
Sevoflurane Induces Neurotoxicity in the Animal Model with Alzheimer’s Disease Neuropathology via Modulating Glutamate Transporter and Neuronal Apoptosis

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
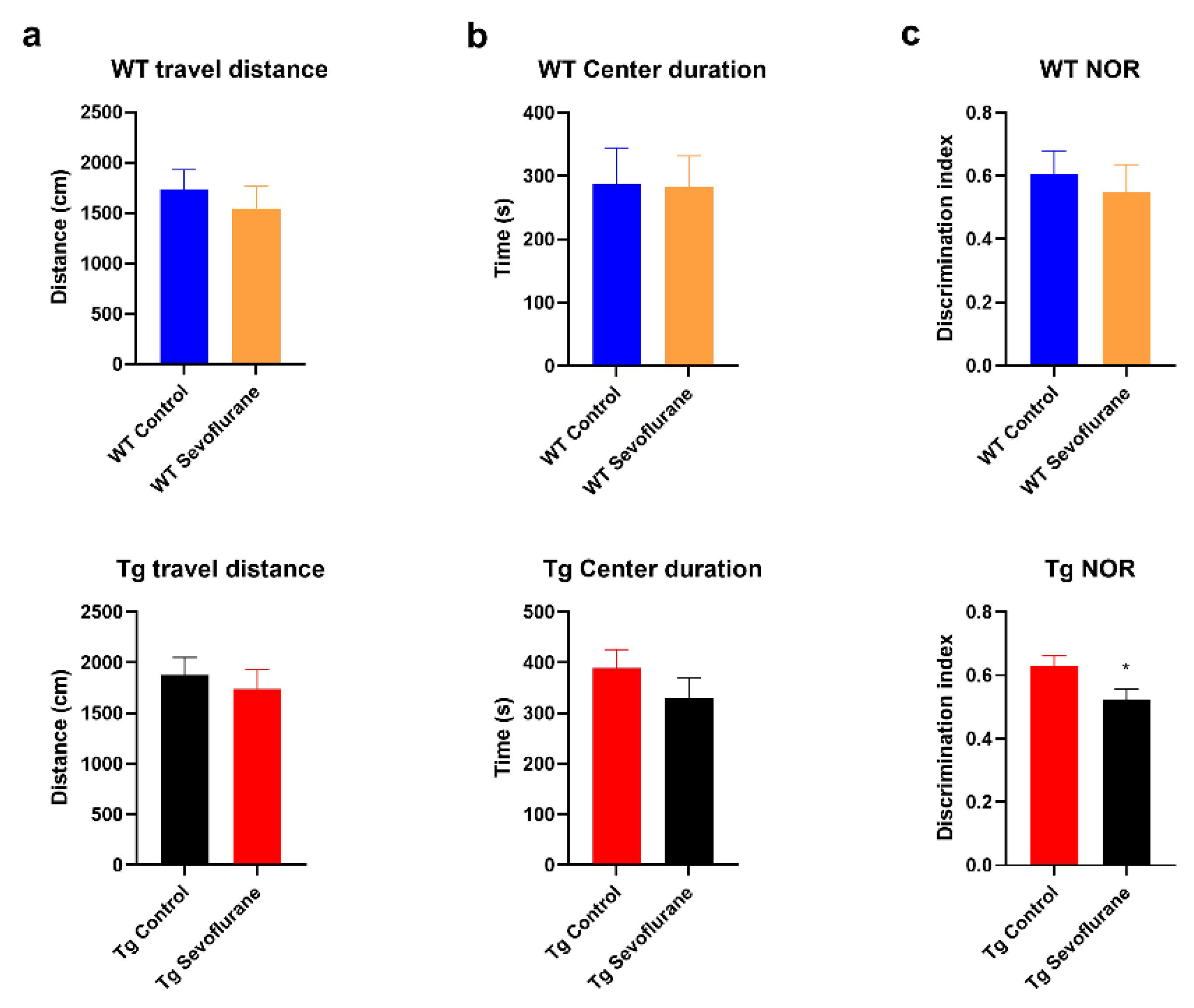
2.1. Cognitive Function Was Impaired in 3 × Tg Mice after Sevoflurane Exposure but Not in Wild-Type Mice
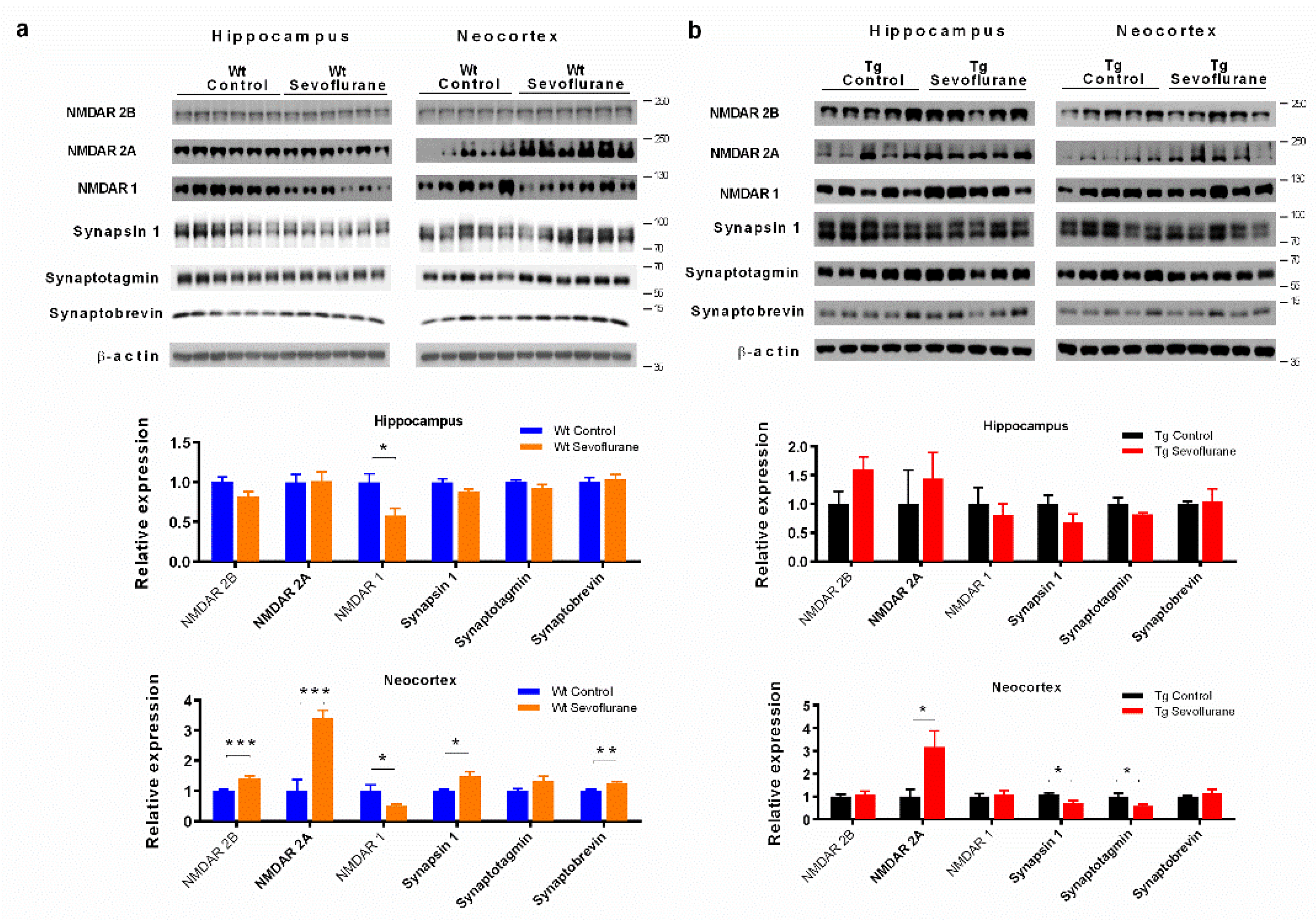
2.2. Differential Effects of Sevoflurane Anesthesia on Synaptic Protein Expression in Wild-Type and 3 × Tg Mice
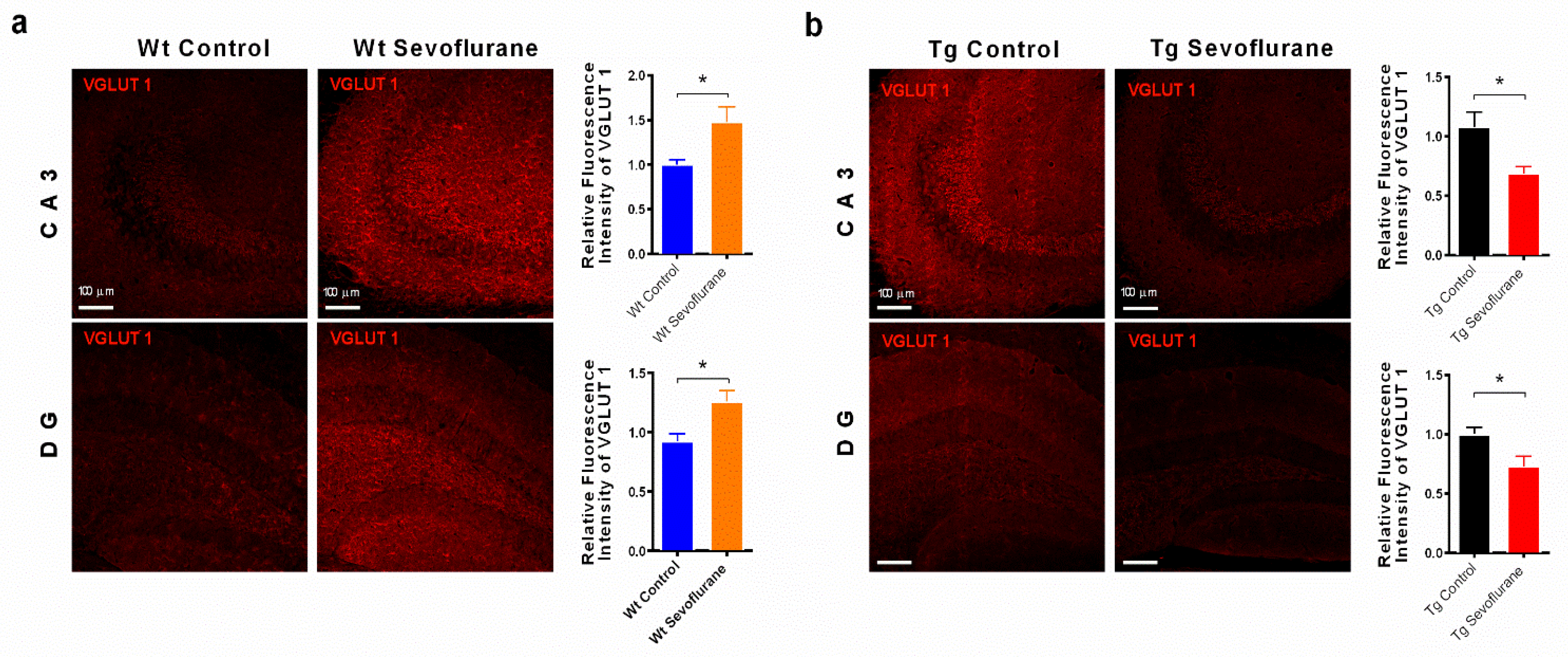
2.3. Sevoflurane Modulated the Expression of Vesicular Glutamate Transporters 1
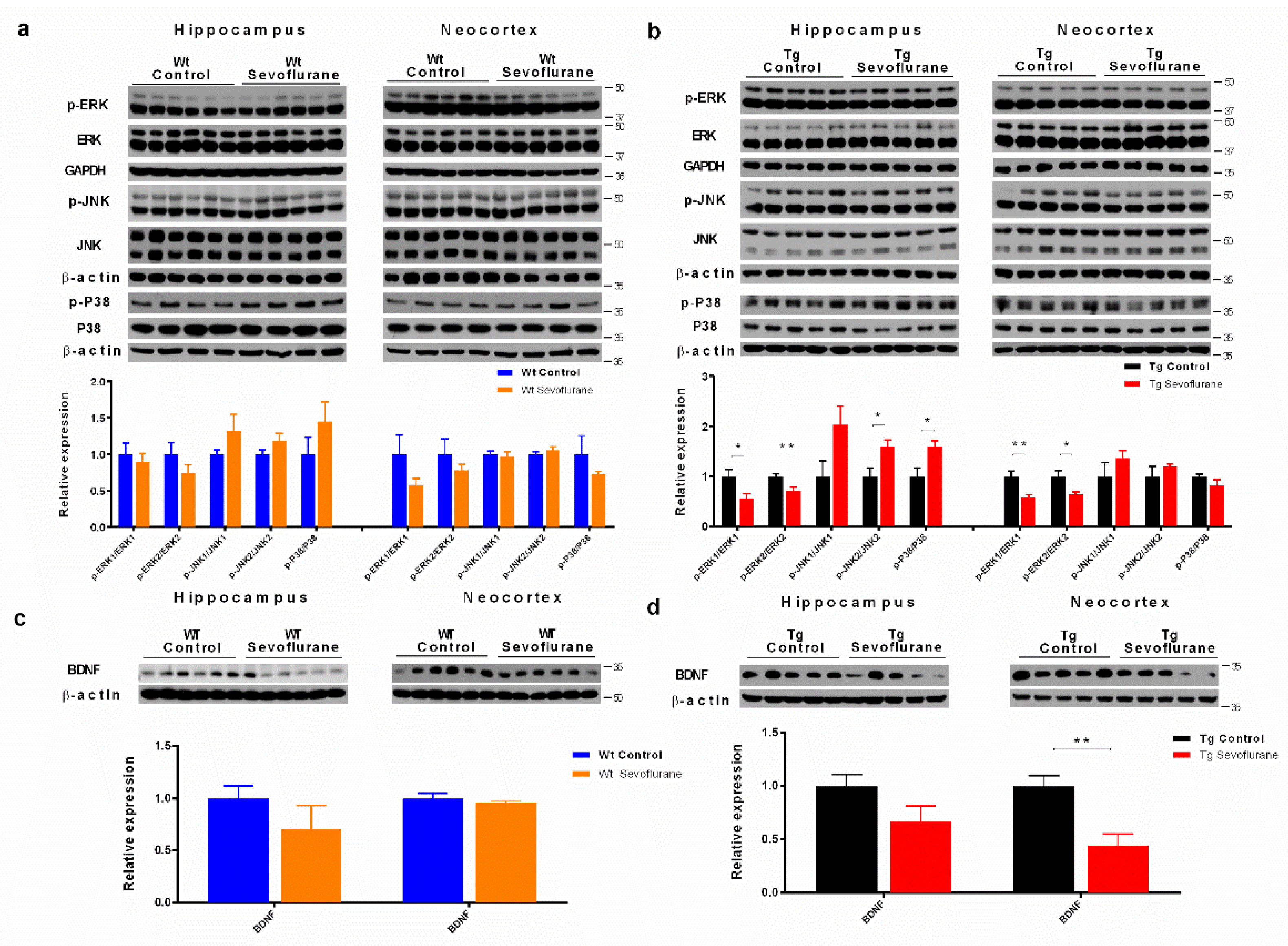
2.4. Sevoflurane Differentially Affected MAPK Signaling Pathways and Neurotrophic Factor Expression in Wild-Type and 3 × Tg Mice
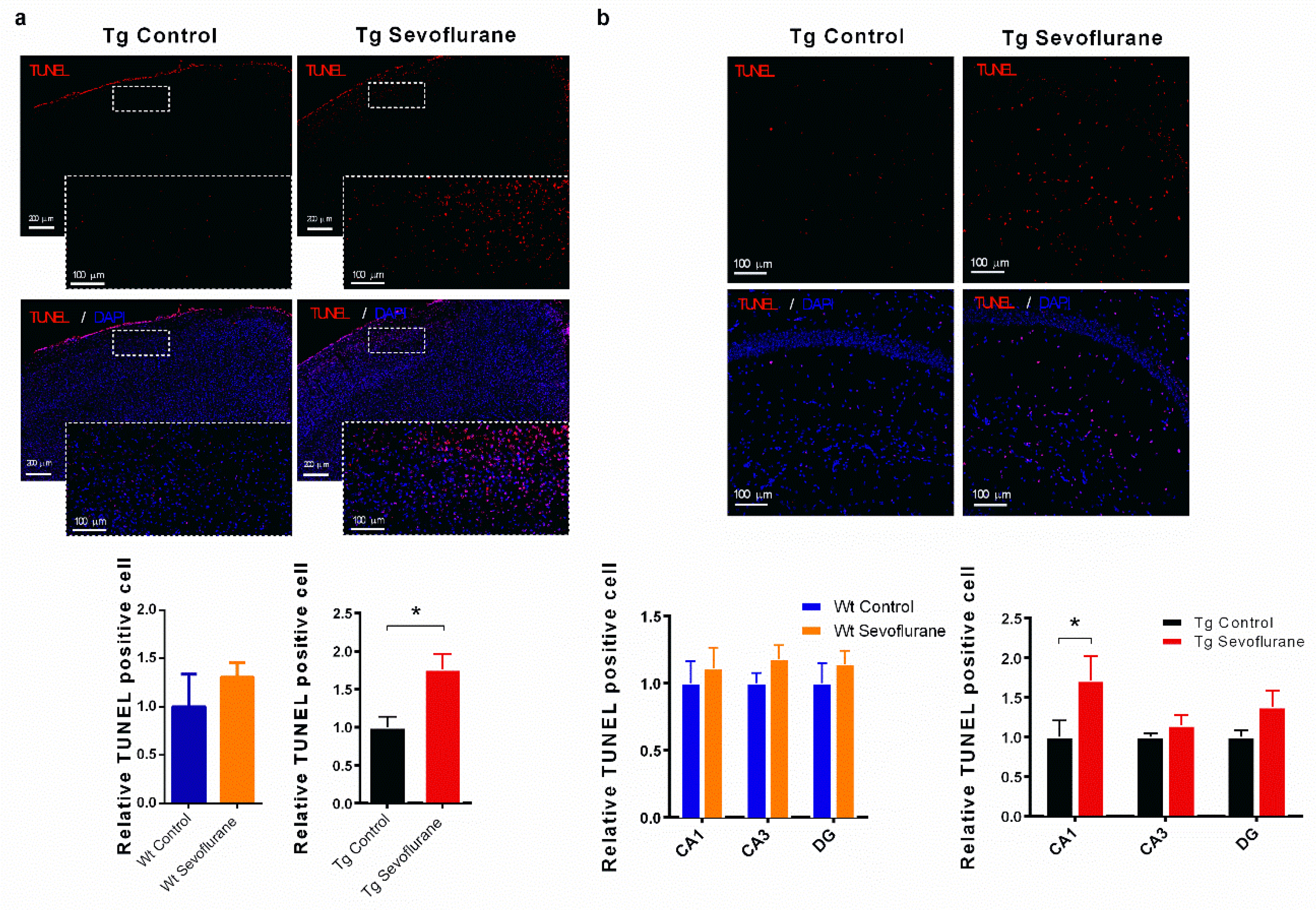
2.5. Sevoflurane Exerted Different Effects on Apoptosis in Hippocampi and Neocortices of Wild-Type and 3 × Tg Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Sevoflurane Exposure
4.3. Open Field Test
4.4. Novel Object Recognition Test
4.5. Preparation of the Whole Tissue Lysate and Synaptosomal Fraction
4.6. SDS-PAGE and Western-Blot Analysis
4.7. Immunofluorescence Staining
4.8. TUNEL Assay
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Subramaniyan, S.; Terrando, N. Neuroinflammation and Perioperative Neurocognitive Disorders. Anesth. Analg. 2019, 128, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Herrera, L.; Ostroff, R.D.; Rogers, S.A. Sevoflurane: Approaching the ideal inhalational anesthetic a pharmacologic, pharmacoeconomic, and clinical review. CNS Drug Rev. 2001, 7, 48–120. [Google Scholar] [CrossRef]
- Shi, C.X.; Jin, J.; Wang, X.Q.; Song, T.; Li, G.H.; Li, K.Z.; Ma, J.H. Sevoflurane attenuates brain damage through inhibiting autophagy and apoptosis in cerebral ischemia-reperfusion rats. Mol. Med. Rep. 2020, 21, 123–130. [Google Scholar] [CrossRef]
