
Molecular Mechanism of Induction of Bone Growth by the C-Type Natriuretic Peptide




Abstract
1. Introduction
2. C-Type Natriuretic Peptide and Nitric Oxide as Bone Growth Regulators
3. CNP/NPR-B/cGMP Pathway
4. CNP/NPR-B/cGMP/MAPK Pathway
5. CNP/NPR-B/cGMP/pCREB Pathway
6. C-Type Natriuretic Peptide as Extracellular Matrix Regulator
7. Synergic Effect of OP-1 and CNP
8. Survival of Animals with C-Type Natriuretic Peptide
9. C-Type Natriuretic Peptide as a Treatment
10. Discussion
- CNP produced in the growth plate is a potent positive regulator of linear growth.
- Reduced intracellular CNP pathway activity may increase CNP production; a negative feedback loop regulates CNP
- In a group of skeletal dysplasias, elevated plasma levels of NTproCNP indicate the presence of tissue resistance to CNP.
- Long-term CNP therapy for achondroplasia and other skeletal disorders remains unknown.
- The interaction between the CNP/NPR-B pathway and other pathways should be explored to elucidate the bone growth.
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Long, F.; Ornitz, D.M. Development of the Endochondral Skeleton. Cold Spring Harb. Perspect. Biol. 2013, 5, a008334. [Google Scholar] [CrossRef] [PubMed]
- Salhotra, A.; Shah, H.N.; Levi, B.; Longaker, M.T. Mechanisms of Bone Development and Repair. Nat. Rev. Mol. Cell Biol. 2020, 21, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Santhekadur, P.K.; Kumar, D.P.; Seneshaw, M.; Mirshahi, F.; Sanyal, A.J. The Multifaceted Role of Natriuretic Peptides in Metabolic Syndrome. Biomed. Pharmacother. 2017, 92, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, H.; Chong, C.K.; Veress, A.T. Cardiac Atrial Factor—an Endogenous Diuretic? Can. J. Physiol. Pharmacol. 1981, 59, 1278–1279. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Nishikimi, T.; Kuwahara, K. Atrial and Brain Natriuretic Peptides: Hormones Secreted from the Heart. Peptides 2019, 111, 18–25. [Google Scholar] [CrossRef]
- Collins, S. A Heart–Adipose Tissue Connection in the Regulation of Energy Metabolism. Nat. Rev. Endocrinol. 2014, 10, 157–163. [Google Scholar] [CrossRef]
- Berglund, H.; Nyquist, O.; Beermann, B.; Jensen-Urstad, M.; Theodorsson, E. Influence of Angiotensin Converting Enzyme Inhibition on Relation of Atrial Natriuretic Peptide Concentration to Atrial Pressure in Heart Failure. Heart 1994, 72, 521–527. [Google Scholar] [CrossRef][Green Version]
- Kotby, A.A.; Taman, K.H.; Sedky, H.T.A.; Habeeb, N.M.; El-Hadidi, E.S.; Yosseif, H.S. Atrial Natriuretic Peptide as a Marker of Heart Failure in Children with Left Ventricular Volume Overload. J. Paediatr. Child Health 2013, 49, 43–47. [Google Scholar] [CrossRef]
- Asakura, M.; Jiyoong, K.; Minamino, T.; Shintani, Y.; Asanuma, H.; Kitakaze, M.; The J-WIND Investigators. Rationale and Design of a Large-Scale Trial Using Atrial Natriuretic Peptide (ANP) as an Adjunct to Percutaneous Coronary Intervention for ST-Segment Elevation Acute Myocardial Infarction-Japan-Working Groups of Acute Myocardial Infarction for the Reduction of Necrotic Damage by ANP (J-WIND-ANP). Circ. J. 2004, 68, 95–100. [Google Scholar] [CrossRef]
- Nishikimi, T.; Mori, Y.; Ishimura, K.; Tadokoro, K.; Yagi, H.; Yabe, A.; Horinaka, S.; Matsuoka, H. Association of Plasma Atrial Natriuretic Peptide, n-Terminal Proatrial Natriuretic Peptide, and Brain Natriuretic Peptide Levels with Coronary Artery Stenosis in Patients with Normal Left Ventricular Systolic Function. Am. J. Med. 2004, 116, 517–523. [Google Scholar] [CrossRef]
- Hui, D.; Naberhuis, J.; Dibaj, S.; Naqvi, M.; Liu, D.; Bruera, E. Association Between Plasma Brain Natriuretic Peptide and Overall Survival in Patients With Advanced Cancer: Preliminary Findings. J. Pain Symptom Manag. 2019, 58, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.B.; McKenna, K.; Osman, M.; Tormey, W.P.; McDonald, D.; Thompson, C.J. Atrial Natriuretic Peptide Increases Urinary Albumin Excretion in People with Normoalbuminuric Type-2 Diabetes. Ir. J. Med. Sci. 2007, 176, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, E.B.; Bacevicius, E.; Bech, J.N.; Solling, K.; Pedersen, H.B. Abnormal Rhythmic Oscillations of Atrial Natriuretic Peptide and Brain Natriuretic Peptide in Chronic Renal Failure. Clin. Sci. 2006, 110, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Katan, M.; Fluri, F.; Schuetz, P.; Morgenthaler, N.G.; Zweifel, C.; Bingisser, R.; Kappos, L.; Steck, A.; Engelter, S.T.; Müller, B.; et al. Midregional Pro-Atrial Natriuretic Peptide and Outcome in Patients With Acute Ischemic Stroke. J. Am. Coll. Cardiol. 2010, 56, 1045–1053. [Google Scholar] [CrossRef]
- Zois, N.E.; Terzic, D.; Færch, K.; Plomgaard, P.; Hansen, J.S.; Rossing, P.; Goetze, J.P. Effect of Pancreatic Hormones on Pro-Atrial Natriuretic Peptide in Humans. EBioMedicine 2017, 17, 88–94. [Google Scholar] [CrossRef]
- Jujić, A.; Nilsson, P.M.; Persson, M.; Holst, J.J.; Torekov, S.S.; Lyssenko, V.; Groop, L.; Melander, O.; Magnusson, M. Atrial Natriuretic Peptide in the High Normal Range Is Associated With Lower Prevalence of Insulin Resistance. J. Clin. Endocrinol. Metab. 2016, 101, 1372–1380. [Google Scholar] [CrossRef]
- Parcha, V.; Patel, N.; Gutierrez, O.M.; Li, P.; Gamble, K.L.; Musunuru, K.; Margulies, K.B.; Cappola, T.P.; Wang, T.J.; Arora, G.; et al. Chronobiology of Natriuretic Peptides and Blood Pressure in Lean and Obese Individuals. J. Am. Coll. Cardiol. 2021, 77, 2291–2303. [Google Scholar] [CrossRef]
- Molina, C.R.; Fowler, M.B.; Mccrory, S.; Peterson, C.; Myers, B.D.; Schroeder, J.S.; Murad, F. Hemodynamic, Renal and Endocrine Effects of Atrial Natriuretic Peptide Infusion in Severe Heart Failure. J. Am. Coll. Cardiol. 1988, 12, 175–186. [Google Scholar] [CrossRef]
