
Targeting Energy Metabolism in Cancer Treatment

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
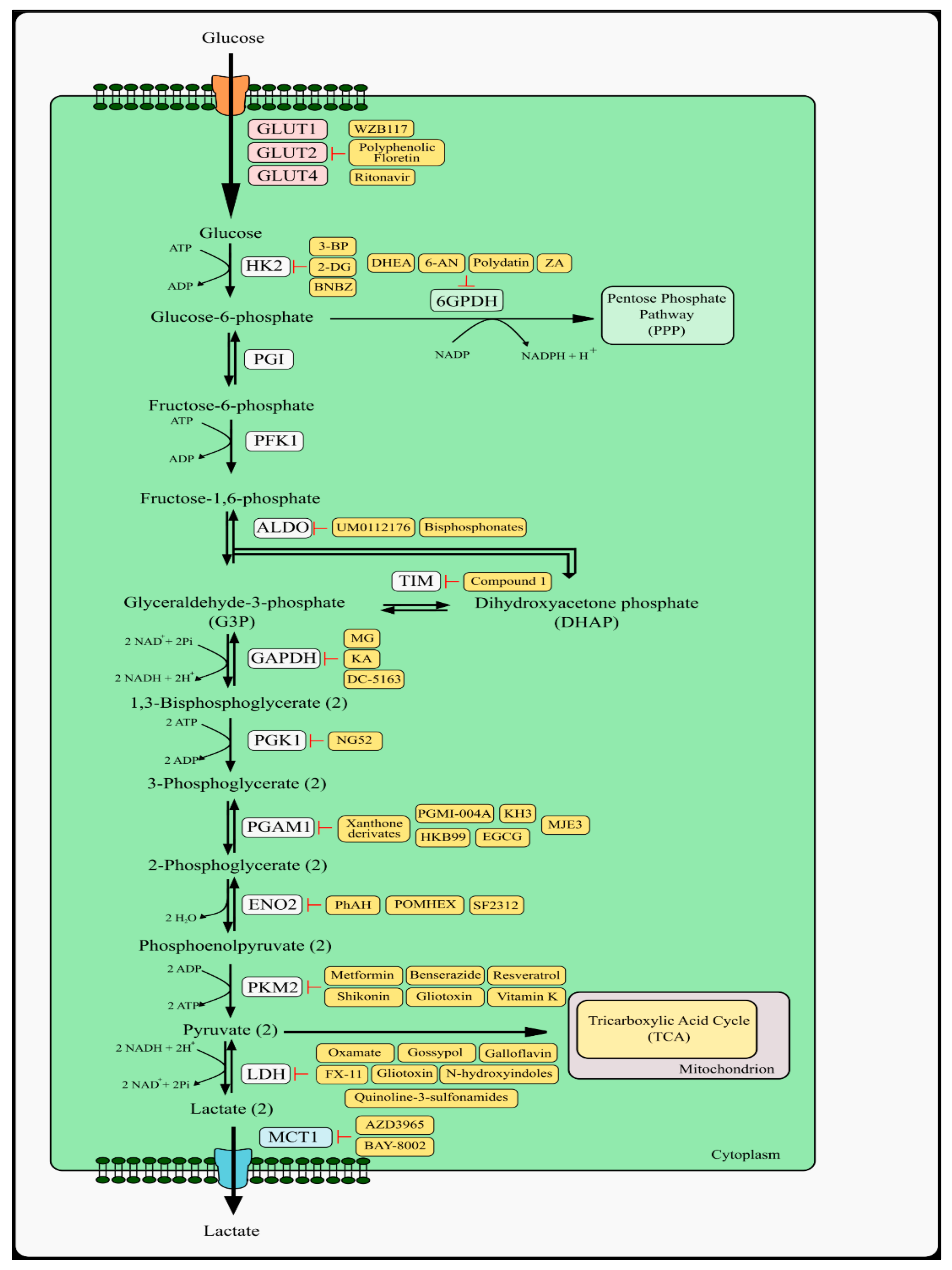
2. Glycolysis Pathway and Therapeutic Targets
2.1. Glucose Transporters
2.2. Hexokinase Type 2 and Phosphoglucose Isomerase
2.3. Aldolase
2.4. Glyceraldehyde 3-Phosphate Dehydrogenase and Phosphoglycerate Kinase 1
2.5. Phosphoglycerate Mutase 1
2.6. Pyruvate Kinase
3. Pyruvate and Its Further Fate
3.1. Lactate Dehydrogenase
3.2. Monocarboxylate Transporter 4
3.3. Mitochondrial Pyruvate Carriers and Pyruvate Dehydrogenase
4. The Pentose Phosphate Pathway
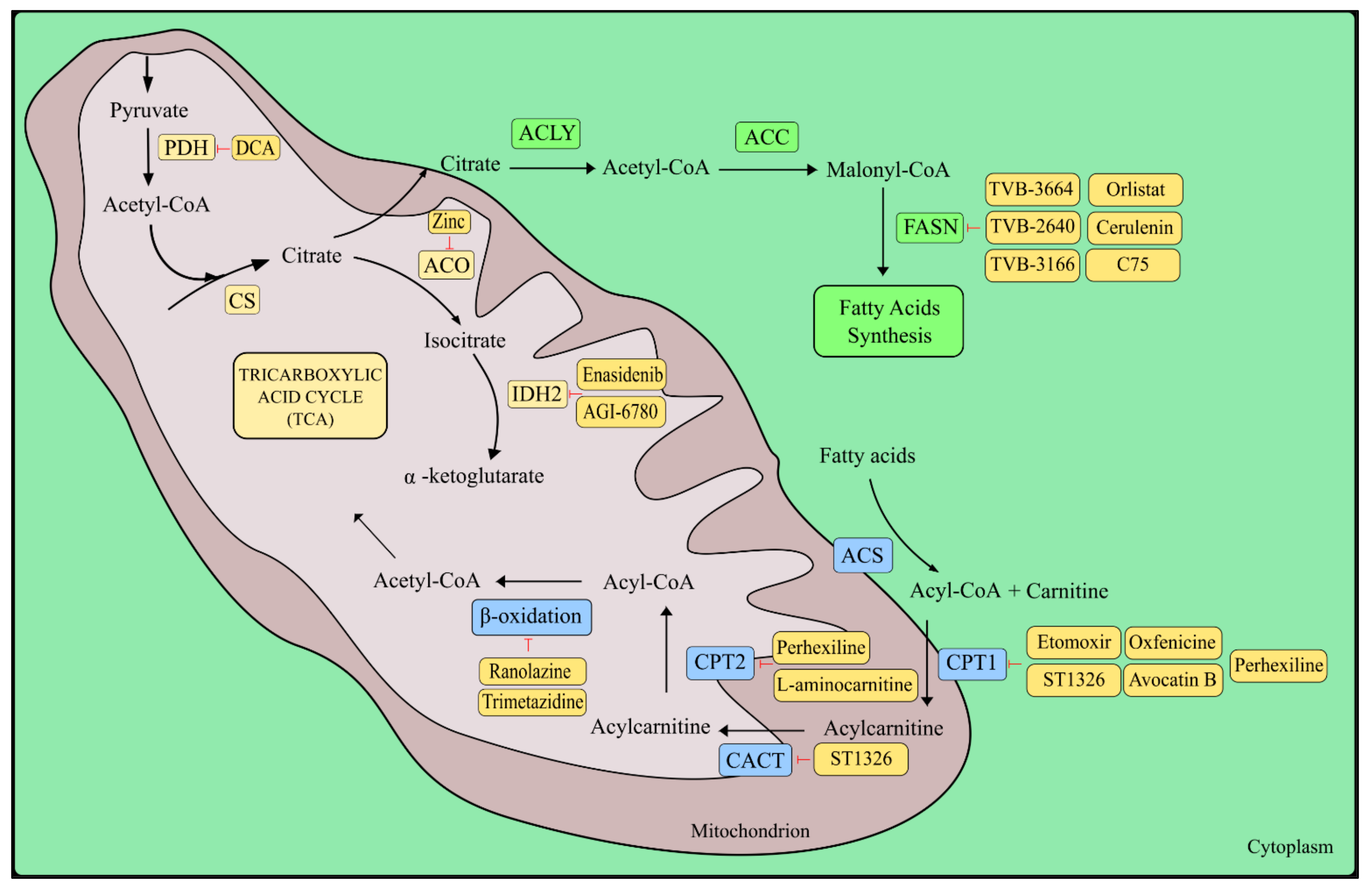
5. The Krebs Cycle
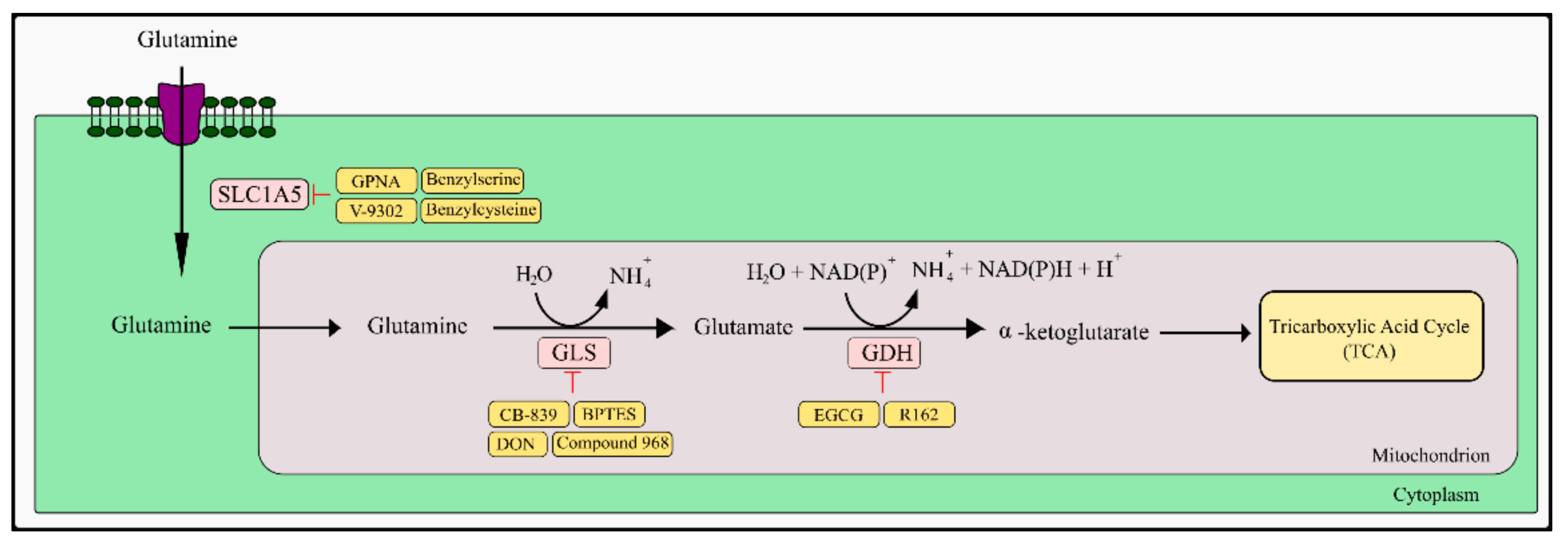
5.1. Glutamine and Fatty Acids as TCA Anaplerotic Substances
5.2. Fatty Acids
5.3. Isocitrate Dehydrogenase
6. Mitochondria as a Therapeutic Target in Cancer Treatment—Mitocans
- Class 1: hexokinase inhibitors.
- Class 2: compounds targeting Bcl-2 family proteins.
- Class 3: thiol redox inhibitors.
- Class 4: VDAC/ANT-targeting drugs.
- Class 5: electron transport chain-targeting drugs.
- Class 6: lipophilic cations targeting inner membrane.
- Class 7: TCA cycle-targeting drugs.
- Class 8: mtDNA-targeting drugs [353].
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Pucci, C.; Martinelli, C.; Ciofani, G. Innovative Approaches for Cancer Treatment: Current Perspectives and New Challenges. Ecancer 2019, 13, 961. [Google Scholar] [CrossRef] [PubMed]
- Ros, S.; Schulze, A. Balancing Glycolytic Flux: The Role of 6-Phosphofructo-2-Kinase/Fructose 2,6-Bisphosphatases in Cancer Metabolism. Cancer Metab. 2013, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Maguire, S.L.; Leonidou, A.; Wai, P.; Marchiò, C.; Ng, C.K.; Sapino, A.; Salomon, A.; Reis-Filho, J.S.; Weigelt, B.; Natrajan, R.C. SF3B1 Mutations Constitute a Novel Therapeutic Target in Breast Cancer. J. Pathol. 2015, 235, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.-P.; Booker, R.C.; Brosseau, J.-P.; Chen, Z.; Mo, J.; Tchegnon, E.; Wang, Y.; Clapp, D.W.; Le, L.Q. Contributions of Inflammation and Tumor Microenvironment to Neurofibroma Tumorigenesis. J. Clin. Investig. 