- Liu, H.; Chen, B.; Guo, B.; Deng, X.; Wang, B.; Dou, X. Postconditioning with Sevoflurane or Propofol Alleviates Lipopolysaccharide-Induced Neuroinflammation but Exerts Dissimilar Effects on the NR2B Subunit and Cognition. Mol. Neurobiol. 2021, 58, 4251–4267. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, L.; Wang, J.; Zhang, M.; Zhang, Y.; Hu, X. Sevoflurane postconditioning improves spatial learning and memory ability involving mitochondrial permeability transition pore in hemorrhagic shock and resuscitation rats. Brain Behav. 2020, 10, e01501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, F.H.; Li, J.; Li, K.Z.; Xie, Y.G.; Zhao, X.L. Effects of sevoflurane exposure during different stages of pregnancy on the brain development of rat offspring. J. Anesth. 2021, 35, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, L.; Li, L.; Li, X.; Huang, B.; Zhou, C.; Zhang, Z.; Wang, C.; Dong, P.; Zhang, X. Sevoflurane induces exaggerated and persistent cognitive decline in a type II diabetic rat model by aggregating hippocampal inflammation. Front. Pharmacol. 2017, 8, 886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neag, M.-A.; Mitre, A.-O.; Catinean, A.; Mitre, C.-I. An Overview on the Mechanisms of Neuroprotection and Neurotoxicity of Isoflurane and Sevoflurane in Experimental Studies. Brain Res. Bull. 2020, 165, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Millar, K.; Asbury, A.J.; Murray, G.D. Pre-existing cognitive impairment as a factor influencing outcome after cardiac surgery. BJA Br. J. Anaesth. 2001, 86, 63–67. [Google Scholar] [CrossRef] [Green Version]
- Silbert, B.; Evered, L.; Scott, D.A.; McMahon, S.; Choong, P.; Ames, D.; Maruff, P.; Jamrozik, K. Preexisting cognitive impairment is associated with postoperative cognitive dysfunction after hip joint replacement surgery. Anesthesiol. J. Am. Soc. Anesthesiol. 2015, 122, 1224–1234. [Google Scholar] [CrossRef]