- Tanaka, T.; Tsutamoto, T.; Sakai, H.; Nishiyama, K.; Fujii, M.; Yamamoto, T.; Horie, M. Effect of Atrial Natriuretic Peptide on Adiponectin in Patients with Heart Failure. Eur. J. Heart Fail. 2008, 10, 360–366. [Google Scholar] [CrossRef]
- Valsson, F.; Lundin, S.; Kirno, K.; Hedner, T.; Houltz, E.; Saito, Y.; Ricksten, S.-E. Atrial Natriuretic Peptide Attenuates Pacing-Induced Myocardial Ischemia During General Anesthesia in Patients with Coronary Artery Disease. Anesth. Analg. 1999, 88, 279–285. [Google Scholar] [CrossRef]
- Nojiri, T.; Yamamoto, K.; Maeda, H.; Takeuchi, Y.; Funakoshi, Y.; Inoue, M.; Okumura, M. Effect of Low-Dose Human Atrial Natriuretic Peptide on Postoperative Atrial Fibrillation in Patients Undergoing Pulmonary Resection for Lung Cancer: A Double-Blind, Placebo-Controlled Study. J. Thorac. Cardiovasc. Surg. 2012, 143, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Kitakaze, M.; Asakura, M.; Kim, J.; Shintani, Y.; Asanuma, H.; Hamasaki, T.; Seguchi, O.; Myoishi, M.; Minamino, T.; Ohara, T.; et al. Human Atrial Natriuretic Peptide and Nicorandil as Adjuncts to Reperfusion Treatment for Acute Myocardial Infarction (J-WIND): Two Randomised Trials. Lancet 2007, 370, 1483–1493. [Google Scholar] [CrossRef]
- Sezai, A.; Wakui, S.; Akiyama, K.; Hata, M.; Yoshitake, I.; Unosawa, S.; Shiono, M.; Hirayama, A. Myocardial Protective Effect of Human Atrial Natriuretic Peptide in Cardiac Surgery—“HANP Shot” in Clinical Safety Trial. Circ. J. 2011, 75, 2144–2150. [Google Scholar] [CrossRef] [PubMed]
- Nojiri, T.; Yamamoto, H.; Hamasaki, T.; Onda, K.; Ohshima, K.; Shintani, Y.; Okumura, M.; Kangawa, K. A Multicenter Randomized Controlled Trial of Surgery Alone or Surgery with Atrial Natriuretic Peptide in Lung Cancer Surgery: Study Protocol for a Randomized Controlled Trial. Trials 2017, 18, 183. [Google Scholar] [CrossRef]
- Hayashida, N.; Chihara, S.; Kashikie, H.; Tayama, E.; Yokose, S.; Akasu, K.; Aoyagi, S. Effects of Intraoperative Administration of Atrial Natriuretic Peptide. Ann. Thorac. Surg. 2000, 70, 1319–1326. [Google Scholar] [CrossRef]
- Park, K.; Itoh, H.; Yamahara, K.; Sone, M.; Miyashita, K.; Oyamada, N.; Sawada, N.; Taura, D.; Inuzuka, M.; Sonoyama, T.; et al. Therapeutic Potential of Atrial Natriuretic Peptide Administration on Peripheral Arterial Diseases. Endocrinology 2008, 149, 483–491. [Google Scholar] [CrossRef]
- Groban, L.; Cowley, A.W.; Ebert, T.J. Atrial Natriuretic Peptide Augments Forearm Capillary Filtration in Humans. Am. J. Physiol.-Heart Circ. Physiol. 1990, 259, H258–H263. [Google Scholar] [CrossRef]
- Kasama, S.; Toyama, T.; Hatori, T.; Sumino, H.; Kumakura, H.; Takayama, Y.; Ichikawa, S.; Suzuki, T.; Kurabayashi, M. Effects of Intravenous Atrial Natriuretic Peptide on Cardiac Sympathetic Nerve Activity and Left Ventricular Remodeling in Patients With First Anterior Acute Myocardial Infarction. J. Am. Coll. Cardiol. 2007, 49, 667–674. [Google Scholar] [CrossRef]
- Moriyama, T.; Hagihara, S.; Shiramomo, T.; Nagaoka, M.; Iwakawa, S.; Kanmura, Y. The Protective Effect of Human Atrial Natriuretic Peptide on Renal Damage during Cardiac Surgery. J. Anesth. 2017, 31, 163–169. [Google Scholar] [CrossRef]
- Mitaka, C.; Ohnuma, T.; Murayama, T.; Kunimoto, F.; Nagashima, M.; Takei, T.; Iguchi, N.; Tomita, M. Effects of Low-Dose Atrial Natriuretic Peptide Infusion on Cardiac Surgery–Associated Acute Kidney Injury: A Multicenter Randomized Controlled Trial. J. Crit. Care 2017, 38, 253–258. [Google Scholar] [CrossRef]
- Rahman, S.N.; Kim, G.E.; Mathew, A.S.; Goldberg, C.A.; Allgren, R.; Schrier, R.W.; Conger, J.D. Effects of Atrial Natriuretic Peptide in Clinical Acute Renal Failure. Kidney Int. 1994, 45, 1731–1738. [Google Scholar] [CrossRef] [PubMed]
- Swärd, K.; Valsson, F.; Odencrants, P.; Samuelsson, O.; Ricksten, S.-E. Recombinant Human Atrial Natriuretic Peptide in Ischemic Acute Renal Failure: A Randomized Placebo-Controlled Trial*. Crit. Care Med. 2004, 32, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Mitaka, C.; Kudo, T.; Jibiki, M.; Sugano, N.; Inoue, Y.; Makita, K.; Imai, T. Effects of Human Atrial Natriuretic Peptide on Renal Function in Patients Undergoing Abdominal Aortic Aneurysm Repair*. Crit. Care Med. 2008, 36, 745–751. [Google Scholar] [CrossRef]
- Koda, M.; Sakamoto, A.; Ogawa, R. Effects of Atrial Natriuretic Peptide at a Low Dose on Water and Electrolyte Metabolism during General Anesthesia. J. Clin. Anesth. 2005, 17, 3–7. [Google Scholar] [CrossRef]
- Swärd, K.; Valson, F.; Ricksten, S.-E. Long-Term Infusion of Atrial Natriuretic Peptide (ANP) Improves Renal Blood Flow and Glomerular Filtration Rate in Clinical Acute Renal Failure. Acta Anaesthesiol. Scand. 2001, 45, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Takeda, T.; Nishizawa, Y.; Ogino, T.; Miyoshi, N.; Matsuda, C.; Yamamoto, H.; Mizushima, T.; Doki, Y.; Eguchi, H. Phase I Study of the Administration of Low-Dose Perioperative Human Atrial Natriuretic Peptide in Patients With Resectable Colorectal Cancer. Anticancer Res. 2020, 40, 5301–5307. [Google Scholar] [CrossRef]
- van der Zander, K. Effects of Brain Natriuretic Peptide on Forearm Vasculature: Comparison with Atrial Natriuretic Peptide. Cardiovasc. Res. 1999, 44, 595–600. [Google Scholar] [CrossRef][Green Version]
- Mori, Y.; Kamada, T.; Ochiai, R. Reduction in the Incidence of Acute Kidney Injury after Aortic Arch Surgery with Low-Dose Atrial Natriuretic Peptide. Eur. J. Anaesthesiol. 2014, 31, 381–387. [Google Scholar] [CrossRef]
- Moro, C.; Polak, J.; Hejnova, J.; Klimcakova, E.; Crampes, F.; Stich, V.; Lafontan, M.; Berlan, M. Atrial Natriuretic Peptide Stimulates Lipid Mobilization during Repeated Bouts of Endurance Exercise. Am. J. Physiol.-Endocrinol. Metab. 2006, 290, E864–E869. [Google Scholar] [CrossRef]