2018, 128, 2848–2861. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. Uber Phosphorylierung Im Licht. J. Gen. Physiol. 1962, 45, 17–20. [Google Scholar] [CrossRef]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic Suppression Dramatically Changes the Intracellular Metabolic Profile of Multiple Cancer Cell Lines in a Mitochondrial Metabolism-Dependent Manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef]
- Rajendran, J.G.; Krohn, K.A. F-18 Fluoromisonidazole for Imaging Tumor Hypoxia: Imaging the Microenvironment for Personalized Cancer Therapy. Semin. Nucl. Med. 2015, 45, 151–162. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, Q.; Lian, X.; Jiang, P.; Cui, J. Hypoxia-Inducible Factor-1α (HIF-1α) Promotes Hypoxia-Induced Invasion and Metastasis in Ovarian Cancer by Targeting Matrix Metallopeptidase 13 (MMP13). Med. Sci. Monit. 2019, 25, 7202–7208. [Google Scholar] [CrossRef]
- Faes, S.; Uldry, E.; Planche, A.; Santoro, T.; Pythoud, C.; Demartines, N.; Dormond, O. Acidic PH Reduces VEGF-Mediated Endothelial Cell Responses by Downregulation of VEGFR-2; Relevance for Anti-Angiogenic Therapies. Oncotarget 2016, 7, 86026–86038. [Google Scholar] [CrossRef]
- Bomanji, J.; Costa, D.; Ell, P. Clinical Role of Positron Emission Tomography in Oncology. Lancet Oncol. 2001, 2, 157–164. [Google Scholar] [CrossRef]
- Locasale, J.W.; Cantley, L.C. Altered Metabolism in Cancer. BMC Biol. 2010, 8, 88. [Google Scholar] [CrossRef]
- Epstein, T.; Xu, L.; Gillies, R.J.; Gatenby, R.A. Separation of Metabolic Supply and Demand: Aerobic Glycolysis as a Normal Physiological Response to Fluctuating Energetic Demands in the Membrane. Cancer Metab. 2014, 2, 7. [Google Scholar] [CrossRef]
- Weinhouse, S. The Warburg Hypothesis Fifty Years Later. Z. Krebsforsch. 1976, 87, 115–126. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s Contributions to Current Concepts of Cancer Metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, E.K.J.; Ribeiro, M.J.; Stoot, J.H.M.B.; McCready, V.R.; Bourguignon, M.; Mazière, B. FDG Accumulation and Tumor Biology. Nucl. Med. Biol. 1998, 25, 317–322. [Google Scholar] [CrossRef]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic Coupling and the Reverse Warburg Effect in Cancer: Implications for Novel Biomarker and Anticancer Agent Development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef]