- Stephan, B.C.; Hunter, S.; Harris, D.; Llewellyn, D.J.; Siervo, M.; Matthews, F.E.; Brayne, C. The neuropathological profile of mild cognitive impairment (MCI): A systematic review. Mol. Psychiatry 2012, 17, 1056–1076. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-X.; Xiong, B.; Xu, W.; Wei, H.-H.; Qu, W.-M.; Hong, Z.-Y.; Huang, Z.-L. Activation of parabrachial nucleus glutamatergic neurons accelerates reanimation from sevoflurane anesthesia in mice. Anesthesiology 2019, 130, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Chen, Y.; Sun, Z.; Shi, Z.; Qin, J.; Lu, J.; Li, C.; Ma, D.; Zhou, L.; Song, X. Prenatal sevoflurane exposure causes neuronal excitatory/inhibitory imbalance in the prefrontal cortex and neurofunctional abnormality in rats. Neurobiol. Dis. 2020, 146, 105121. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Centeno, C.; Repici, M.; Chatton, J.; Riederer, B.; Bonny, C.; Nicod, P.; Price, M.; Clarke, P.; Papa, S.; Franzoso, G. Role of the JNK pathway in NMDA-mediated excitotoxicity of cortical neurons. Cell Death Differ. 2007, 14, 240. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.; Manadas, B.; Melo, C.; Gomes, J.; Mendes, C.; Graos, M.; Carvalho, R.; Carvalho, A.; Duarte, C. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 2005, 12, 1329. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.C.; Rosario, E.R.; Chang, L.; Stanczyk, F.Z.; Oddo, S.; LaFerla, F.M.; Pike, C.J. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J. Neurosci. 2007, 27, 13357–13365. [Google Scholar] [CrossRef]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–62. [Google Scholar]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar]
- Bergmann, A.; Tugentman, M.; Shilo, B.-Z.; Steller, H. Regulation of cell number by MAPK-dependent control of apoptosis: A mechanism for trophic survival signaling. Dev. Cell 2002, 2, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Haddad, J.J. N-methyl-D-aspartate (NMDA) and the regulation of mitogen-activated protein kinase (MAPK) signaling pathways: A revolving neurochemical axis for therapeutic intervention? Prog. Neurobiol. 2005, 77, 252–282. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Finkbeiner, S.; Arnold, D.B.; Shaywitz, A.J.; Greenberg, M.E. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 1998, 20, 709–726. [Google Scholar] [CrossRef] [Green Version]
- Le Freche, H.; Brouillette, J.; Fernandez-Gomez, F.J.; Patin, P.; Caillierez, R.; Zommer, N.; Sergeant, N.; Buee-Scherrer, V.; Lebuffe, G.; Blum, D.; et al. Tau phosphorylation and sevoflurane anesthesia: An association to postoperative cognitive impairment. Anesthesiology 2012, 116, 779–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Zhang, G.; Zhang, B.; Moir, R.D.; Xia, W.; Marcantonio, E.R.; Culley, D.J.; Crosby, G.; Tanzi, R.E.; Xie, Z. The common inhalational anesthetic sevoflurane induces apoptosis and increases beta-amyloid protein levels. Arch. Neurol. 2009, 66, 620–631. [Google Scholar] [CrossRef]
- Steinmetz, J.; Christensen, K.B.; Lund, T.; Lohse, N.; Rasmussen, L.S.; Group, I. Long-term consequences of postoperative cognitive dysfunction. Anesthesiology 2009, 110, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Kapogiannis, D.; Schwartz, J.B.; Lobach, I.V.; Goetzl, L.; Abner, E.L.; Jicha, G.A.; Karydas, A.M.; Boxer, A.; Miller, B.L. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 4141–4148. [Google Scholar] [CrossRef] [Green Version]
- Clare, R.; King, V.G.; Wirenfeldt, M.; Vinters, H.V. Synapse loss in dementias. J. Neurosci. Res. 2010, 88, 2083–2090. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef]
- Wang, S.-H.; Morris, R.G. Hippocampal-neocortical interactions in memory formation, consolidation, and reconsolidation. Annu. Rev. Psychol. 2010, 61, 49–79. [Google Scholar] [CrossRef]
- Bykhovskaia, M. Synapsin regulation of vesicle organization and functional pools. Semin Cell Dev. Biol 2011, 22, 387–392. [Google Scholar] [CrossRef]
- Südhof, T.C. A molecular machine for neurotransmitter release: Synaptotagmin and beyond. Nat. Med. 2013, 19, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Cesca, F.; Baldelli, P.; Valtorta, F.; Benfenati, F. The synapsins: Key actors of synapse function and plasticity. Prog. Neurobiol. 2010, 91, 313–348. [Google Scholar] [CrossRef]
- De Jong, A.P.; Meijer, M.; Saarloos, I.; Cornelisse, L.N.; Toonen, R.F.; Sorensen, J.B.; Verhage, M. Phosphorylation of synaptotagmin-1 controls a post-priming step in PKC-dependent presynaptic plasticity. Proc. Natl. Acad. Sci. USA 2016, 113, 5095–5100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cull-Candy, S.; Brickley, S.; Farrant, M. NMDA receptor subunits: Diversity, development and disease. Curr. Opin. Neurobiol. 2001, 11, 327–335. [Google Scholar] [CrossRef]
- Brewer, L.D.; Thibault, O.; Staton, J.; Thibault, V.; Rogers, J.T.; Garcia-Ramos, G.; Kraner, S.; Landfield, P.W.; Porter, N.M. Increased vulnerability of hippocampal neurons with age in culture: Temporal association with increases in NMDA receptor current, NR2A subunit expression and recruitment of L-type calcium channels. Brain Res. 2007, 1151, 20–31. [Google Scholar] [CrossRef]
- Cui, Z.; Feng, R.; Jacobs, S.; Duan, Y.; Wang, H.; Cao, X.; Tsien, J.Z. Increased NR2A: NR2B ratio compresses long-term depression range and constrains long-term memory. Sci. Rep. 2013, 3, 1036. [Google Scholar] [CrossRef] [Green Version]
- Bellocchio, E.E.; Reimer, R.J.; Fremeau, R.T.; Edwards, R.H. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 2000, 289, 957–960. [Google Scholar] [CrossRef]