- Cargill, R.I.; Lipworth, B.J. Pulmonary Vasorelaxant Activity of Atrial Natriuretic Peptide and Brain Natriuretic Peptide in Humans. Thorax 1995, 50, 183–185. [Google Scholar] [CrossRef]
- Mukoyama, M.; Nakao, K.; Saito, Y.; Ogawa, Y.; Hosoda, K.; Suga, K.; Shirakami, G.; Jougasaki, M.; Imura, H. Increased Human Brain Natriuretic Peptide in Congestive Heart Failure. N. Engl. J. Med. 1990, 323, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Mukoyama, M.; Nakao, K.; Hosoda, K.; Suga, S.; Saito, Y.; Ogawa, Y.; Shirakami, G.; Jougasaki, M.; Obata, K.; Yasue, H. Brain Natriuretic Peptide as a Novel Cardiac Hormone in Humans. Evidence for an Exquisite Dual Natriuretic Peptide System, Atrial Natriuretic Peptide and Brain Natriuretic Peptide. J. Clin. Investig. 1991, 87, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Maisel, A.S.; Krishnaswamy, P.; Nowak, R.M.; McCord, J.; Hollander, J.E.; Duc, P.; Omland, T.; Storrow, A.B.; Abraham, W.T.; Wu, A.H.B.; et al. Rapid Measurement of B-Type Natriuretic Peptide in the Emergency Diagnosis of Heart Failure. N. Engl. J. Med. 2002, 347, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Maisel, A.; Barnard, D.; Jaski, B.; Frivold, G.; Marais, J.; Azer, M.; Miyamoto, M.I.; Lombardo, D.; Kelsay, D.; Borden, K.; et al. Primary Results of the HABIT Trial (Heart Failure Assessment With BNP in the Home). J. Am. Coll. Cardiol. 2013, 61, 1726–1735. [Google Scholar] [CrossRef]
- Maisel, A.S.; Shah, K.S.; Barnard, D.; Jaski, B.; Frivold, G.; Marais, J.; Azer, M.; Miyamoto, M.I.; Lombardo, D.; Kelsay, D.; et al. How B-Type Natriuretic Peptide (BNP) and Body Weight Changes Vary in Heart Failure With Preserved Ejection Fraction Compared With Reduced Ejection Fraction: Secondary Results of the HABIT (HF Assessment With BNP in the Home) Trial. J. Card. Fail. 2016, 22, 283–293. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, J.; Bolun, Z.; Pei, M.; Ma, B.; Tong, Q.; Yin, H.; Zhang, Y.; You, L.; Xie, R. Comparison of Cardiac Function between Left Bundle Branch Pacing and Right Ventricular Outflow Tract Septal Pacing in the Short-Term: A Registered Controlled Clinical Trial. Int. J. Cardiol. 2021, 322, 70–76. [Google Scholar] [CrossRef]
- Passino, C.; del Ry, S.; Severino, S.; Gabutti, A.; Prontera, C.; Clerico, A.; Giannessi, D.; Emdin, M. C-Type Natriuretic Peptide Expression in Patients with Chronic Heart Failure: Effects of Aerobic Training. Eur. J. Cardiovasc. Prev. Rehabil. 2008, 15, 168–172. [Google Scholar] [CrossRef]
- Mann, D.L.; Greene, S.J.; Givertz, M.M.; Vader, J.M.; Starling, R.C.; Ambrosy, A.P.; Shah, P.; McNulty, S.E.; Mahr, C.; Gupta, D.; et al. Sacubitril/Valsartan in Advanced Heart Failure With Reduced Ejection Fraction. JACC Heart Fail. 2020, 8, 789–799. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, X.; Li, J.; Xu, M.; Ren, X.; Liu, H.; Xu, L.; Shao, J. Astragaloside IV Reduces OxLDL-Induced BNP Overexpression by Regulating HDAC. J. Healthc. Eng. 2021, 2021, 3433615. [Google Scholar] [CrossRef]
- Morbach, C.; Buck, T.; Rost, C.; Peter, S.; Günther, S.; Störk, S.; Prettin, C.; Erbel, R.; Ertl, G.; Angermann, C.E. Point-of-Care B-Type Natriuretic Peptide and Portable Echocardiography for Assessment of Patients with Suspected Heart Failure in Primary Care: Rationale and Design of the Three-Part Handheld-BNP Program and Results of the Training Study. Clin. Res. Cardiol. 2018, 107, 95–107. [Google Scholar] [CrossRef]
- Hu, J.; Wan, Q.; Zhang, Y.; Zhou, J.; Li, M.; Jiang, L.; Yuan, F. Efficacy and Safety of Early Ultrafiltration in Patients with Acute Decompensated Heart Failure with Volume Overload: A Prospective, Randomized, Controlled Clinical Trial. BMC Cardiovasc. Disord. 2020, 20, 447. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R.; Silva Enciso, J.; Gersh, B.J.; Grady, C.; Rice, M.M.; Singh, S.; Sopko, G.; Boineau, R.; Rosenberg, Y.; Greenberg, B.H. Detection and Management of Geographic Disparities in the TOPCAT Trial. JACC Basic Transl. Sci. 2016, 1, 180–189. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Karlström, P.; Dahlström, U.; Boman, K.; Alehagen, U. Responder to BNP-Guided Treatment in Heart Failure. The Process of Defining a Responder. Scand. Cardiovasc. J. 2015, 49, 316–324. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mueller, C.; Scholer, A.; Laule-Kilian, K.; Martina, B.; Schindler, C.; Buser, P.; Pfisterer, M.; Perruchoud, A.P. Use of B-Type Natriuretic Peptide in the Evaluation and Management of Acute Dyspnea. N. Engl. J. Med. 2004, 350, 647–654. [Google Scholar] [CrossRef]
- Motamed, H.; Forouzan, A.; Heybar, H.; Khorasani, M.J.; Hesam, S. Bronchodilatory Effects of B-Type Natriuretic Peptide in Acute Asthma Attacks: A Randomized Controlled Clinical Trial. Adv. Respir. Med. 2020, 88, 531–538. [Google Scholar] [CrossRef]
- Ogawa, S.; Takeuchi, K.; Ito, S. Plasma BNP Levels in the Treatment of Type 2 Diabetes with Pioglitazone. J. Clin. Endocrinol. Metab. 2003, 88, 3993–3996. [Google Scholar] [CrossRef]
- Stavrakis, S.; Pakala, A.; Thadani, U.; Thomas, J.; Chaudhry, M.A. Obesity, Brain Natriuretic Peptide Levels and Mortality in Patients Hospitalized With Heart Failure and Preserved Left Ventricular Systolic Function. Am. J. Med. Sci. 2013, 345, 211–217. [Google Scholar] [CrossRef]
- Pitt, B.; Kober, L.; Ponikowski, P.; Gheorghiade, M.; Filippatos, G.; Krum, H.; Nowack, C.; Kolkhof, P.; Kim, S.-Y.; Zannad, F. Safety and Tolerability of the Novel Non-Steroidal Mineralocorticoid Receptor Antagonist BAY 94-8862 in Patients with Chronic Heart Failure and Mild or Moderate Chronic Kidney Disease: A Randomized, Double-Blind Trial. Eur. Heart J. 2013, 34, 2453–2463. [Google Scholar] [CrossRef]