- Mercier, I.; Casimiro, M.C.; Wang, C.; Rosenberg, A.L.; Quong, J.; Minkeu, A.; Allen, K.G.; Danilo, C.; Sotgia, F.; Bonuccelli, G.; et al. Human Breast Cancer-Associated Fibroblasts (CAFs) Show Caveolin-1 down-Regulation and RB Tumor Suppressor Functional Inactivation: Implications for the Response to Hormonal Therapy. Cancer Biol. Ther. 2008, 7, 1212–1225. [Google Scholar] [CrossRef]
- Melvin, A.; Mudie, S.; Rocha, S. Mechanism of Hypoxia-Induced NFκB. Cell Cycle 2011, 10, 879–882. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and Lactate “Fuel” Tumor Growth and Metastasis: Evidence That Epithelial Cancer Cells Use Oxidative Mitochondrial Metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Corchado-Cobos, R.; García-Sancha, N.; Mendiburu-Eliçabe, M.; Gómez-Vecino, A.; Jiménez-Navas, A.; Pérez-Baena, M.J.; Holgado-Madruga, M.; Mao, J.-H.; Cañueto, J.; Castillo-Lluva, S.; et al. Pathophysiological Integration of Metabolic Reprogramming in Breast Cancer. Cancers 2022, 14, 322. [Google Scholar] [CrossRef] [PubMed]
- Marín, D.; Sabater, B. The Cancer Warburg Effect May Be a Testable Example of the Minimum Entropy Production Rate Principle. Phys. Biol. 2017, 14, 024001. [Google Scholar] [CrossRef] [PubMed]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef]
- Amoêdo, N.D.; Valencia, J.P.; Rodrigues, M.F.; Galina, A.; Rumjanek, F.D. How Does the Metabolism of Tumour Cells Differ from That of Normal Cells. Biosci. Rep. 2013, 33, e00080. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug Resistance and Combating Drug Resistance in Cancer. CDR 2019, 2, 141–160. [Google Scholar] [CrossRef]
- Kunkel, M.; Reichert, T.E.; Benz, P.; Lehr, H.-A.; Jeong, J.-H.; Wieand, S.; Bartenstein, P.; Wagner, W.; Whiteside, T.L. Overexpression of Glut-1 and Increased Glucose Metabolism in Tumors Are Associated with a Poor Prognosis in Patients with Oral Squamous Cell Carcinoma. Cancer 2003, 97, 1015–1024. [Google Scholar] [CrossRef]
- Haskins, C.; Cohen, J.; Kotecha, R.; Kaiser, A. Low Carbohydrate Diets in Cancer Therapeutics: Current Evidence. Front. Nutr. 2021, 8, 662952. [Google Scholar] [CrossRef]
- Klement, R.J.; Kämmerer, U. Is There a Role for Carbohydrate Restriction in the Treatment and Prevention of Cancer? Nutr. Metab. 2011, 8, 75. [Google Scholar] [CrossRef]
- Li, Y.-L.; Weng, H.-C.; Hsu, J.-L.; Lin, S.-W.; Guh, J.-H.; Hsu, L.-C. The Combination of MK-2206 and WZB117 Exerts a Synergistic Cytotoxic Effect Against Breast Cancer Cells. Front. Pharmacol. 2019, 10, 1311. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A Small-Molecule Inhibitor of Glucose Transporter 1 Downregulates Glycolysis, Induces Cell-Cycle Arrest, and Inhibits Cancer Cell Growth In Vitro and In Vivo. Mol. Cancer 2012, 11, 1672–1682. [Google Scholar] [CrossRef]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting Cellular Metabolism to Improve Cancer Therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef]
- Wu, K.-H.; Ho, C.-T.; Chen, Z.-F.; Chen, L.-C.; Whang-Peng, J.; Lin, T.-N.; Ho, Y.-S. The Apple Polyphenol Phloretin Inhibits Breast Cancer Cell Migration and Proliferation via Inhibition of Signals by Type 2 Glucose Transporter. J. Food Drug Anal. 2018, 26, 221–231. [Google Scholar] [CrossRef]
- Murad, H.; Hawat, M.; Ekhtiar, A.; AlJapawe, A.; Abbas, A.; Darwish, H.; Sbenati, O.; Ghannam, A. Induction of G1-Phase Cell Cycle Arrest and Apoptosis Pathway in MDA-MB-231 Human Breast Cancer Cells by Sulfated Polysaccharide Extracted from Laurencia Papillosa. Cancer Cell Int. 2016, 16, 39. [Google Scholar] [CrossRef]
- Tu, S.-H.; Chen, L.-C.; Ho, Y.-S. An Apple a Day to Prevent Cancer Formation: Reducing Cancer Risk with Flavonoids. J. Food Drug Anal. 2017, 25, 119–124. [Google Scholar] [CrossRef]
- Lin, S.-T.; Tu, S.-H.; Yang, P.-S.; Hsu, S.-P.; Lee, W.-H.; Ho, C.-T.; Wu, C.-H.; Lai, Y.-H.; Chen, M.-Y.; Chen, L.-C. Apple Polyphenol Phloretin Inhibits Colorectal Cancer Cell Growth via Inhibition of the Type 2 Glucose Transporter and Activation of P53-Mediated Signaling. J. Agric. Food Chem. 2016, 64, 6826–6837. [Google Scholar] [CrossRef]
- Akins, N.S.; Nielson, T.C.; Le, H.V. Inhibition of Glycolysis and Glutaminolysis: An Emerging Drug Discovery Approach to Combat Cancer. CTMC 2018, 18, 494–504. [Google Scholar] [CrossRef]
- Wei, C.; Bajpai, R.; Sharma, H.; Heitmeier, M.; Jain, A.D.; Matulis, S.M.; Nooka, A.K.; Mishra, R.K.; Hruz, P.W.; Schiltz, G.E.; et al. Development of GLUT4-Selective Antagonists for Multiple Myeloma Therapy. Eur. J. Med. Chem. 2017, 139, 573–586. [Google Scholar] [CrossRef]
- Dalva-Aydemir, S.; Bajpai, R.; Martinez, M.; Adekola, K.U.A.; Kandela, I.; Wei, C.; Singhal, S.; Koblinski, J.E.; Raje, N.S.; Rosen, S.T.; et al. Targeting the Metabolic Plasticity of Multiple Myeloma with FDA-Approved Ritonavir and Metformin. Clin. Cancer Res. 2015, 21, 1161–1171. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S. Molecular Intricacies of Aerobic Glycolysis in Cancer: Current Insights into the Classic Metabolic Phenotype. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 667–682. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, N.; Zhao, J.; Shi, S. High Expression of Hexokinase 2 Promotes Lung Cancer Proliferation and Metastasis. Arch. Med. Sci. 2020, 16, 1–13. [Google Scholar] [CrossRef]
- Ros, S.; Schulze, A. Glycolysis Back in the Limelight: Systemic Targeting of HK2 Blocks Tumor Growth. Cancer Discov. 2013, 3, 1105–1107. [Google Scholar] [CrossRef]
- Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef]
- Gong, L.; Wei, Y.; Yu, X.; Peng, J.; Leng, X. 3-Bromopyruvic Acid, A Hexokinase II Inhibitor, Is an Effective Antitumor Agent on the Hepatoma Cells: In Vitro and in Vivo Findings. ACAMC 2014, 14, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, N.; Chen, J.; Shen, J. Emerging Glycolysis Targeting and Drug Discovery from Chinese Medicine in Cancer Therapy. Evid. Based Complement. Altern. Med. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.C.; Krishan, A.; Lampidis, T.J. Greater Cell Cycle Inhibition and Cytotoxicity Induced by 2-Deoxy-d-Glucose in Tumor Cells Treated under Hypoxic vs Aerobic Conditions. Cancer Chemother. Pharmacol. 2004, 53, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Deslandes, E.; Villedieu, M.; Poulain, L.; Duval, M.; Gauduchon, P.; Schwartz, L.; Icard, P. Effect of 2-Deoxy-D-Glucose on Various Malignant Cell Lines In Vitro. Anticancer Res. 2006, 26, 3561–3566. [Google Scholar] [PubMed]
- Azevedo-Silva, J.; Queirós, O.; Baltazar, F.; Ułaszewski, S.; Goffeau, A.; Ko, Y.H.; Pedersen, P.L.; Preto, A.; Casal, M. The Anticancer Agent 3-Bromopyruvate: A Simple but Powerful Molecule Taken from the Lab to the Bedside. J. Bioenerg. Biomembr. 2016, 48, 349–362. [Google Scholar] [CrossRef]
- Linke, C.; Wösle, M.; Harder, A. Anti-Cancer Agent 3-Bromopyruvate Reduces Growth of MPNST and Inhibits Metabolic Pathways in a Representative in-Vitro Model. BMC Cancer 2020, 20, 896. [Google Scholar] [CrossRef]
- Philippe, I.; Xiao-Dong, Z.; Edwige, L.; Marie-Hélène, L.; Stéphane, A.; Hubert, L.; Laurent, P. Experimental Results Using 3-Bromopyruvate in Mesothelioma: In Vitro and in Vivo Studies. J. Bioenerg. Biomembr. 2012, 44, 81–90. [Google Scholar] [CrossRef]
- Zou, X.; Zhang, M.; Sun, Y.; Zhao, S.; Wei, Y.; Zhang, X.; Jiang, C.; Liu, H. Inhibitory Effects of 3-Bromopyruvate in Human Nasopharyngeal Carcinoma Cells. Oncol. Rep. 2015, 34, 1895–1904. [Google Scholar] [CrossRef]
- Sayed, S.M.E.; Mohamed, W.G.; Seddik, M.-A.H.; Ahmed, A.-S.A.; Mahmoud, A.G.; Amer, W.H.; Nabo, M.M.H.; Hamed, A.R.; Ahmed, N.S.; Abd-Allah, A.A.-R. Safety and Outcome of Treatment of Metastatic Melanoma Using 3-Bromopyruvate: A Concise Literature Review and Case Study. Chin. J. Cancer 2014, 33, 356–364. [Google Scholar] [CrossRef]
- El Sayed, S.M. Enhancing Anticancer Effects, Decreasing Risks and Solving Practical Problems Facing 3-Bromopyruvate in Clinical Oncology: 10 Years of Research. Experience. IJN 2018, 13, 4699–4709. [Google Scholar] [CrossRef]
- Ko, Y.H.; Verhoeven, H.A.; Lee, M.J.; Corbin, D.J.; Vogl, T.J.; Pedersen, P.L. A Translational Study “Case Report” on the Small Molecule “Energy Blocker” 3-Bromopyruvate (3BP) as a Potent Anticancer Agent: From Bench Side to Bedside. J. Bioenerg. Biomembr. 2012, 44, 163–170. [Google Scholar] [CrossRef]
- