
A Novel Rodent Model of Hypertensive Cerebral Small Vessel Disease with White Matter Hyperintensities and Peripheral Oxidative Stress

 , ,
, , 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
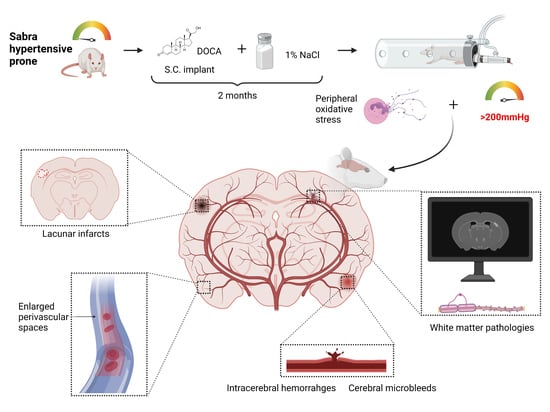
1. Introduction
2. Results
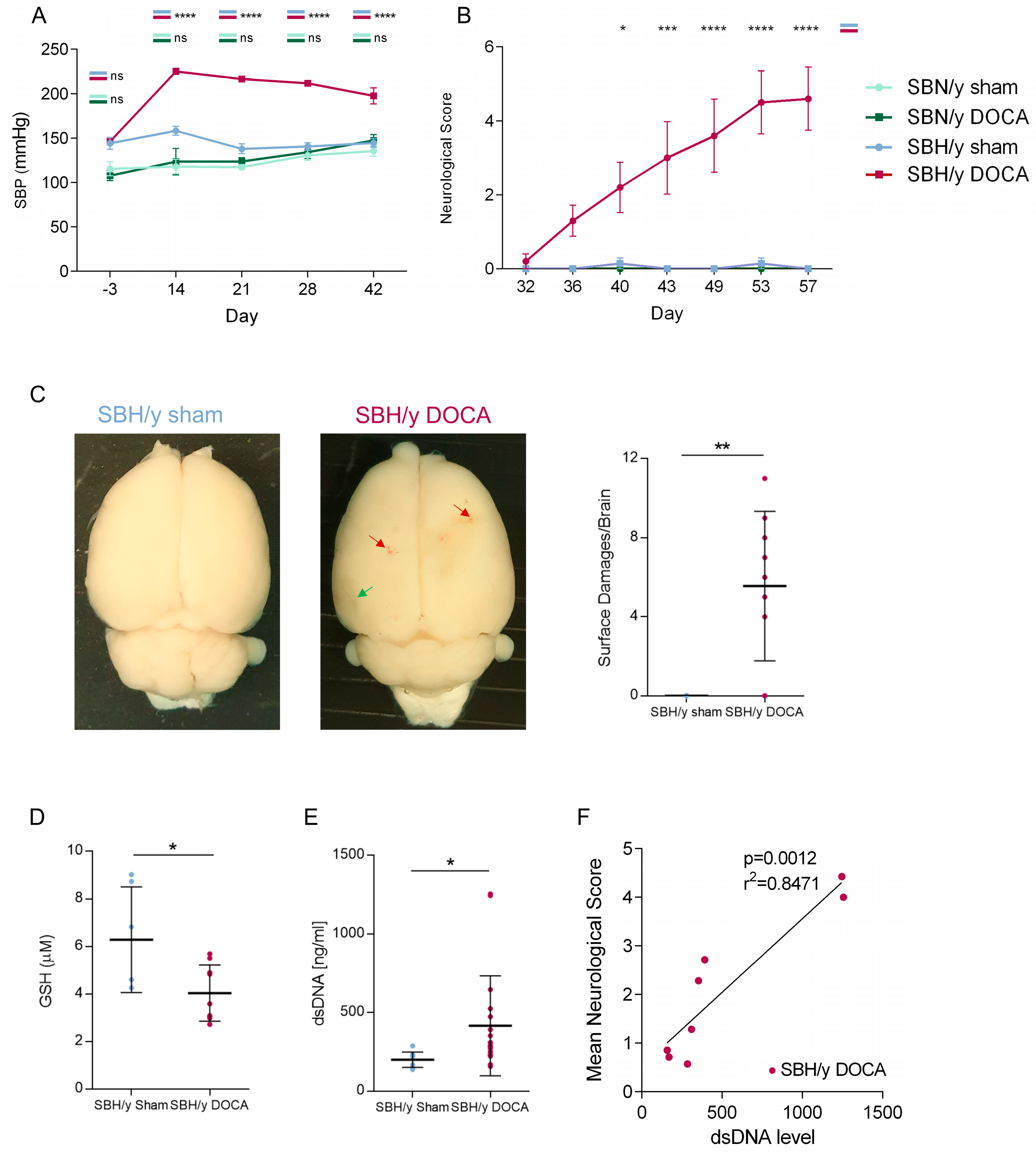
2.1. SBH/y Rats Develop High SBP and Neurological Deficits Following DOCA Administration
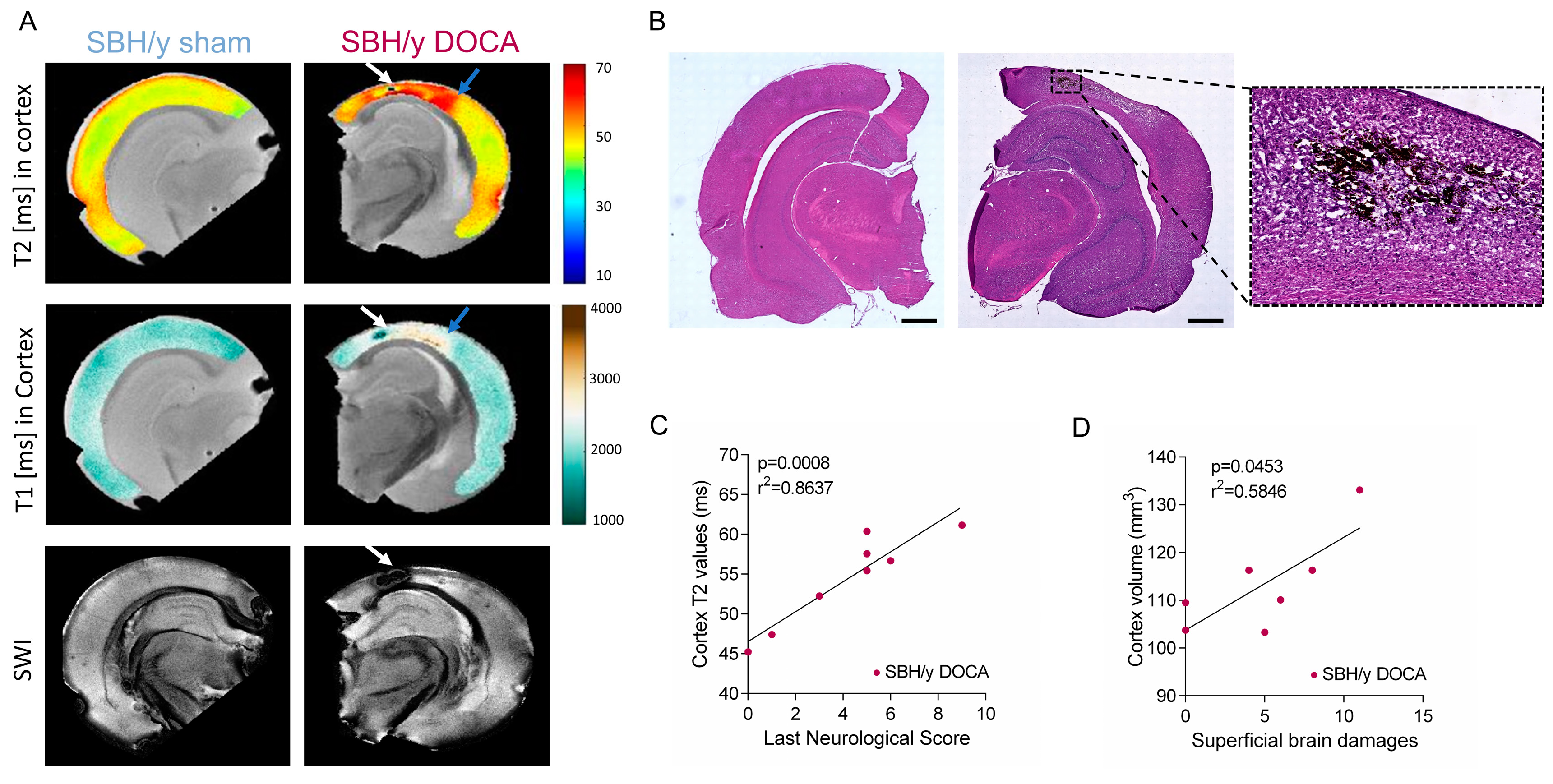
2.2. SBH/y Rats on MRI
2.3. Pathological Assessment of SBH/y-DOCA Brains
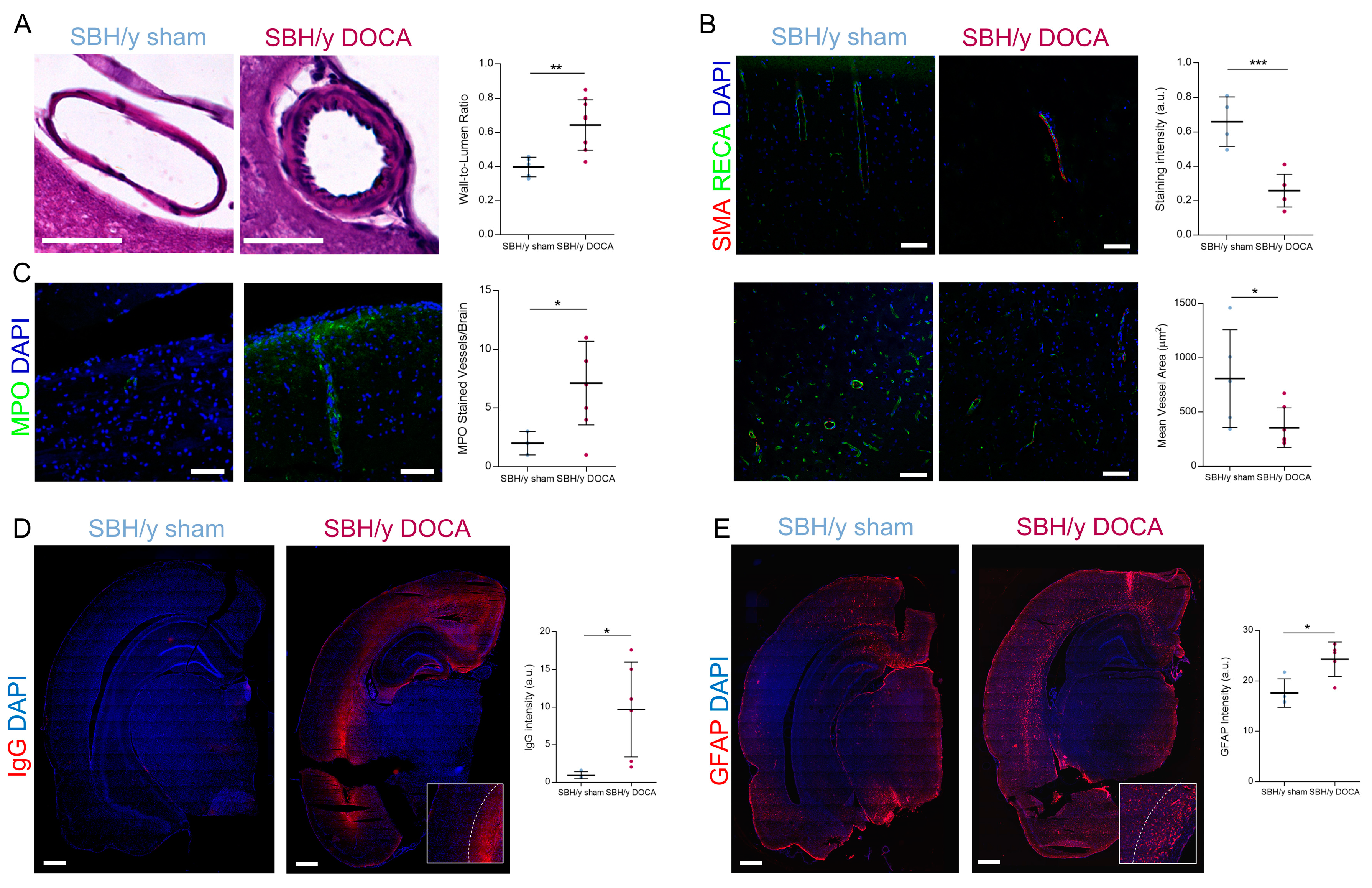
2.4. Blood Vessel Pathology in SBH/y-DOCA Rats
2.5. BBB Permeability and White Matter Inflammation
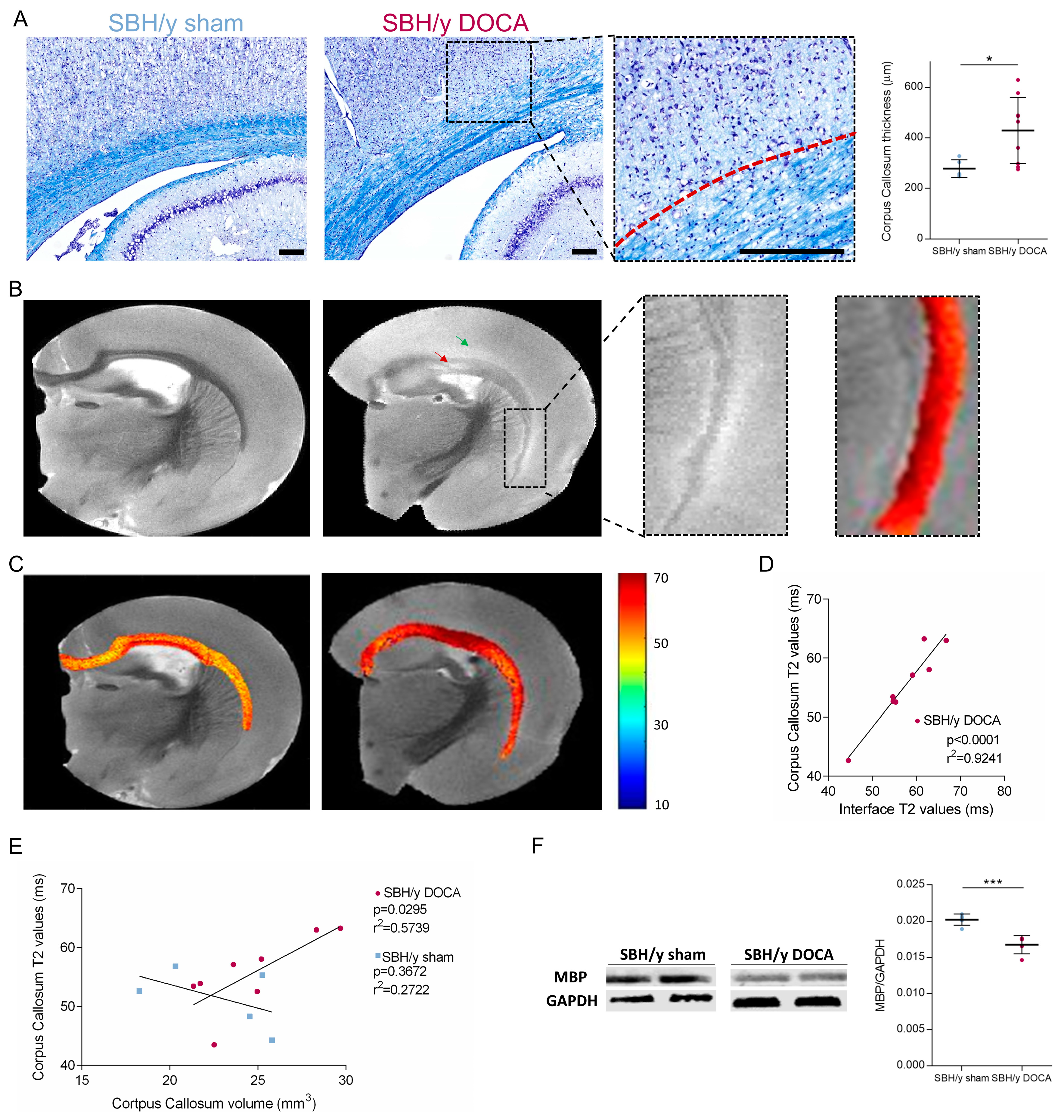
2.6. White Matter Pathology in SBH/y-DOCA Brains
2.7. Cognitive Decline in SBH/y DOCA Rats
3. Discussion
4. Materials and Methods
4.1. Animal Housing
4.2. Experimental Design and Induction of Hypertension
4.3. Systolic Blood Pressure Measurements
4.4. Neurological Deficits Score
4.5. Behavioral Examinations
4.6. Novel Object Recognition (NOR) Test
4.7. Contextual Fear Conditioning
4.8. Tissue Collection and Sample Preparation
4.9. Protein Extraction and Western Blot
4.10. Histological Stainings
4.11. Immunohistochemistry
4.12. Microscopy and Image Analysis
4.13. NETosis Levels Measurement
4.14. Glutathione (GSH) Levels Measurement
4.15. MRI
4.15.1. Imaging Protocols
4.15.2. MRI Data Analysis
4.16. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wardlaw, J.M.; Smith, C.; Dichgans, M. Mechanisms of sporadic cerebral small vessel disease: Insights from neuroimaging. Lancet Neurol. 2013, 12, 483–497. [Google Scholar] [CrossRef]
- Shindo, A.; Ishikawa, H.; Ii, Y.; Niwa, A.; Tomimoto, H. Clinical Features and Experimental Models of Cerebral Small Vessel Disease. Front. Aging Neurosci. 2020, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Pantoni, L. Cerebral small vessel disease: From pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010, 9, 689–701. [Google Scholar] [CrossRef]
- Joutel, A. Prospects for diminishing the impact of nonamyloid small-vessel diseases of the brain. Annu. Rev. Pharmacol. Toxicol. 2020, 6, 437–456. [Google Scholar] [CrossRef] [PubMed]
- Litak, J.; Mazurek, M.; Kulesza, B.; Szmygin, P.; Litak, J.; Kamieniak, P.; Grochowski, C. Cerebral small vessel disease. Int J. Mol. Sci. 2020, 21, 9729. [Google Scholar] [CrossRef]
- Cuadrado-Godia, E.; Dwivedi, P.; Sharma, S.; Santiago, A.O.; Gonzalez, M.B.; Laird, J.; Turk, M.; Suri, H.S.; Nikolaides, A.; Saba, L.; et al. Cerebral small vessel disease: A review focusing on pathophysiology, biomarkers, and machine learning strategies. J. Stroke 2018, 20, 302–320. [Google Scholar] [CrossRef]
- Cipolla, M.J.; Liebeskind, D.S.; Chan, S.L. The importance of comorbidities in ischemic stroke: Impact of hypertension on the cerebral circulation. J. Cereb Blood Flow Metab. 2018, 38, 2129–2149. [Google Scholar] [CrossRef]
- Iadecola, C.; Davisson, R.L. Hypertension and Cerebrovascular Dysfunction. Cell Metab. 2008, 7, 476–484. [Google Scholar] [CrossRef]
- Pires, P.W.; Dams Ramos, C.M.; Matin, N.; Dorrance, A.M. The effects of hypertension on the cerebral circulation. Am. J. Physiol.–Hear. Circ. Physiol. 2013, 304, 1598–1614. [Google Scholar] [CrossRef]
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y.; et al. Oxidative Stress-Mediated Blood-Brain Barrier (BBB) Disruption in Neurological Diseases. Oxid. Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef]
- González, J. Essential hypertension and oxidative stress: New insights. World J. Cardiol. 2014, 6, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular oxidative stress: Impact and therapeutic approaches. Front. Physiol. 2018, 4, 1668. [Google Scholar] [CrossRef] [PubMed]
- Grochowski, C.; Litak, J.; Kamieniak, P.; Maciejewski, R. Oxidative stress in cerebral small vessel disease. Role of reactive species. Free Radic. Res. 2018, 52, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, D.P.; Gotto, A.M. Biological relevance of inflammation and oxidative stress in the pathogenesis of arterial diseases. Am. J. Pathol. 2013, 182, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, A.H.; Markus, H.S. Do in vivo experimental models reflect human cerebral small vessel disease? A systematic review. J. Cereb. Blood Flow Metab. 2008, 28, 1877–1891. [Google Scholar] [CrossRef] [PubMed]
- De Silva, T.M.; Miller, A.A. Cerebral small vessel disease: Targeting oxidative stress as a novel therapeutic strategy? Front. Pharmacol. 2016, 17, 61. [Google Scholar] [CrossRef]
- Mustapha, M.; Nassir, C.M.N.C.M.; Aminuddin, N.; Safri, A.A.; Ghazali, M.M. Cerebral Small Vessel Disease (CSVD)–Lessons from the Animal Models. Front. Physiol. 2019, 24, 1317. [Google Scholar] [CrossRef]
- Mekler, J.; Yagil, Y.; Ben-Ishay, D. Renal response to acute saline loading in Sabra hypertension-prone and-resistant rats. Experientia 1985, 41, 923–924. [Google Scholar] [CrossRef]
- Gross, V.; Lippoldt, A.; Yagil, C.; Yagil, Y.; Luft, F.C. Pressure natriuresis in salt-sensitive and salt-resistant Sabra rats. Hypertension 1997, 29, 1252–1259. [Google Scholar] [CrossRef]
- Yagil, C.; Sapojnikov, M.; Katni, G.; Ilan, Z.; Zangen, S.W.; Rosenmann, E.; Yagil, Y. Proteinuria and glomerulosclerosis in the Sabra genetic rat model of salt susceptibility. Physiol. Genomics 2002, 9, 167–178. [Google Scholar] [CrossRef][Green Version]
- Sela, S.; Mazor, R.; Amsalam, M.; Yagil, C.; Yagil, Y.; Kristal, B. Primed polymorphonuclear leukocytes, oxidative stress, and inflammation antecede hypertension in the Sabra rat. Hypertension 2004, 44, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Brittain, J.F.; McCabe, C.; Khatun, H.; Kaushal, N.; Bridges, L.R.; Holmes, W.M.; Barrick, T.R.; Graham, D.; Dominiczak, A.F.; Macrae, M.; et al. An MRI-histological study of white matter in stroke-free SHRSP. J. Cereb. Blood Flow Metab. 2013, 33, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Sironi, L.; Guerrini, U.; Tremoli, E.; Miller, I.; Gelosa, P.; Lascialfari, A.; Zucca, I.; Eberini, I.; Gemeiner, M.; Paoletti, R.; et al. Analysis of pathological events at the onset of brain damage in stroke-prone rats: A proteomics and magnetic resonance imaging approach. J. Neurosci. Res. 2004, 78, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Schmidt, H.; Haybaeck, J.; Loitfelder, M.; Weis, S.; Cavalieri, M.; Seiler, S.; Enzinger, C.; Ropele, S.; Erkinjuntti, T.; et al. Heterogeneity in age-related white matter changes. Acta Neuropathol. 2011, 122, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Fazekas, F.; Kleinert, R.; Offenbacher, H.; Schmidt, R.; Kleinert, G.; Payer, F.; Radner, H.; Lechner, H. Pathologic correlates of incidental mri white matter signal hyperintensities. Neurology 1993, 43, 1683–1689. [Google Scholar] [CrossRef] [PubMed]
- Bueche, C.Z.; Hawkes, C.; Garz, C.; Vielhaber, S.; Attems, J.; Knight, R.T.; Reymann, K.; Heinze, H.-J.; Carare, R.O.; Schreiber, S. Hypertension drives parenchymal β-amyloid accumulation in the brain parenchyma. Ann. Clin. Transl. Neurol. 2014, 1, 124–129. [Google Scholar] [CrossRef]
- Werring, D.J. Cerebral Microbleeds: Pathophysiology to Clinical Practice; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar] [CrossRef]
- Schreiber, S.; Bueche, C.Z.; Garz, C.; Kropf, S.; Angenstein, F.; Goldschmind, J.; Neumann, J.; Heinze, H.-J.; Goertler, M.; Reymann, K.G.; et al. The pathologic cascade of cerebrovascular lesions in SHRSP: Is erythrocyte accumulation an early phase. J. Cereb. Blood Flow Metab. 2012, 32, 278–290. [Google Scholar] [CrossRef]
- Blanco, P.J.; Müller, L.O.; Spence, J.D. Blood pressure gradients in cerebral arteries: A clue to pathogenesis of cerebral small vessel disease. Stroke Vasc. Neurol. 2017, 2, 108–117. [Google Scholar] [CrossRef]
- Kang, L.; Yu, H.; Yang, X.; Zhu, Y.; Bai, X.; Wang, R.; Cao, Y.; Xu, H.; Luo, H.; Lu, L.; et al. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat. Commun. 2020, 11, 2488. [Google Scholar] [CrossRef]
- Vallés, J.; Lago, A.; Santos, M.T.; Latorre, A.-M.; Tembl, J.I.; Salom, J.B.; Nieves, C.; Moscardó, A. Neutrophil extracellular traps are increased in patients with acute ischemic stroke: Prognostic significance. Thromb. Haemost. 2017, 117, 1919–1929. [Google Scholar] [CrossRef]
- Zhou, P.; Li, T.; Jin, J.; Liu, Y.; Li, B.; Sun, Q.; Tian, J.; Zhao, H.; Liu, Z.; Ma, S.; et al. Interactions between neutrophil extracellular traps and activated platelets enhance procoagulant activity in acute stroke patients with ICA occlusion. EBioMedicine 2020, 53, 102671. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.W.S.; Westra, J.; Limburg, P.C.; Bijl, M.; Kallenberg, C.G.M. HMGB1 in vascular diseases: Its role in vascular inflammation and atherosclerosis. Autoimmun Rev. 2012, 11, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Mori, S.; Takahashi, H.K.; Tomono, Y.; Wake, H.; Kanke, T.; Sato, Y.; Hiraga, N.; Adachi, N.; Yoshino, T.; et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007, 21, 3904–3916. [Google Scholar] [CrossRef]
- Kim, S.W.; Lee, J.K. Role of HMGB1 in the Interplay between NETosis and Thrombosis in Ischemic Stroke: A Review. Cells 2020, 9, 1794. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799. [Google Scholar] [CrossRef] [PubMed]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biocim. Biophys. Acta Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef] [PubMed]
- Slaman, M.M.; Kitchen, P.; Halsey, A.; Wang, M.X.; Törnroth-Horsefield, S.; Conner, A.C.; Badaut, J.; Iliff, J.J.; Bill, R.M. Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain 2021, 145, 64–75. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Iliff, J.J.; Bill, R.M. Aquaporin 4 and lymphatic flow have central roles in brain fluid homeostasis. Nat. Rev. Neurosci. 2021, 22, 650–651. [Google Scholar] [CrossRef]
- Markou, A.; Unger, L.; Abir-Awan, M.; Saadallah, A.; Halsey, A.; Balklava, Z.; Conner, M.; Törnroth-Horsefield, S.; Greenhill, S.D.; Conner, A.; et al. Molecular mechanisms governing aquaporin relocalization. Biocim. Biophys. Acta Biomembr. 2022, 1864, 183853. [Google Scholar] [CrossRef]
- Slaman, M.M.; Kitchen, P.; Yool, A.J.; Bill, R.M. Recent breakthroughs and future directions in drugging aquaporins. Trends Pharmacol. Sci. 2022, 43, 30–42. [Google Scholar] [CrossRef]
- Wagner, K.; Unger, L.; Salman, M.M.; Kitchen, P.; Bill, R.M.; Yool, A.J. Signaling Mechanisms and Pharmacological Modulators Governing Diverse Aquaporin Functions in Human Health and Disease. Int. J. Mol. Sci. 2022, 23, 1388. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Al-Obaidi, Z.; Kitchen, P.; Loreto, A.; Bill, R.M.; Martins, R.W. Advances in Applying Computer-Aided Drug Design for Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4688. [Google Scholar] [CrossRef] [PubMed]
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering 2021, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.J.; Hatcher, J.; Virley, D.; Nelson, P.; Irving, E.; Hadingham, S.J.; Parsons, A.A. Functional assessments in mice and rats after focal stroke. Neuropharmacology 2000, 39, 806–816. [Google Scholar] [CrossRef]
- Shmueli, K.; De Zwart, J.A.; Van Gelderen, P.; Li, T.Q.; Dodd, S.J.; Duyn, J.H. Magnetic susceptibility mapping of brain tissue in vivo using MRI phase data. Magn. Reason. Med. 2009, 62, 1510–1522. [Google Scholar] [CrossRef]
- Chary, K.; Nissi, M.J.; Nykänen, O.; Manninen, E.; Rey, R.I.; Shmueli, K.; Sierra, A.; Gröhn, O. Quantitative susceptibility mapping of the rat brain after traumatic brain injury. NMR Biomed. 2021, 34, e4438. [Google Scholar] [CrossRef]
- Hsieh, M.C.; Tsai, C.Y.; Liao, M.C.; Yang, J.L.; Su, C.H.; Chen, J.H. Quantitative susceptibility mapping-based microscopy of magnetic resonance venography (QSM-mMRV) for in vivo morphologically and functionally assessing cerebromicrovasculature in rat stroke model. PLoS ONE 2016, 11, e0149602. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guy, R.; Volkman, R.; Wilczynski, E.; Yagil, C.; Yagil, Y.; Findler, M.; Auriel, E.; Nevo, U.; Offen, D. A Novel Rodent Model of Hypertensive Cerebral Small Vessel Disease with White Matter Hyperintensities and Peripheral Oxidative Stress. Int. J. Mol. Sci. 2022, 23, 5915. https://doi.org/10.3390/ijms23115915
Guy R, Volkman R, Wilczynski E, Yagil C, Yagil Y, Findler M, Auriel E, Nevo U, Offen D. A Novel Rodent Model of Hypertensive Cerebral Small Vessel Disease with White Matter Hyperintensities and Peripheral Oxidative Stress. International Journal of Molecular Sciences. 2022; 23(11):5915. https://doi.org/10.3390/ijms23115915
Chicago/Turabian StyleGuy, Reut, Rotem Volkman, Ella Wilczynski, Chana Yagil, Yoram Yagil, Michael Findler, Eitan Auriel, Uri Nevo, and Daniel Offen. 2022. "A Novel Rodent Model of Hypertensive Cerebral Small Vessel Disease with White Matter Hyperintensities and Peripheral Oxidative Stress" International Journal of Molecular Sciences 23, no. 11: 5915. https://doi.org/10.3390/ijms23115915
APA StyleGuy, R., Volkman, R., Wilczynski, E., Yagil, C., Yagil, Y., Findler, M., Auriel, E., Nevo, U., & Offen, D. (2022). A Novel Rodent Model of Hypertensive Cerebral Small Vessel Disease with White Matter Hyperintensities and Peripheral Oxidative Stress. International Journal of Molecular Sciences, 23(11), 5915. https://doi.org/10.3390/ijms23115915