- Koomen, J.; Stevens, J.; Bakris, G.; Correa-Rotter, R.; Hou, F.F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.; Parving, H.; et al. Inter-individual Variability in Atrasentan Exposure Partly Explains Variability in Kidney Protection and Fluid Retention Responses: A Post Hoc Analysis of the SONAR Trial. Diabetes Obes. Metab. 2021, 23, 561–568. [Google Scholar] [CrossRef]
- Qin, W.; Bai, W.; Liu, K.; Liu, Y.; Meng, X.; Zhang, K.; Zhang, M. Clinical Course and Risk Factors of Disease Deterioration in Critically Ill Patients with COVID-19. Hum. Gene Ther. 2021, 32, 310–315. [Google Scholar] [CrossRef]
- Inohara, T.; Kim, S.; Pieper, K.; Blanco, R.G.; Allen, L.A.; Fonarow, G.C.; Gersh, B.J.; Ezekowitz, M.D.; Kowey, P.R.; Reiffel, J.A.; et al. B-Type Natriuretic Peptide, Disease Progression and Clinical Outcomes in Atrial Fibrillation. Heart 2018, 105, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.; Hazama, T.; Nakano, K.; Urae, K.; Moriyama, T.; Ariyoshi, T.; Kurokawa, Y.; Kodama, G.; Wada, Y.; Yano, J.; et al. Effects of Reducing L-Carnitine Supplementation on Carnitine Kinetics and Cardiac Function in Hemodialysis Patients: A Multicenter, Single-Blind, Placebo-Controlled, Randomized Clinical Trial. Nutrients 2021, 13, 1900. [Google Scholar] [CrossRef] [PubMed]
- Seki, N.; Matsumoto, T.; Fukazawa, M. Relationship Between the Brain Natriuretic Peptide (BNP) Level and Prognosis of Diabetic Nephropathy with Microalbuminuria: A 7-Year Follow-Up Study. Horm. Metab. Res. 2018, 50, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Lok, D.J.; Klip, I.T.; Voors, A.A.; Lok, S.I.; Bruggink-André de la Porte, P.W.; Hillege, H.L.; Jaarsma, T.; van Veldhuisen, D.J.; van der Meer, P. Prognostic Value of N-Terminal pro C-Type Natriuretic Peptide in Heart Failure Patients with Preserved and Reduced Ejection Fraction. Eur. J. Heart Fail. 2014, 16, 958–966. [Google Scholar] [CrossRef]
- Fudim, M.; Sayeed, S.; Xu, H.; Matsouaka, R.A.; Heidenreich, P.A.; Velazquez, E.J.; Yancy, C.W.; Fonarow, G.C.; Hernandez, A.F.; DeVore, A.D. Representativeness of the PIONEER-HF Clinical Trial Population in Patients Hospitalized With Heart Failure and Reduced Ejection Fraction. Circ. Heart Fail. 2020, 13, e006645. [Google Scholar] [CrossRef]
- Mortensen, S.A.; Rosenfeldt, F.; Kumar, A.; Dolliner, P.; Filipiak, K.J.; Pella, D.; Alehagen, U.; Steurer, G.; Littarru, G.P. The Effect of Coenzyme Q 10 on Morbidity and Mortality in Chronic Heart Failure. JACC Heart Fail. 2014, 2, 641–649. [Google Scholar] [CrossRef]
- Pellicori, P.; Ferreira, J.P.; Mariottoni, B.; Brunner-La Rocca, H.; Ahmed, F.Z.; Verdonschot, J.; Collier, T.; Cuthbert, J.J.; Petutschnigg, J.; Mujaj, B.; et al. Effects of Spironolactone on Serum Markers of Fibrosis in People at High Risk of Developing Heart Failure: Rationale, Design and Baseline Characteristics of a Proof-of-concept, Randomised, Precision-medicine, Prevention Trial. The Heart OMics in AGing (HOMAGE) Trial. Eur. J. Heart Fail. 2020, 22, 1711–1723. [Google Scholar] [CrossRef]
- Pfisterer, M.; Buser, P.; Rickli, H.; Gutmann, M.; Erne, P.; Rickenbacher, P.; Vuillomenet, A.; Jeker, U.; Dubach, P.; Beer, H.; et al. BNP-Guided vs Symptom-Guided Heart Failure Therapy. JAMA 2009, 301, 383. [Google Scholar] [CrossRef]
- Khan, A.; Johnson, D.K.; Carlson, S.; Hocum-Stone, L.; Kelly, R.F.; Gravely, A.A.; Mbai, M.; Green, D.L.; Santilli, S.; Garcia, S.; et al. NT-Pro BNP Predicts Myocardial Injury Post-Vascular Surgery and Is Reduced with CoQ10: A Randomized Double-Blind Trial. Ann. Vasc. Surg. 2020, 64, 292–302. [Google Scholar] [CrossRef]
- Walford, G.A.; Ma, Y.; Christophi, C.A.; Goldberg, R.B.; Jarolim, P.; Horton, E.; Mather, K.J.; Barrett-Connor, E.; Davis, J.; Florez, J.C.; et al. Circulating Natriuretic Peptide Concentrations Reflect Changes in Insulin Sensitivity over Time in the Diabetes Prevention Program. Diabetologia 2014, 57, 935–939. [Google Scholar] [CrossRef]
- Wolsk, E.; Claggett, B.; Pfeffer, M.A.; Diaz, R.; Dickstein, K.; Gerstein, H.C.; Lawson, F.C.; Lewis, E.F.; Maggioni, A.P.; McMurray, J.J.; et al. Role of B-Type Natriuretic Peptide and N-Terminal Prohormone BNP as Predictors of Cardiovascular Morbidity and Mortality in Patients With a Recent Coronary Event and Type 2 Diabetes Mellitus. J. Am. Heart Assoc. 2017, 6, e004743. [Google Scholar] [CrossRef] [PubMed]
- Buckley, L.F.; Canada, J.M.; del Buono, M.G.; Carbone, S.; Trankle, C.R.; Billingsley, H.; Kadariya, D.; Arena, R.; van Tassell, B.W.; Abbate, A. Low NT-ProBNP Levels in Overweight and Obese Patients Do Not Rule out a Diagnosis of Heart Failure with Preserved Ejection Fraction. ESC Heart Fail. 2018, 5, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Fontes-Carvalho, R.; Sampaio, F.; Teixeira, M.; Gama, V.; Leite-Moreira, A.F. The Role of a Structured Exercise Training Program on Cardiac Structure and Function after Acute Myocardial Infarction: Study Protocol for a Randomized Controlled Trial. Trials 2015, 16, 90. [Google Scholar] [CrossRef]
- Hillock, R.J.; Frampton, C.M.; Yandle, T.G.; Troughton, R.W.; Lainchbury, J.G.; Richards, A.M. B-Type Natriuretic Peptide Infusions in Acute Myocardial Infarction. Heart 2008, 94, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Hubers, S.A.; Schirger, J.A.; Sangaralingham, S.J.; Chen, Y.; Burnett, J.C., Jr.; Hodge, D.; Chen, H.H. B-Type Natriuretic Peptide and Cardiac Remodelling after Myocardial Infarction: A Randomised Trial. Heart 2021, 107, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Kalra, P.R.; Clague, J.R.; Bolger, A.P.; Anker, S.D.; Poole-Wilson, P.A.; Struthers, A.D.; Coats, A.J. Myocardial Production of C-Type Natriuretic Peptide in Chronic Heart Failure. Circulation 2003, 107, 571–573. [Google Scholar] [CrossRef]
- Yuan, X.; Zhiya, D.; Wenli, L.; Xiuming, W.; Wenxin, S.; Defen, W.; Jihong, N.; Fengsheng, C.; Junqi, W.; Wei, W. Measurement of Amino-Terminal Propeptide of C-Type Natriuretic Peptide in Patients with Idiopathic Short Stature or Isolated Growth Hormone Deficiency. J. Pediatr. Endocrinol. Metab. 2011, 24, 989–994. [Google Scholar] [CrossRef]