- Choi, D.W. Ionic dependence of glutamate neurotoxicity. J. Neurosci. 1987, 7, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Kashani, A.; Lepicard, E.; Poirel, O.; Videau, C.; David, J.P.; Fallet-Bianco, C.; Simon, A.; Delacourte, A.; Giros, B.; Epelbaum, J. Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol. Aging 2008, 29, 1619–1630. [Google Scholar] [CrossRef] [Green Version]
- Soghomonian, J.J.; Martin, D.L. Two isoforms of glutamate decarboxylase: Why? Trends Pharm. Sci. 1998, 19, 500–505. [Google Scholar] [CrossRef]
- Sun, Z.; Satomoto, M.; Adachi, Y.U.; Makita, K. Apocynin preserves glutamatergic neurons in the basolateral amygdala in mice with neonatal sevoflurane exposure. Korean J. Anesth. 2017, 70, 335–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, I.; Blanco, R.; Carmona, M.; Puig, B. Phosphorylated mitogen-activated protein kinase (MAPK/ERK-P), protein kinase of 38 kDa (p38-P), stress-activated protein kinase (SAPK/JNK-P), and calcium/calmodulin-dependent kinase II (CaM kinase II) are differentially expressed in tau deposits in neurons and glial cells in tauopathies. J. Neural. Transm. 2001, 108, 1397–1415. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Zhang, Y.; Zuo, Y.; Israr, M.; Li, B.; Yu, P.; Gao, G.; Chang, Y.Z.; Shi, Z. Transcriptomic analysis reveals the molecular mechanism of Alzheimer-related neuropathology induced by sevoflurane in mice. J. Cell Biochem. 2019, 120, 17555–17565. [Google Scholar] [CrossRef]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [Green Version]
- Ip, Y.T.; Davis, R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)—from inflammation to development. Curr. Opin. Cell Biol. 1998, 10, 205–219. [Google Scholar] [CrossRef]
- Khairova, R.A.; Machado-Vieira, R.; Du, J.; Manji, H.K. A potential role for pro-inflammatory cytokines in regulating synaptic plasticity in major depressive disorder. Int. J. Neuropsychopharmacol. 2009, 12, 561–578. [Google Scholar] [CrossRef]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef] [Green Version]
- Tsuruta, F.; Sunayama, J.; Mori, Y.; Hattori, S.; Shimizu, S.; Tsujimoto, Y.; Yoshioka, K.; Masuyama, N.; Gotoh, Y. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 2004, 23, 1889–1899. [Google Scholar] [CrossRef] [Green Version]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef]
- Shin, D.J.; Germann, A.L.; Johnson, A.D.; Forman, S.A.; Steinbach, J.H.; Akk, G. Propofol is an allosteric agonist with multiple binding sites on concatemeric ternary GABAA receptors. Mol. Pharmacol. 2018, 93, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Mardini, F.; Tang, J.; Li, J.; Arroliga, M.; Eckenhoff, R.; Eckenhoff, M. Effects of propofol and surgery on neuropathology and cognition in the 3xTgAD Alzheimer transgenic mouse model. BJA Br. J. Anaesth. 2017, 119, 472–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodhouse, A.; Fernandez-Martos, C.M.; Atkinson, R.A.K.; Hanson, K.A.; Collins, J.M.; O’Mara, A.R.; Terblanche, N.; Skinner, M.W.; Vickers, J.C.; King, A.E. Repeat propofol anesthesia does not exacerbate plaque deposition or synapse loss in APP/PS1 Alzheimer’s disease mice. BMC Anesthesiol. 2018, 18, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Tovote, P.; Fadok, J.P.; Luthi, A. Neuronal circuits for fear and anxiety. Nat. Rev. Neurosci. 2015, 16, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Tatem, K.S.; Quinn, J.L.; Phadke, A.; Yu, Q.; Gordish-Dressman, H.; Nagaraju, K. Behavioral and locomotor measurements using an open field activity monitoring system for skeletal muscle diseases. J. Vis. Exp. 2014, 91, e51785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.M.T.; Abulimiti, A.; Wong, B.S.H.; Zhao, G.D.; Xiong, S.H.; Zhao, M.M.; Wang, Y.; Chen, Y.; Wang, J.; Zhang, Y. Sigesbeckia orientalis L. Derived Active Fraction Ameliorates Perioperative Neurocognitive Disorders Through Alleviating Hippocampal Neuroinflammation. Front. Pharmacol. 2022, 13, 846631. [Google Scholar] [CrossRef]
- Chu, J.M.T.; Xiong, W.; Linghu, K.G.; Liu, Y.; Zhang, Y.; Zhao, G.D.; Irwin, M.G.; Wong, G.T.C.; Yu, H. Siegesbeckia Orientalis L. Extract Attenuates Postoperative Cognitive Dysfunction, Systemic Inflammation, and Neuroinflammation. Exp. Neurobiol. 2018, 27, 564–573. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.; Chu, J.M.T.; Liu, Y.; Kwong, V.S.W.; Chang, R.C.C.; Wong, G.T.C. Sevoflurane Induces Neurotoxicity in the Animal Model with Alzheimer’s Disease Neuropathology via Modulating Glutamate Transporter and Neuronal Apoptosis. Int. J. Mol. Sci. 2022, 23, 6250. https://doi.org/10.3390/ijms23116250
Huang C, Chu JMT, Liu Y, Kwong VSW, Chang RCC, Wong GTC. Sevoflurane Induces Neurotoxicity in the Animal Model with Alzheimer’s Disease Neuropathology via Modulating Glutamate Transporter and Neuronal Apoptosis. International Journal of Molecular Sciences. 2022; 23(11):6250. https://doi.org/10.3390/ijms23116250
Chicago/Turabian StyleHuang, Chunxia, John Man Tak Chu, Yan Liu, Vivian Suk Wai Kwong, Raymond Chuen Chung Chang, and Gordon Tin Chun Wong. 2022. "Sevoflurane Induces Neurotoxicity in the Animal Model with Alzheimer’s Disease Neuropathology via Modulating Glutamate Transporter and Neuronal Apoptosis" International Journal of Molecular Sciences 23, no. 11: 6250. https://doi.org/10.3390/ijms23116250
APA StyleHuang, C., Chu, J. M. T., Liu, Y., Kwong, V. S. W., Chang, R. C. C., & Wong, G. T. C. (2022). Sevoflurane Induces Neurotoxicity in the Animal Model with Alzheimer’s Disease Neuropathology via Modulating Glutamate Transporter and Neuronal Apoptosis. International Journal of Molecular Sciences, 23(11), 6250. https://doi.org/10.3390/ijms23116250