; Dejana, E.; et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 2007, 204, 2349–2362. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Brakenhielm, E.; González, A.; Díez, J. Role of Cardiac lymphatics in myocardial edema and fibrosis: JACC review topic of the week. J. Am. Coll. Cardiol. 2020, 76, 735–744. [Google Scholar] [CrossRef]
- Tanaka, M.; Iwakiri, Y. Lymphatics in the liver. Curr. Opin. Immunol. 2018, 53, 137–142. [Google Scholar] [CrossRef]
- Juneja, P.; Tripathi, D.M.; Kaur, S. Revisiting the gut-liver axis: Gut lymphatic system in liver cirrhosis and portal hypertension. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 322, G473–G479. [Google Scholar] [CrossRef]
- Brakenhielm, E.; Alitalo, K. Cardiac lymphatics in health and disease. Nat. Rev. Cardiol. 2019, 16, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; De la Cruz, E.; Gu, X.; Balint, L.; Oxendine-Burns, M.; Terrones, T.; Ma, W.; Kuo, H.H.; Lantz, C.; Bansal, T.; et al. Lymphoangiocrine signals promote cardiac growth and repair. Nature 2020, 588, 705–711. [Google Scholar] [CrossRef]
- Oliver, G.; Kipnis, J.; Randolph, G.J.; Harvey, N.L. The lymphatic vasculature in the 21st Century: Novel functional roles in homeostasis and disease. Cell 2020, 182, 270–296. [Google Scholar] [CrossRef]
- Starling, E.H. On the absorption of fluids from the connective tissue spaces. J. Physiol. 1896, 19, 312–326. [Google Scholar] [CrossRef]
- Triacca, V.; Güç, E.; Kilarski, W.W.; Pisano, M.; Swartz, M.A. Transcellular Pathways in lymphatic endothelial cells regulate changes in solute transport by fluid stress. Circ. Res. 2017, 120, 1440–1452. [Google Scholar] [CrossRef]
- Yang, V.V.; O’Morchoe, P.J.; O’Morchoe, C.C. Transport of protein across lymphatic endothelium in the rat kidney. Microvasc. Res. 1981, 21, 75–91. [Google Scholar] [CrossRef]
- Albertine, K.H.; O’Morchoe, C.C. Renal lymphatic ultrastructure and translymphatic transport. Microvasc. Res. 1980, 19, 338–351. [Google Scholar] [CrossRef]
- Miteva, D.O.; Rutkowski, J.M.; Dixon, J.B.; Kilarski, W.; Shields, J.D.; Swartz, M.A. Transmural flow modulates cell and fluid transport functions of lymphatic endothelium. Circ. Res. 2010, 106, 920–931. [Google Scholar] [CrossRef]
- Jannaway, M.; Scallan, J.P. VE-Cadherin and vesicles differentially regulate lymphatic vascular permeability to solutes of various sizes. Front. Physiol. 2021, 12, 687563. [Google Scholar] [CrossRef]
- Ingber, D.E. Cellular mechanotransduction: Putting all the pieces together again. Fed. Am. Soc. Exp. Biol. J. 2006, 20, 811–827. [Google Scholar] [CrossRef]
- Chiu, J.J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef] [Green Version]
- Breslin, J.W.; Kurtz, K.M. Lymphatic endothelial cells adapt their barrier function in response to changes in shear stress. Lymphat. Res. Biol. 2009, 7, 229–237. [Google Scholar] [CrossRef]
- Mukherjee, A.; Dixon, J.B. Mechanobiology of lymphatic vessels. In Vascular Mechanobiology in Physiology and Disease; Springer: Cham, Switzerland, 2021; pp. 191–239. [Google Scholar]
- Tammela, T.; Alitalo, K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell 2010, 140, 460–476. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Yu, Z.; Liu, N. Comparison of approaches for microscopic imaging of skin lymphatic vessels. Scanning 2012, 34, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A.; Kaipainen, A.; Netti, P.A.; Brekken, C.; Boucher, Y.; Grodzinsky, A.J.; Jain, R.K. Mechanics of interstitial-lymphatic fluid transport: Theoretical foundation and experimental validation. J. Biomech. 1999, 32, 1297–1307. [Google Scholar] [CrossRef]
- Skobe, M.; Detmar, M. Structure, function, and molecular control of the skin lymphatic system. J. Investig. Dermatol. Symp. Proc. 2000, 5, 14–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cromer, W.E.; Zawieja, S.D.; Tharakan, B.; Childs, E.W.; Newell, M.K.; Zawieja, D.C. The effects of inflammatory cytokines on lymphatic endothelial barrier function. Angiogenesis 2014, 17, 395–406. [Google Scholar] [CrossRef] [Green Version]
- Chaitanya, G.V.; Franks, S.E.; Cromer, W.; Wells, S.R.; Bienkowska, M.; Jennings, M.H.; Ruddell, A.; Ando, T.; Wang, Y.; Gu, Y.; et al. Differential cytokine responses in human and mouse lymphatic endothelial cells to cytokines in vitro. Lymphat. Res. Biol. 2010, 8, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Jackson, D.G. Hyaluronan and its receptors: Key mediators of immune cell entry and trafficking in the lymphatic system. Cells 2021, 10, 2061. [Google Scholar] [CrossRef]
- Tang, T.H.; Alonso, S.; Ng, L.F.; Thein, T.L.; Pang, V.J.; Leo, Y.S.; Lye, D.C.; Yeo, T.W. Increased serum hyaluronic acid and heparan sulfate in dengue fever: Association with plasma leakage and disease severity. Sci Rep. 2017, 7, 46191. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Kolliopoulos, C.; Huang, C.H.; Tenhunen, J.; Heldin, C.H.; Chen, Y.H.; Heldin, P. High levels of serum hyaluronan is an early predictor of dengue warning signs and perturbs vascular integrity. EBioMedicine 2019, 48, 425–441. [Google Scholar] [CrossRef] [Green Version]
- Becker, B.F.; Jacob, M.; Leipert, S.; Salmon, A.H.; Chappell, D. Degradation of the endothelial glycocalyx in clinical settings: Searching for the sheddases. Br. J. Clin. Pharmacol. 2015, 80, 389–402. [Google Scholar] [CrossRef]
- Kajiya, K.; Hirakawa, S.; Detmar, M. Vascular endothelial growth factor—A mediates ultraviolet B-induced impairment of lymphatic vessel function. Am. J. Pathol. 2006, 169, 1496–1503. [Google Scholar] [CrossRef] [Green Version]
- Schwager, S.; Detmar, M. Inflammation and lymphatic function. Front. Immunol. 2019, 10, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunstfeld, R.; Hirakawa, S.; Hong, Y.K.; Schacht, V.; Lange-Asschenfeldt, B.; Velasco, P.; Lin, C.; Fiebiger, E.; Wei, X.; Wu, Y.; et al. Induction of cutaneous delayed-type hypersensitivity reactions in VEGF-A transgenic mice results in chronic skin inflammation associated with persistent lymphatic hyperplasia. Blood 2004, 104, 1048–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, S.; Nath, S.; Meininger, C.J.; Gashev, A.A. Emerging roles of mast cells in the regulation of lymphatic immuno-physiology. Front. Immunol. 2020, 11, 1234. [Google Scholar] [CrossRef] [PubMed]
- Kunder, C.A.; St John, A.L.; Abraham, S.N. Mast cell modulation of the vascular and lymphatic endothelium. Blood 2011, 118, 5383–5393. [Google Scholar] [CrossRef]
- Yao, L.C.; Baluk, P.; Srinivasan, R.S.; Oliver, G.; McDonald, D.M. Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am. J. Pathol. 2012, 180, 2561–2575. [Google Scholar] [CrossRef] [Green Version]
- Herrera, M.; Molina, P.; Souza-Smith, F.M. Ethanol-induced lymphatic endothelial cell permeability via MAP-kinase regulation. Am. J. Physiol. Cell. Physiol. 2021, 321, C104–C116. [Google Scholar] [CrossRef]
- Baranwal, G.; Creed, H.A.; Cromer, W.E.; Wang, W.; Upchurch, B.D.; Smithhart, M.C.; Vadlamani, S.S.; Clark, M.C.; Busbuso, N.C.; Blais, S.N.; et al. Dichotomous effects on lymphatic transport with loss of caveolae in mice. Acta Physiol. 2021, 232, e13656. [Google Scholar] [CrossRef]
- Meens, M.J.; Sabine, A.; Petrova, T.V.; Kwak, B.R. Connexins in lymphatic vessel physiology and disease. FEBS Lett. 2014, 588, 1271–1277. [Google Scholar] [CrossRef]
- Chen, C.Y.; Bertozzi, C.; Zou, Z.; Yuan, L.; Lee, J.S.; Lu, M.; Stachelek, S.J.; Srinivasan, S.; Guo, L.; Vicente, A.; et al. Blood flow reprograms lymphatic vessels to blood vessels. J. Clin. Investig. 2012, 122, 2006–2017. [Google Scholar] [CrossRef] [Green Version]
- Kalucka, J.; Teuwen, L.A.; Geldhof, V.; Carmeliet, P. How to cross the lymphatic fence: Lessons from solute transport. Circ. Res. 2017, 120, 1376–1378. [Google Scholar] [CrossRef] [Green Version]
- Geng, X.; Yanagida, K.; Akwii, R.G.; Choi, D.; Chen, L.; Ho, Y.; Cha, B.; Mahamud, M.R.; Berman de Ruiz, K.; Ichise, H.; et al. S1PR1 regulates the quiescence of lymphatic vessels by inhibiting laminar shear stress-dependent VEGF-C signaling. JCI Insight 2020, 5, e137652. [Google Scholar] [CrossRef] [PubMed]
- Ducoli, L.; Detmar, M. Beyond PROX1: Transcriptional, epigenetic, and noncoding RNA regulation of lymphatic identity and function. Dev. Cell 2021, 56, 406–426. [Google Scholar] [CrossRef] [PubMed]
- Scavelli, C.; Weber, E.; Aglianò, M.; Cirulli, T.; Nico, B.; Vacca, A.; Ribatti, D. Lymphatics at the crossroads of angiogenesis and lymphangiogenesis. J. Anat. 2004, 204, 433–449. [Google Scholar] [CrossRef] [PubMed]
- Alitalo, K.; Carmeliet, P. Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell 2002, 1, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.; Park, E.; Jung, E.; Seong, Y.J.; Hong, M.; Lee, S.; Burford, J.; Gyarmati, G.; Peti-Peterdi, J.; Srikanth, S.; et al. ORAI1 activates proliferation of lymphatic endothelial cells in response to laminar flow through krüppel-like factors 2 and 4. Circ. Res. 2017, 120, 1426–1439. [Google Scholar] [CrossRef]
- Choi, D.; Park, E.; Jung, E.; Seong, Y.J.; Yoo, J.; Lee, E.; Hong, M.; Lee, S.; Ishida, H.; Burford, J.; et al. Laminar flow downregulates notch activity to promote lymphatic sprouting. J. Clin. Investig. 2017, 127, 1225–1240. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kataru, R.P.; Koh, G.Y. Inflammation-associated lymphangiogenesis: A double-edged sword? J. Clin. Investig. 2014, 124, 936–942. [Google Scholar] [CrossRef]
- Kim, K.E.; Koh, Y.J.; Jeon, B.H.; Jang, C.; Han, J.; Kataru, R.P.; Schwendener, R.A.; Kim, J.M.; Koh, G.Y. Role of CD11b+ macrophages in intraperitoneal lipopolysaccharide-induced aberrant lymphangiogenesis and lymphatic function in the diaphragm. Am. J. Pathol. 2009, 175, 1733–1745. [Google Scholar] [CrossRef] [Green Version]
- Karaman, S.; Hollmén, M.; Yoon, S.Y.; Alkan, H.F.; Alitalo, K.; Wolfrum, C.; Detmar, M. Transgenic overexpression of VEGF-C induces weight gain and insulin resistance in mice. Sci. Rep. 2016, 6, 31566. [Google Scholar] [CrossRef] [Green Version]
- Proulx, S.T.; Luciani, P.; Alitalo, A.; Mumprecht, V.; Christiansen, A.J.; Huggenberger, R.; Leroux, J.C.; Detmar, M. Non-invasive dynamic near-infrared imaging and quantification of vascular leakage in vivo. Angiogenesis 2013, 16, 525–540. [Google Scholar] [CrossRef] [Green Version]
- Glinton, K.E.; Ma, W.; Lantz, C.W.; Grigoryeva, L.S.; DeBerge, M.; Liu, X.; Febbraio, M.; Kahn, M.; Oliver, G.; Thorp, E.B. Macrophage-produced VEGFC is induced by efferocytosis to ameliorate cardiac injury and inflammation. J. Clin. Investig. 2022, 132, e140685. [Google Scholar] [CrossRef] [PubMed]
- Karnezis, T.; Shayan, R.; Caesar, C.; Roufail, S.; Harris, N.C.; Ardipradja, K.; Zhang, Y.F.; Williams, S.P.; Farnsworth, R.H.; Chai, M.G.; et al. VEGF-D promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell 2012, 21, 181–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cursiefen, C.; Chen, L.; Borges, L.P.; Jackson, D.; Cao, J.; Radziejewski, C.; D’Amore, P.A.; Dana, M.R.; Wiegand, S.J.; Streilein, J.W. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J. Clin. Investig. 2004, 113, 1040–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M.; Zheng, Y.; Hirashima, M.; Suda, T.; Morita, Y.; Ooehara, J.; Ema, H.; Fong, G.H.; Shibuya, M. VEGFR1 tyrosine kinase signaling promotes lymphangiogenesis as well as angiogenesis indirectly via macrophage recruitment. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 658–664. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Chen, L.; Li, Q.; Liang, X.; Wang, K.; Zhang, Y.; Li, Y.; Liu, Y.; Xu, G. CD137L-macrophage induce lymphatic endothelial cells autophagy to promote lymphangiogenesis in renal fibrosis. Int. J. Biol. Sci. 2022, 18, 1171–1187. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, C.; Li, L.; Liang, X.; Cheng, P.; Li, Q.; Chang, X.; Wang, K.; Huang, S.; Li, Y.; et al. Lymphangiogenesis in renal fibrosis arises from macrophages via VEGF-C/VEGFR3-dependent autophagy and polarization. Cell Death Dis. 2021, 12, 109. [Google Scholar] [CrossRef]
- Rutkowski, J.M.; Moya, M.; Johannes, J.; Goldman, J.; Swartz, M.A. Secondary lymphedema in the mouse tail: Lymphatic hyperplasia, VEGF-C upregulation, and the protective role of MMP-9. Microvasc. Res. 2006, 72, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Gardenier, J.C.; Hespe, G.E.; Kataru, R.P.; Savetsky, I.L.; Torrisi, J.S.; Nores, G.D.G.; Dayan, J.J.; Chang, D.; Zampell, J.; Martínez-Corral, I.; et al. Diphtheria toxin-mediated ablation of lymphatic endothelial cells results in progressive lymphedema. JCI Insight 2016, 1, e84095. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; Cheng, G.; Conner, D.A.; Huang, Y.; Kucherlapati, R.S.; Munn, L.L.; Ruddle, N.H.; Jain, R.K.; Fukumura, D.; Padera, T.P. Impaired lymphatic contraction associated with immunosuppression. Proc. Natl. Acad. Sci. USA 2011, 108, 18784–18789. [Google Scholar] [CrossRef] [Green Version]
- Sano, M.; Sasaki, T.; Hirakawa, S.; Sakabe, J.; Ogawa, M.; Baba, S.; Zaima, N.; Tanaka, H.; Inuzuka, K.; Yamamoto, N.; et al. Lymphangiogenesis and angiogenesis in abdominal aortic aneurysm. PLoS ONE 2014, 9, e89830. [Google Scholar] [CrossRef]
- Houck, K.A.; Leung, D.W.; Rowland, A.M.; Winer, J.; Ferrara, N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 1992, 267, 26031–26037. [Google Scholar] [CrossRef]
- Tan, K.W.; Chong, S.Z.; Wong, F.H.; Evrard, M.; Tan, S.M.; Keeble, J.; Kemeny, D.M.; Ng, L.G.; Abastado, J.P.; Angeli, V. Neutrophils contribute to inflammatory lymphangiogenesis by increasing VEGF-A bioavailability and secreting VEGF-D. Blood 2013, 122, 3666–3677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakovija, A.; Chtanova, T. Neutrophil Interactions with the lymphatic system. Cells 2021, 10, 2016. [Google Scholar] [CrossRef] [PubMed]
- Marone, G.; Varricchi, G.; Loffredo, S.; Granata, F. Mast cells and basophils in inflammatory and tumor angiogenesis and lymphangiogenesis. Eur. J. Pharmacol. 2016, 778, 146–151. [Google Scholar] [CrossRef]
- Keser, S.H.; Kandemir, N.O.; Ece, D.; Gecmen, G.G.; Gul, A.E.; Barisik, N.O.; Sensu, S.; Buyukuysal, C.; Barut, F. Relationship of mast cell density with lymphangiogenesis and prognostic parameters in breast carcinoma. Kaohsiung J. Med. Sci. 2017, 33, 171–180. [Google Scholar] [CrossRef]
- Munn, L.L.; Padera, T.P. Imaging the lymphatic system. Microvasc. Res. 2014, 96, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Hägerling, R.; Pollmann, C.; Andreas, M.; Schmidt, C.; Nurmi, H.; Adams, R.H.; Alitalo, K.; Andresen, V.; Schulte-Merker, S.; Kiefer, F. A novel multistep mechanism for initial lymphangiogenesis in mouse embryos based on ultramicroscopy. Eur. Mol. Biol. Organ. J. 2013, 32, 629–644. [Google Scholar] [CrossRef] [Green Version]
- Hägerling, R.; Drees, D.; Scherzinger, A.; Dierkes, C.; Martin-Almedina, S.; Butz, S.; Gordon, K.; Schäfers, M.; Hinrichs, K.; Ostergaard, P.; et al. VIPAR, a quantitative approach to 3D histopathology applied to lymphatic malformations. JCI Insight 2017, 2, e93424. [Google Scholar] [CrossRef]
- Jafree, D.J.; Moulding, D.; Kolatsi-Joannou, M.; Perretta Tejedor, N.; Price, K.L.; Milmoe, N.J.; Walsh, C.L.; Correra, R.M.; Winyard, P.J.; Harris, P.C.; et al. Spatiotemporal dynamics and heterogeneity of renal lymphatics in mammalian development and cystic kidney disease. eLife 2019, 8, 48183. [Google Scholar] [CrossRef]
- McElroy, M.; Hayashi, K.; Garmy-Susini, B.; Kaushal, S.; Varner, J.A.; Moossa, A.R.; Hoffman, R.M.; Bouvet, M. Fluorescent LYVE-1 antibody to image dynamically lymphatic trafficking of cancer cells in vivo. J. Surg. Res. 2009, 151, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Redder, E.; Kirschnick, N.; Bobe, S.; Hägerling, R.; Hansmeier, N.R.; Kiefer, F. Vegfr3-tdTomato, a reporter mouse for microscopic visualization of lymphatic vessel by multiple modalities. PLoS ONE 2021, 16, e0249256. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Putra, I.; Huang, Y.H.; Chang, M.; Han, K.; Zhong, W.; Gao, X.; Wang, S.; Dugas-Ford, J.; Nguyen, T.; et al. Limited versus total epithelial debridement ocular surface injury: Live fluorescence imaging of hemangiogenesis and lymphangiogenesis in Prox1-GFP/Flk1: Myr-mCherry mice. Biochim. Biophys. Acta 2016, 1860, 2148–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, H.A.; Kwon, S.; Hall, M.A.; Rasmussen, J.C.; Aldrich, M.B.; Sevick-Muraca, E.M. Non-invasive optical imaging of the lymphatic vasculature of a mouse. J. Vis. Exp. 2013, 4326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Sevick-Muraca, E.M. A review of performance of near-infrared fluorescence imaging devices used in clinical studies. Br. J. Radiol. 2015, 88, 20140547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scallan, J.P.; Wolpers, J.H.; Davis, M.J. Constriction of isolated collecting lymphatic vessels in response to acute increases in downstream pressure. J. Physiol. 2013, 591, 443–459. [Google Scholar] [CrossRef]
- Bell, R.D.; Keyl, M.J.; Shrader, F.R.; Jones, E.W.; Henry, L.P. Renal lymphatics: The internal distribution. Nephron 1968, 5, 454–463. [Google Scholar] [CrossRef]
- Tanabe, M.; Shimizu, A.; Masuda, Y.; Kataoka, M.; Ishikawa, A.; Wakamatsu, K.; Mii, A.; Fujita, E.; Higo, S.; Kaneko, T.; et al. Development of lymphatic vasculature and morphological characterization in rat kidney. Clin. Exp. Nephrol. 2012, 16, 833–842. [Google Scholar] [CrossRef]
- O’Morchoe, C.C.; O’Morchoe, P.J.; Heney, N.M. Renal hilar lymph. Effects of diuresis on flow and composition in dogs. Circ. Res. 1970, 26, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Bivol, L.M.; Iversen, B.M.; Hultström, M.; Wallace, P.W.; Reed, R.K.; Wiig, H.; Tenstad, O. Unilateral renal ischaemia in rats induces a rapid secretion of inflammatory markers to renal lymph and increased capillary permeability. J. Physiol. 2016, 594, 1709–1726. [Google Scholar] [CrossRef] [Green Version]
- Donnan, M.D.; Kenig-Kozlovsky, Y.; Quaggin, S.E. The lymphatics in kidney health and disease. Nat. Rev. Nephrol. 2021, 17, 655–675. [Google Scholar] [CrossRef]
- Sakamoto, I.; Ito, Y.; Mizuno, M.; Suzuki, Y.; Sawai, A.; Tanaka, A.; Maruyama, S.; Takei, Y.; Yuzawa, Y.; Matsuo, S. Lymphatic vessels develop during tubulointerstitial fibrosis. Kidney Int. 2009, 75, 828–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarjou, A.; Black, L.M.; Bolisetty, S.; Traylor, A.M.; Bowhay, S.A.; Zhang, M.Z.; Harris, R.C.; Agarwal, A. Dynamic signature of lymphangiogenesis during acute kidney injury and chronic kidney disease. Lab. Investig. 2019, 99, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Randolph, G.J.; Ivanov, S.; Zinselmeyer, B.H.; Scallan, J.P. The lymphatic system: Integral roles in immunity. Annu. Rev. Immunol. 2017, 35, 31–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoja, C.; Benigni, A.; Remuzzi, G. Cellular responses to protein overload: Key event in renal disease progression. Curr. Opin. Nephrol. Hypertens. 2004, 13, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Boor, P.; Ostendorf, T.; Floege, J. Renal fibrosis: Novel insights into mechanisms and therapeutic targets. Nat. Rev. Nephrol. 2010, 6, 643–656. [Google Scholar] [CrossRef]
- Vigl, B.; Aebischer, D.; Nitschké, M.; Iolyeva, M.; Röthlin, T.; Antsiferova, O.; Halin, C. Tissue inflammation modulates gene expression of lymphatic endothelial cells and dendritic cell migration in a stimulus-dependent manner. Blood 2011, 118, 205–215. [Google Scholar] [CrossRef]
- Kerjaschki, D.; Huttary, N.; Raab, I.; Regele, H.; Bojarski-Nagy, K.; Bartel, G.; Kröber, S.M.; Greinix, H.; Rosenmaier, A.; Karlhofer, F.; et al. Lymphatic endothelial progenitor cells contribute to de novo lymphangiogenesis in human renal transplants. Nat. Med. 2006, 12, 230–234. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, C.; Cho, Y.P.; Lee, E.; Kim, H.; Kim, P.; Yun, S.H.; Yoon, Y.S. Podoplanin-expressing cells derived from bone marrow play a crucial role in postnatal lymphatic neovascularization. Circulation 2010, 122, 1413–1425. [Google Scholar] [CrossRef]
- Hur, J.; Jang, J.H.; Oh, I.Y.; Choi, J.I.; Yun, J.Y.; Kim, J.; Choi, Y.E.; Ko, S.B.; Kang, J.A.; Kang, J.; et al. Human podoplanin-positive monocytes and platelets enhance lymphangiogenesis through the activation of the podoplanin/CLEC-2 axis. Mol. Ther. 2014, 22, 1518–1529. [Google Scholar] [CrossRef] [Green Version]
- Bourne, J.H.; Beristain-Covarrubias, N.; Zuidscherwoude, M.; Campos, J.; Di, Y.; Garlick, E.; Colicchia, M.; Terry, L.V.; Thomas, S.G.; Brill, A.; et al. CLEC-2 Prevents accumulation and retention of inflammatory macrophages during murine peritonitis. Front. Immunol. 2021, 12, 693974. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ito, Y.; Mizuno, M.; Kinashi, H.; Sawai, A.; Noda, Y.; Mizuno, T.; Shimizu, H.; Fujita, Y.; Matsui, K.; et al. Transforming growth factor-β induces vascular endothelial growth factor-C expression leading to lymphangiogenesis in rat unilateral ureteral obstruction. Kidney Int. 2012, 81, 865–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, G.; Yao, Y.; Yang, Q.; Wang, M.; Wang, Y.; Wu, J.; Wang, P.; Li, Y.; Zhu, F.; Yang, J.; et al. Lymphangiogenesis in kidney and lymph node mediates renal inflammation and fibrosis. Sci. Adv. 2019, 5, eaaw5075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, S.; Nakano, T.; Torisu, K.; Tsuchimoto, A.; Eriguchi, M.; Haruyama, N.; Masutani, K.; Tsuruya, K.; Kitazono, T. Vascular endothelial growth factor-C ameliorates renal interstitial fibrosis through lymphangiogenesis in mouse unilateral ureteral obstruction. Lab. Investig. 2017, 97, 1439–1452. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, S.; Poosti, F.; Kramer, A.B.; Mirković, K.; Kwakernaak, A.J.; Hovingh, M.; Slagman, M.C.; Sjollema, K.A.; de Borst, M.H.; Navis, G.; et al. Proteinuria triggers renal lymphangiogenesis prior to the development of interstitial fibrosis. PLoS ONE 2012, 7, e50209. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Hwang, S.D.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Choi, B.S.; Kim, Y.S.; Kim, H.W.; Park, C.W. Attenuated lymphatic proliferation ameliorates diabetic nephropathy and high-fat diet-induced renal lipotoxicity. Sci. Rep. 2019, 9, 1994. [Google Scholar] [CrossRef]
- Hwang, S.D.; Song, J.H.; Kim, Y.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Hong, Y.A.; Chung, S.; Choi, B.S.; Kim, Y.S.; et al. Inhibition of lymphatic proliferation by the selective VEGFR-3 inhibitor SAR131675 ameliorates diabetic nephropathy in db/db mice. Cell Death Dis. 2019, 10, 219. [Google Scholar] [CrossRef]
- Kinashi, H.; Falke, L.L.; Nguyen, T.Q.; Bovenschen, N.; Aten, J.; Leask, A.; Ito, Y.; Goldschmeding, R. Connective tissue growth factor regulates fibrosis-associated renal lymphangiogenesis. Kidney Int. 2017, 92, 850–863. [Google Scholar] [CrossRef] [Green Version]
- Yokoi, H.; Mukoyama, M.; Nagae, T.; Mori, K.; Suganami, T.; Sawai, K.; Yoshioka, T.; Koshikawa, M.; Nishida, T.; Takigawa, M.; et al. Reduction in connective tissue growth factor by antisense treatment ameliorates renal tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2004, 15, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- Guha, M.; Xu, Z.G.; Tung, D.; Lanting, L.; Natarajan, R. Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J. 2007, 21, 3355–3368. [Google Scholar] [CrossRef]
- Reddy, S.T.; Rehor, A.; Schmoekel, H.G.; Hubbell, J.A.; Swartz, M.A. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J. Control. Release 2006, 112, 26–34. [Google Scholar] [CrossRef]
- Haraldsson, B.; Nyström, J.; Deen, W.M. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol. Rev. 2008, 88, 451–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolla, V.; Nizamutdinova, I.T.; Scharf, B.; Clement, C.C.; Maejima, D.; Akl, T.; Nagai, T.; Luciani, P.; Leroux, J.C.; Halin, C.; et al. Aging-related anatomical and biochemical changes in lymphatic collectors impair lymph transport, fluid homeostasis, and pathogen clearance. Aging Cell 2015, 14, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Lopes, T.G.; De Souza, M.L.; Da Silva, V.D.; Dos Santos, M.; Da Silva, W.I.C.; Itaquy, T.P.; Garbin, H.I.; Veronese, F.V. Markers of renal fibrosis: How do they correlate with podocyte damage in glomerular diseases? PLoS ONE 2019, 14, e0217585. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shelton, E.L.; Crescenzi, R.; Colvin, D.C.; Kirabo, A.; Zhong, J.; Delpire, E.J.; Yang, H.C.; Kon, V. Kidney Injury Causes Accumulation of Renal Sodium That Modulates Renal Lymphatic Dynamics. Int. J. Mol. Sci. 2022, 23, 1428. [Google Scholar] [CrossRef]
- Wiig, H.; Schröder, A.; Neuhofer, W.; Jantsch, J.; Kopp, C.; Karlsen, T.V.; Boschmann, M.; Goss, J.; Bry, M.; Rakova, N.; et al. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J. Clin. Investig. 2013, 123, 2803–2815. [Google Scholar] [CrossRef]
- Scallan, J.P.; Hill, M.A.; Davis, M.J. Lymphatic vascular integrity is disrupted in type 2 diabetes due to impaired nitric oxide signalling. Cardiovasc. Res. 2015, 107, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhuge, Z.; Carvalho, L.; Braga, V.A.; Lucena, R.B.; Li, S.; Schiffer, T.A.; Han, H.; Weitzberg, E.; Lundberg, J.O.; et al. Inorganic nitrate and nitrite ameliorate kidney fibrosis by restoring lipid metabolism via dual regulation of AMP-activated protein kinase and the AKT-PGC1α pathway. Redox Biol. 2022, 51, 102266. [Google Scholar] [CrossRef]
- Joukov, V.; Kumar, V.; Sorsa, T.; Arighi, E.; Weich, H.; Saksela, O.; Alitalo, K. A recombinant mutant vascular endothelial growth factor-C that has lost vascular endothelial growth factor receptor-2 binding, activation, and vascular permeability activities. J. Biol. Chem. 1998, 273, 6599–6602. [Google Scholar] [CrossRef] [Green Version]
- Lopez Gelston, C.A.; Balasubbramanian, D.; Abouelkheir, G.R.; Lopez, A.H.; Hudson, K.R.; Johnson, E.R.; Muthuchamy, M.; Mitchell, B.M.; Rutkowski, J.M. Enhancing renal lymphatic expansion prevents hypertension in mice. Circ. Res. 2018, 122, 1094–1101. [Google Scholar] [CrossRef] [Green Version]
- Yazdani, S.; Hijmans, R.S.; Poosti, F.; Dam, W.; Navis, G.; Van Goor, H.; Van den Born, J. Targeting tubulointerstitial remodeling in proteinuric nephropathy in rats. Dis. Model. Mech. 2015, 8, 919–930. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Hiremath, C.; Patterson, Q.; Vora, S.; Shang, Z.; Jamieson, A.R.; Fiolka, R.; Dean, K.M.; Dellinger, M.T.; Marciano, D.K. Heterozygous mutation of Vegfr3 reduces renal lymphatics without renal dysfunction. J. Am. Soc. Nephrol. 2021, 32, 3099–3113. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.M.; Zhang, D.; Camporez, J.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 2018, 24, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Yang, H.C.; Yermalitsky, V.; Shelton, E.L.; Otsuka, T.; Wiese, C.B.; May-Zhang, L.S.; Banan, B.; Abumrad, N.; Huang, J.; et al. Kidney injury-mediated disruption of intestinal lymphatics involves dicarbonyl-modified lipoproteins. Kidney Int. 2021, 100, 585–596. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Method | Description | Tissues |
|---|---|---|
| Digital three-dimensional reconstruction | Combination of immuno-staining, optical clearance and confocal microscopy imaging to achieve three-dimensional images of isolated tissues | skin; kidney; heart |
| Fluorescent dyes with/without transgenic mice | Transgenic mice expressed mOrange, tdTomato or green fluorescent protein under Prox1 transcriptional control to better visualize lymphatic network | adrenal medulla; skin; cornea; tumor; lung, kidney, heart, diaphragm, intestine, mesentery, liver; ocular surface; |
| Cannulation | Cannulation of isolated lymphatic vessels can be used to study contractility of lymphatic vessels under multiple controlled experimental conditions | mesentery; heart; kidney |
| Strategy | Model | Mechanism and Dosage | Results |
|---|---|---|---|
| VEGF-C-156S | UUO | Recombinant VEGF-C (Cys156Ser) is a selective VEGF-C agonist 10 μg/100 g BW per day for 14 days; intraperitoneal injection | ① Enhanced proliferation, not dilatation, of LVs ② Suppressed interstitial renal fibrosis; decreased collagen I level ③ Attenuated immune cells infiltration |
| LYVE-1-Cre/iDTR mice | UUO IRI | LYVE1+ LVs could be ablated in a DT-dependent manner 1.25 ng/g BW single dose; intravenous injection | ① Attenuated lymphocyte expansion, perirenal lymphadenectasis, and splenomegaly ② Suppressed interstitial inflammatory infiltration and renal fibrosis |
| Soluble VEGFR-3 or LYVE-1 fusion constructs | UUO IRI | Inhibit lymphangiogenesis by suppressing VEGF-C/D-VEGFR-3 and FGF2-LYVE1 signaling pathway 0.5 μg/g BW 24 h before UUO or IRI; tail vein injection | ① Decreased LV density in kidney and RDLN ② Ameliorated renal inflammation ③ Ameliorated renal fibrosis |
| KidVD+ mice | Salt-sensitive hypertension L-NAME–Induced hypertension | Transgenic mice with kidney specific overexpression of VEGF in doxycycline-dependent manner 1 week before L-NAME administration; continued L-NAME and doxycycline for 3 weeks; oral administration | ① Augmented renal lymphangiogenesis and prevented development of hypertension ② Renal lymphatic expansion ③ Reduced immune cells accumulation |
| IMC-3C5 | Adriamycin rats | Anti-VEGFR-3 antibody 40 mg/kg BW; 3 times/week for 6 weeks; intraperitoneal injection | ① Prevented LVs formation ② Did not effect inflammatory, fibrotic level |
| Vegfr3Chy/+ mice | Cisplatin-mediated injury | Transgenic mice with abrogated kinase ability of Vegfr3 | ① No change in the magnitude of renal dysfunction ② Increased renal LVs density due to the loss of cortex |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Yu, C. Lymphangiogenesis and Lymphatic Barrier Dysfunction in Renal Fibrosis. Int. J. Mol. Sci. 2022, 23, 6970. https://doi.org/10.3390/ijms23136970
Liu J, Yu C. Lymphangiogenesis and Lymphatic Barrier Dysfunction in Renal Fibrosis. International Journal of Molecular Sciences. 2022; 23(13):6970. https://doi.org/10.3390/ijms23136970
Chicago/Turabian StyleLiu, Jing, and Chen Yu. 2022. "Lymphangiogenesis and Lymphatic Barrier Dysfunction in Renal Fibrosis" International Journal of Molecular Sciences 23, no. 13: 6970. https://doi.org/10.3390/ijms23136970
APA StyleLiu, J., & Yu, C. (2022). Lymphangiogenesis and Lymphatic Barrier Dysfunction in Renal Fibrosis. International Journal of Molecular Sciences, 23(13), 6970. https://doi.org/10.3390/ijms23136970