- Kawakami, R.; Lee, C.Y.W.; Scott, C.; Bailey, K.R.; Schirger, J.A.; Chen, H.H.; Benike, S.L.; Cannone, V.; Martin, F.L.; Sangaralingham, S.J.; et al. A Human Study to Evaluate Safety, Tolerability, and Cyclic GMP Activating Properties of Cenderitide in Subjects With Stable Chronic Heart Failure. Clin. Pharmacol. Ther. 2018, 104, 546–552. [Google Scholar] [CrossRef]
- Igaki, T.; Itoh, H.; Suga, S.; Hama, N.; Ogawa, Y.; Komatsu, Y.; Mukoyama, M.; Sugawara, A.; Yoshimasa, T.; Tanaka, I.; et al. C-Type Natriuretic Peptide in Chronic Renal Failure and Its Action in Humans. Kidney Int. Suppl. 1996, 55, S144–S147. [Google Scholar]
- Barletta, G.; Lazzeri, C.; Vecchiarino, S.; del Bene, R.; Messeri, G.; dello Sbarba, A.; Mannelli, M.; la Villa, G. Low-Dose C-Type Natriuretic Peptide Does Not Affect Cardiac and Renal Function in Humans. Hypertension 1998, 31, 802–808. [Google Scholar] [CrossRef][Green Version]
- Savarirayan, R.; Irving, M.; Bacino, C.A.; Bostwick, B.; Charrow, J.; Cormier-Daire, V.; le Quan Sang, K.-H.; Dickson, P.; Harmatz, P.; Phillips, J.; et al. C-Type Natriuretic Peptide Analogue Therapy in Children with Achondroplasia. N. Engl. J. Med. 2019, 381, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Savarirayan, R.; Tofts, L.; Irving, M.; Wilcox, W.R.; Bacino, C.A.; Hoover-Fong, J.; Font, R.U.; Harmatz, P.; Rutsch, F.; Bober, M.B.; et al. Safe and Persistent Growth-Promoting Effects of Vosoritide in Children with Achondroplasia: 2-Year Results from an Open-Label, Phase 3 Extension Study. Genet. Med. 2021, 23, 2443–2447. [Google Scholar] [CrossRef] [PubMed]
- Kellner, M.; Yassouridis, A.; Hua, Y.; Wendrich, M.; Jahn, H.; Wiedemann, K. Intravenous C-Type Natriuretic Peptide Augments Behavioral and Endocrine Effects of Cholecystokinin Tetrapeptide in Healthy Men. J. Psychiatr. Res. 2002, 36, 1–6. [Google Scholar] [CrossRef]
- Potter, L.R.; Yoder, A.R.; Flora, D.R.; Antos, L.K.; Dickey, D.M. Natriuretic Peptides: Their Structures, Receptors, Physiologic Functions and Therapeutic Applications. In cGMP: Generators, Effectors and Therapeutic Implications; Springer: Berlin/Heidelberg, Germany, 2009; pp. 341–366. [Google Scholar]
- Sudoh, T.; Minamino, N.; Kangawa, K.; Matsuo, H. C-Type Natriuretic Peptide (CNP): A New Member of Natriuretic Peptide Family Identified in Porcine Brain. Biochem. Biophys. Res. Commun. 1990, 168, 863–870. [Google Scholar] [CrossRef]
- Moyes, A.J.; Hobbs, A.J. C-Type Natriuretic Peptide: A Multifaceted Paracrine Regulator in the Heart and Vasculature. Int. J. Mol. Sci. 2019, 20, 2281. [Google Scholar] [CrossRef]
- Fujii, T.; Komatsu, Y.; Yasoda, A.; Kondo, E.; Yoshioka, T.; Nambu, T.; Kanamoto, N.; Miura, M.; Tamura, N.; Arai, H.; et al. Circulating C-Type Natriuretic Peptide (CNP) Rescues Chondrodysplastic CNP Knockout Mice from Their Impaired Skeletal Growth and Early Death. Endocrinology 2010, 151, 4381–4388. [Google Scholar] [CrossRef]
- Nakao, K.; Osawa, K.; Yasoda, A.; Yamanaka, S.; Fujii, T.; Kondo, E.; Koyama, N.; Kanamoto, N.; Miura, M.; Kuwahara, K.; et al. The Local CNP/GC-B System in Growth Plate Is Responsible for Physiological Endochondral Bone Growth. Sci. Rep. 2015, 5, 10554. [Google Scholar] [CrossRef]
- Breinholt, V.M.; Rasmussen, C.E.; Mygind, P.H.; Kjelgaard-Hansen, M.; Faltinger, F.; Bernhard, A.; Zettler, J.; Hersel, U. TransCon CNP, a Sustained-Release C-Type Natriuretic Peptide Prodrug, a Potentially Safe and Efficacious New Therapeutic Modality for the Treatment of Comorbidities Associated with Fibroblast Growth Factor Receptor 3–Related Skeletal Dysplasias. J. Pharmacol. Exp. Ther. 2019, 370, 459–471. [Google Scholar] [CrossRef]
- Yamashita, T.; Fujii, T.; Yamauchi, I.; Ueda, Y.; Hirota, K.; Kanai, Y.; Yasoda, A.; Inagaki, N. C-Type Natriuretic Peptide Restores Growth Impairment Under Enzyme Replacement in Mice With Mucopolysaccharidosis VII. Endocrinology 2020, 161, bqaa008. [Google Scholar] [CrossRef]
- Chen, W.X.; Liu, H.H.; Li, R.X.; Mammadov, G.; Wang, J.J.; Liu, F.F.; Samadli, S.; Wu, Y.F.; Zhang, D.D.; Luo, H.H.; et al. C-Type Natriuretic Peptide Stimulates Osteoblastic Proliferation and Collagen-X Expression but Suppresses Fibroblast Growth Factor-23 Expression in Vitro. Pediatr. Rheumatol. 2020, 18, 46. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.Y.Y.; Blaser, M.C.; Mirzaei, Z.; Zhong, X.; Simmons, C.A. Inhibition of Pathological Differentiation of Valvular Interstitial Cells by C-Type Natriuretic Peptide. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1881–1889. [Google Scholar] [CrossRef][Green Version]
- Holliday, L.S.; Dean, A.D.; Greenwald, J.E.; Gluck, S.L. C-Type Natriuretic Peptide Increases Bone Resorption in 1,25-Dihydroxyvitamin D3-Stimulated Mouse Bone Marrow Cultures. J. Biol. Chem. 1995, 270, 18983–18989. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, H.; Inoue, A.; Yamaguchi, A.; Yokose, S.; Furuya, M.; Tanaka, S.; Hirose, S. CGMP Produced in Response to ANP and CNP Regulates Proliferation and Differentiation of Osteoblastic Cells. Am. J. Physiol.-Cell Physiol. 1996, 270, C1311–C1318. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.C.; Agoston, H.; Beier, F. Nitric Oxide, C-Type Natriuretic Peptide and CGMP as Regulators of Endochondral Ossification. Dev. Biol. 2008, 319, 171–178. [Google Scholar] [CrossRef]
- Armour, K.E.; Armour, K.J.; Gallagher, M.E.; Gödecke, A.; Helfrich, M.H.; Reid, D.M.; Ralston, S.H. Defective Bone Formation and Anabolic Response to Exogenous Estrogen in Mice with Targeted Disruption of Endothelial Nitric Oxide Synthase**This Study Was Supported by Grants from the Arthritis Research Campaign (UK) and the Medical Research Council (UK). Endocrinology 2001, 142, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, J.; Buttery, L.; O’Shaughnessy, M.; Afzal, F.; Fernandez de Marticorena, I.; Hukkanen, M.; Huang, P.; MacIntyre, I.; Polak, J. Endothelial Nitric Oxide Synthase Gene-Deficient Mice Demonstrate Marked Retardation in Postnatal Bone Formation, Reduced Bone Volume, and Defects in Osteoblast Maturation and Activity. Am. J. Pathol. 2001, 158, 247–257. [Google Scholar] [CrossRef]
- van’T Hof, R.J.; Ralston, S.H. Nitric Oxide and Bone. Immunology 2001, 103, 255–261. [Google Scholar] [CrossRef]
- Mancini, L.; Moradi-Bidhendi, N.; Becherini, L.; Martineti, V.; MacIntyre, I. The Biphasic Effects of Nitric Oxide in Primary Rat Osteoblasts Are CGMP Dependent. Biochem. Biophys. Res. Commun. 2000, 274, 477–481. [Google Scholar] [CrossRef]
- Kiemer, A.K.; Vollmar, A.M. Effects of Different Natriuretic Peptides on Nitric Oxide Synthesis in Macrophages 1. Endocrinology 1997, 138, 4282–4290. [Google Scholar] [CrossRef][Green Version]
- Hoshino, M.; Kaneko, K.; Miyamoto, Y.; Yoshimura, K.; Suzuki, D.; Akaike, T.; Sawa, T.; Ida, T.; Fujii, S.; Ihara, H.; et al. 8-Nitro-CGMP Promotes Bone Growth through Expansion of Growth Plate Cartilage. Free Radic. Biol. Med. 2017, 110, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Meyerhoff, M.E.; Howland, J.L. Methods in Nitric Oxide Research. Biochem. Educ. 1997, 25, 184–185. [Google Scholar] [CrossRef]
- Kiemer, A.K.; Angelika, M. Vollmar Induction of L-Arginine Transport Is Inhibited by Atrial Natriuretic Peptide: A Peptide Hormone as a Novel Regulator of Inducible Nitric-Oxide Synthase Substrate Availability. Mol. Pharmacol. 2001, 60, 421–426. [Google Scholar] [PubMed]
- Peake, N.J.; Hobbs, A.J.; Pingguan-Murphy, B.; Salter, D.M.; Berenbaum, F.; Chowdhury, T.T. Role of C-Type Natriuretic Peptide Signalling in Maintaining Cartilage and Bone Function. Osteoarthr. Cartil. 2014, 22, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.R.; Hunter, T. Activation of Protein Kinase C Stimulates the Dephosphorylation of Natriuretic Peptide Receptor-B at a Single Serine Residue. J. Biol. Chem. 2000, 275, 31099–31106. [Google Scholar] [CrossRef]
- Abbey, S.E.; Potter, L.R. Lysophosphatidic Acid Inhibits C-Type Natriuretic Peptide Activation of Guanylyl Cyclase-B. Endocrinology 2003, 144, 240–246. [Google Scholar] [CrossRef]
- Potter, L.R. Guanylyl Cyclase Structure, Function and Regulation. Cell. Signal. 2011, 23, 1921–1926. [Google Scholar] [CrossRef]
- Schulz, S.; Singh, S.; Bellet, R.A.; Singh, G.; Tubb, D.J.; Chin, H.; Garbers, D.L. The Primary Structure of a Plasma Membrane Guanylate Cyclase Demonstrates Diversity within This New Receptor Family. Cell 1989, 58, 1155–1162. [Google Scholar] [CrossRef]
- Antos, L.K.; Abbey-Hosch, S.E.; Flora, D.R.; Potter, L.R. ATP-Independent Activation of Natriuretic Peptide Receptors. J. Biol. Chem. 2005, 280, 26928–26932. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. Fibroblast Growth Factors. Genome Biol. 2001, 2, reviews3005.1. [Google Scholar] [CrossRef]
- Wilke, T.A.; Gubbels, S.; Schwartz, J.; Richman, J.M. Expression of Fibroblast Growth Factor Receptors (FGFR1, FGFR2, FGFR3) in the Developing Head and Face. Dev. Dyn. 1997, 210, 41–52. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Legeai-Mallet, L. Achondroplasia: Development, Pathogenesis, and Therapy. Dev. Dyn. 2017, 246, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Göke, M.; Tsunekawa, S.; Podolsky, D.K. Signal Transduction Pathway of Human Fibroblast Growth Factor Receptor 3. J. Biol. Chem. 1997, 272, 6621–6628. [Google Scholar] [CrossRef]
- Su, N.; Sun, Q.; Li, C.; Lu, X.; Qi, H.; Chen, S.; Yang, J.; Du, X.; Zhao, L.; He, Q.; et al. Gain-of-Function Mutation in FGFR3 in Mice Leads to Decreased Bone Mass by Affecting Both Osteoblastogenesis and Osteoclastogenesis. Hum. Mol. Genet. 2010, 19, 1199–1210. [Google Scholar] [CrossRef]
- Ozasa, A.; Komatsu, Y.; Yasoda, A.; Miura, M.; Sakuma, Y.; Nakatsuru, Y.; Arai, H.; Itoh, N.; Nakao, K. Complementary Antagonistic Actions between C-Type Natriuretic Peptide and the MAPK Pathway through FGFR-3 in ATDC5 Cells. Bone 2005, 36, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Krejci, P.; Masri, B.; Fontaine, V.; Mekikian, P.B.; Weis, M.; Prats, H.; Wilcox, W.R. Interaction of Fibroblast Growth Factor and C-Natriuretic Peptide Signaling in Regulation of Chondrocyte Proliferation and Extracellular Matrix Homeostasis. J. Cell Sci. 2005, 118, 5089–5100. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, D.; Wu, Y.; Luo, H.; Jiang, G.; Xu, Y.; Wu, Y.; Xia, X.; Wei, W.; Hu, B.; et al. Fibroblast Growth Factor-23 May Serve as a Novel Biomarker for Renal Osteodystrophy Progression. Int. J. Mol. Med. 2018, 43, 535–546. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, J.; Chen, F. The Effects of Elevated Fibroblast Growth Factor 23 on Mandibular Growth in Rats. Arch. Oral Biol. 2018, 95, 156–164. [Google Scholar] [CrossRef]
- Zheng, Q.; Zhou, G.; Morello, R.; Chen, Y.; Garcia-Rojas, X.; Lee, B. Type X Collagen Gene Regulation by Runx2 Contributes Directly to Its Hypertrophic Chondrocyte–Specific Expression in Vivo. J. Cell Biol. 2003, 162, 833–842. [Google Scholar] [CrossRef]
- Tsuji, T.; Kunieda, T. A Loss-of-Function Mutation in Natriuretic Peptide Receptor 2 (Npr2) Gene Is Responsible for Disproportionate Dwarfism in Cn/Cn Mouse. J. Biol. Chem. 2005, 280, 14288–14292. [Google Scholar] [CrossRef]
- Pejchalova, K.; Krejci, P.; Wilcox, W.R. C-Natriuretic Peptide: An Important Regulator of Cartilage. Mol. Genet. Metab. 2007, 92, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Zeb, A.; Shinwari, N.; Shah, K.; Gilani, S.Z.T.; Khan, S.; Lee, K.W.; Raza, S.I.; Hussain, S.; Liaqat, K.; Ahmad, W.; et al. Molecular and in Silico Analyses Validates Pathogenicity of Homozygous Mutations in the NPR2 Gene Underlying Variable Phenotypes of Acromesomelic Dysplasia, Type Maroteaux. Int. J. Biochem. Cell Biol. 2018, 102, 76–86. [Google Scholar] [CrossRef]
- Kenis, V.; Melchenko, E.; Mazunin, I.; Pekkinen, M.; Mäkitie, O. A New Family with Epiphyseal Chondrodysplasia Type Miura. Am. J. Med. Genet. Part A 2021, 185, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Kawai, M.; Yamazaki, M.; Tachikawa, K.; Kubota, T.; Ozono, K.; Michigami, T. CREB Activation in Hypertrophic Chondrocytes Is Involved in the Skeletal Overgrowth in Epiphyseal Chondrodysplasia Miura Type Caused by Activating Mutations of Natriuretic Peptide Receptor B. Hum. Mol. Genet. 2019, 28, 1183–1198. [Google Scholar] [CrossRef] [PubMed]
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.-S.; Mirams, M. Endochondral Ossification: How Cartilage Is Converted into Bone in the Developing Skeleton. Int. J. Biochem. Cell Biol. 2008, 40, 46–62. [Google Scholar] [CrossRef]
- Akiyama, H.; Chaboissier, M.-C.; Martin, J.F.; Schedl, A.; de Crombrugghe, B. The Transcription Factor Sox9 Has Essential Roles in Successive Steps of the Chondrocyte Differentiation Pathway and Is Required for Expression of Sox5 and Sox6. Genes Dev. 2002, 16, 2813–2828. [Google Scholar] [CrossRef]
- Yoshida, C.A.; Komori, T. Role of Runx Proteins in Chondrogenesis. Crit. Rev. ™ Eukaryot. Gene Expr. 2005, 15, 243–254. [Google Scholar] [CrossRef]
- Fu, M.; Wang, C.; Li, Z.; Sakamaki, T.; Pestell, R.G. Minireview: Cyclin D1: Normal and Abnormal Functions. Endocrinology 2004, 145, 5439–5447. [Google Scholar] [CrossRef]
- Beier, F.; Ali, Z.; Mok, D.; Taylor, A.C.; Leask, T.; Albanese, C.; Pestell, R.G.; LuValle, P. TGFβ and PTHrP Control Chondrocyte Proliferation by Activating Cyclin D1 Expression. Mol. Biol. Cell 2001, 12, 3852–3863. [Google Scholar] [CrossRef]
- L’Hôte, C.G.M.; Knowles, M.A. Cell Responses to FGFR3 Signalling: Growth, Differentiation and Apoptosis. Exp. Cell Res. 2005, 304, 417–431. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Marie, P.J. Fibroblast Growth Factor Signaling in Skeletal Development and Disease. Genes Dev. 2015, 29, 1463–1486. [Google Scholar] [CrossRef] [PubMed]
- Kuehnl, A.; Pelisek, J.; Pongratz, J.; Eckstein, H.-H. C-Type Natriuretic Peptide and Its Receptors in Atherosclerotic Plaques of the Carotid Artery of Clinically Asymptomatic Patients. Eur. J. Vasc. Endovasc. Surg. 2012, 43, 649–654. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miyazawa, T.; Ogawa, Y.; Chusho, H.; Yasoda, A.; Tamura, N.; Komatsu, Y.; Pfeifer, A.; Hofmann, F.; Nakao, K. Cyclic GMP-Dependent Protein Kinase II Plays a Critical Role in C-Type Natriuretic Peptide-Mediated Endochondral Ossification. Endocrinology 2002, 143, 3604–3610. [Google Scholar] [CrossRef][Green Version]
- Kake, T.; Kitamura, H.; Adachi, Y.; Yoshioka, T.; Watanabe, T.; Matsushita, H.; Fujii, T.; Kondo, E.; Tachibe, T.; Kawase, Y.; et al. Chronically Elevated Plasma C-Type Natriuretic Peptide Level Stimulates Skeletal Growth in Transgenic Mice. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, E1339–E1348. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, P.; Thomas, G.; Sellin, K.; Bessette, M.-C.; Lafrenière, F.; Akhouayri, O.; St-Arnaud, R.; Lanctôt, C. Osteocrin Is a Specific Ligand of the Natriuretic Peptide Clearance Receptor That Modulates Bone Growth. J. Biol. Chem. 2007, 282, 36454–36462. [Google Scholar] [CrossRef]
- Agoston, H.; Khan, S.; James, C.G.; Gillespie, J.R.; Serra, R.; Stanton, L.-A.; Beier, F. C-Type Natriuretic Peptide Regulates Endochondral Bone Growth through P38 MAP Kinase-Dependent and—Independent Pathways. BMC Dev. Biol. 2007, 7, 18. [Google Scholar] [CrossRef]
- Woods, A.; Khan, S.; Beier, F. C-Type Natriuretic Peptide Regulates Cellular Condensation and Glycosaminoglycan Synthesis during Chondrogenesis. Endocrinology 2007, 148, 5030–5041. [Google Scholar] [CrossRef]
- Inoue, A.; Hayakawa, T.; Otsuka, E.; Kamiya, A.; Suzuki, Y.; Hirose, S.; Hagiwara, H. Correlation between Induction of Expression of Biglycan and Mineralization by C-Type Natriuretic Peptide in Osteoblastic Cells. J. Biochem. 1999, 125, 103–108. [Google Scholar] [CrossRef]
- Yeh, L.-C.C.; Adamo, M.L.; Olson, M.S.; Lee, J.C. Osteogenic Protein-1 and Insulin-Like Growth Factor I Synergistically Stimulate Rat Osteoblastic Cell Differentiation and Proliferation 1. Endocrinology 1997, 138, 4181–4190. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, M.; Mundy, G.R. Bone Morphogenetic Proteins. Growth Factors 2004, 22, 233–241. [Google Scholar] [CrossRef]
- Yeh, L.-C.C.; Zavala, M.C.; Lee, J.C. C-Type Natriuretic Peptide Enhances Osteogenic Protein-1-Induced Osteoblastic Cell Differentiation via Smad5 Phosphorylation. J. Cell. Biochem. 2006, 97, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Chusho, H.; Tamura, N.; Ogawa, Y.; Yasoda, A.; Suda, M.; Miyazawa, T.; Nakamura, K.; Nakao, K.; Kurihara, T.; Komatsu, Y.; et al. Dwarfism and Early Death in Mice Lacking C-Type Natriuretic Peptide. Proc. Natl. Acad. Sci. USA 2001, 98, 4016–4021. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Hirota, K.; Yasoda, A.; Takizawa, A.; Morozumi, N.; Nakamura, R.; Yotsumoto, T.; Kondo, E.; Yamashita, Y.; Sakane, Y.; et al. Rats Deficient C-Type Natriuretic Peptide Suffer from Impaired Skeletal Growth without Early Death. PLoS ONE 2018, 13, e0194812. [Google Scholar] [CrossRef] [PubMed]
- Trotter, T.L.; Hall, J.G. Health Supervision for Children With Achondroplasia. Pediatrics 2005, 116, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Yasoda, A.; Komatsu, Y.; Chusho, H.; Miyazawa, T.; Ozasa, A.; Miura, M.; Kurihara, T.; Rogi, T.; Tanaka, S.; Suda, M.; et al. Overexpression of CNP in Chondrocytes Rescues Achondroplasia through a MAPK-Dependent Pathway. Nat. Med. 2004, 10, 80–86. [Google Scholar] [CrossRef]
- Yamanaka, S.; Nakao, K.; Koyama, N.; Isobe, Y.; Ueda, Y.; Kanai, Y.; Kondo, E.; Fujii, T.; Miura, M.; Yasoda, A.; et al. Circulatory CNP Rescues Craniofacial Hypoplasia in Achondroplasia. J. Dent. Res. 2017, 96, 1526–1534. [Google Scholar] [CrossRef]
- Ueda, Y.; Yasoda, A.; Hirota, K.; Yamauchi, I.; Yamashita, T.; Kanai, Y.; Sakane, Y.; Fujii, T.; Inagaki, N. Exogenous C-Type Natriuretic Peptide Therapy for Impaired Skeletal Growth in a Murine Model of Glucocorticoid Treatment. Sci. Rep. 2019, 9, 8547. [Google Scholar] [CrossRef]
- Jackson, D.J.; Bacharier, L.B.; Mauger, D.T.; Boehmer, S.; Beigelman, A.; Chmiel, J.F.; Fitzpatrick, A.M.; Gaffin, J.M.; Morgan, W.J.; Peters, S.P.; et al. Quintupling Inhaled Glucocorticoids to Prevent Childhood Asthma Exacerbations. N. Engl. J. Med. 2018, 378, 891–901. [Google Scholar] [CrossRef]
- Rivkees, S.A.; Danon, M.; Herrin, J. Prednisone Dose Limitation of Growth Hormone Treatment of Steroid-Induced Growth Failure. J. Pediatr. 1994, 125, 322–325. [Google Scholar] [CrossRef]
- Hirota, K.; Furuya, M.; Morozumi, N.; Yoshikiyo, K.; Yotsumoto, T.; Jindo, T.; Nakamura, R.; Murakami, K.; Ueda, Y.; Hanada, T.; et al. Exogenous C-Type Natriuretic Peptide Restores Normal Growth and Prevents Early Growth Plate Closure in Its Deficient Rats. PLoS ONE 2018, 13, e0204172. [Google Scholar] [CrossRef]
- Ueda, Y.; Hirota, K.; Yamauchi, I.; Hakata, T.; Yamashita, T.; Fujii, T.; Yasoda, A.; Inagaki, N. Is C-Type Natriuretic Peptide Regulated by a Feedback Loop? A Study on Systemic and Local Autoregulatory Effect. PLoS ONE 2020, 15, e0240023. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.C.; & Espiner, E.A. Circulating products of C-type natriuretic peptide and links with organ function in health and disease. Peptides 2020, 132, 170363. [Google Scholar] [CrossRef] [PubMed]
- Espiner, E.; Prickett, T.; Olney, R. Plasma C-Type Natriuretic Peptide: Emerging Applications in Disorders of Skeletal Growth. Horm. Res. Paediatr. 2018, 90, 345–357. [Google Scholar] [CrossRef]
- Olney, R.C.; Permuy, J.W.; Prickett, T.C.; Han, J.C.; Espiner, E.A. Amino-terminal propeptide of C-type natriuretic peptide (NTproCNP) predicts height velocity in healthy children. Clin. Endocrinol. 2012, 77, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Namba, N.; Fujiwara, M.; Ohata, Y.; Ishida, H.; Kitaoka, T.; Kubota, T.; Hirai, H.; Higuchi, C.; Tsumaki, N.; et al. An overgrowth disorder associated with excessive production of cGMP due to a gain-of-function mutation of the natriuretic peptide receptor 2 gene. PLoS ONE 2012, 7, e42180. [Google Scholar] [CrossRef] [PubMed]
- Hannema, S.E.; van Duyvenvoorde, H.A.; Premsler, T.; Yang, R.B.; Mueller, T.D.; Gassner, B.; Oberwinkler, H.; Roelfsema, F.; Santen, G.W.; Prickett, T.; et al. An activating mutation in the kinase homology domain of the natriuretic peptide receptor-2 causes extremely tall stature without skeletal deformities. J. Clin. Endocrinol. Metab. 2013, 98, E1988-98. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, M.R. BDNF Alters ERK/p38 MAPK Activity Ratios to Promote Differentiation in Growth Plate Chondrocytes. Mol. Endocrinol. 2012, 26, 1406–1416. [Google Scholar] [CrossRef]
- Mericq, V.; Uyeda, J.A.; Barnes, K.M.; de Luca, F.; Baron, J. Regulation of Fetal Rat Bone Growth by C-Type Natriuretic Peptide and CGMP. Pediatr. Res. 2000, 47, 189. [Google Scholar] [CrossRef]



{kind=link}
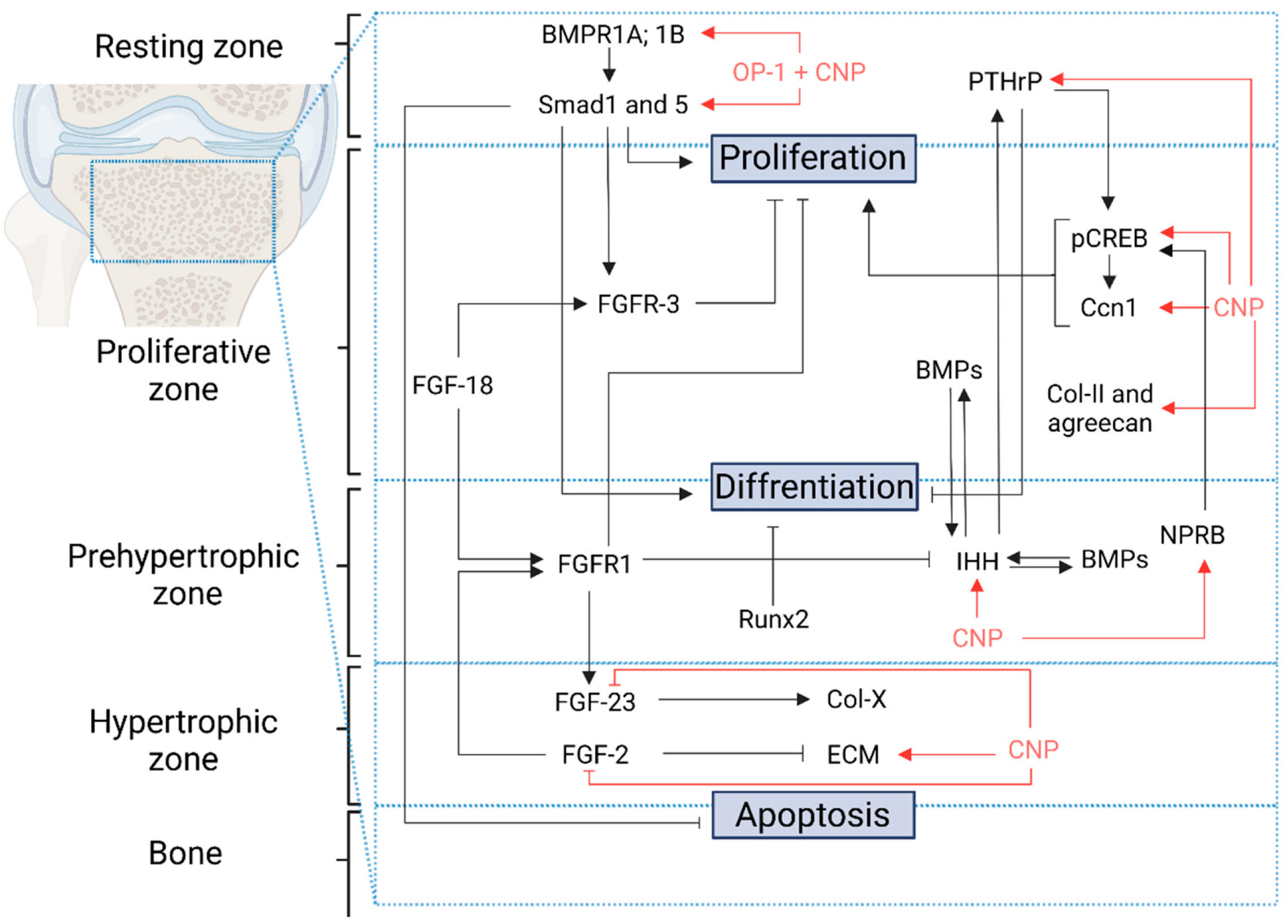{kind=link}
| NTPs | NTPs Used for Diagnosis/Evaluation | NTPs Tested for Diseases as a Treatment | |
|---|---|---|---|
| Final Peptide | Pro-form of the Peptide | ||
| ANP | Heart failure [7,8,9,10] Cancer [11] Type-2 diabetes [12] Chronic renal failure [13] | Heart failure [10,14] Insulin resistance [15,16] Obesity [17] | Heart failure [18,19,20,21,22] Cardioprotective effects during surgery [23,24,25,26,27,28,29,30] Renal failure [31,32,33,34,35,36] Colorectal cancer [37] Forearm vasculature [37] Acute kidney injury [38] Lipid mobilization [39] Pulmonary vasorelaxant [40] |
| BNP | Heart failure [10,41,42,43,44,45,46,47,48,49,50,51,52,53] Acute dyspnea [54] Asthma [55] Type 2 diabetes [56] Cancer [11] Chronic renal failure [13] Obesity [17,57] Chronic kidney disease [58,59] SARS-CoV-2 [60] Atrial fibrillation [61] Hemodialisys patients [62] Diabetic nephropathy [63] | Heart failure [48,64,65,66,67,68,69] Type 2 diabetes [70,71] Obesity [17,72] Chronic kidney disease [58] Cardiac function [73] Stroke [73] | Forearm vasculature [37] Pulmonary vasorelaxant [40] Myocardial infarction [74,75] |
| CNP | Heart failure [47,76] | Heart failure [64] Growth failure [77] | Heart failure (CNP fused to DNP) [78] Renal failure [79,80] Achondroplasia [81,82] Anxiety [83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rintz, E.; Węgrzyn, G.; Fujii, T.; Tomatsu, S. Molecular Mechanism of Induction of Bone Growth by the C-Type Natriuretic Peptide. Int. J. Mol. Sci. 2022, 23, 5916. https://doi.org/10.3390/ijms23115916
Rintz E, Węgrzyn G, Fujii T, Tomatsu S. Molecular Mechanism of Induction of Bone Growth by the C-Type Natriuretic Peptide. International Journal of Molecular Sciences. 2022; 23(11):5916. https://doi.org/10.3390/ijms23115916
Chicago/Turabian StyleRintz, Estera, Grzegorz Węgrzyn, Toshihito Fujii, and Shunji Tomatsu. 2022. "Molecular Mechanism of Induction of Bone Growth by the C-Type Natriuretic Peptide" International Journal of Molecular Sciences 23, no. 11: 5916. https://doi.org/10.3390/ijms23115916
APA StyleRintz, E., Węgrzyn, G., Fujii, T., & Tomatsu, S. (2022). Molecular Mechanism of Induction of Bone Growth by the C-Type Natriuretic Peptide. International Journal of Molecular Sciences, 23(11), 5916. https://doi.org/10.3390/ijms23115916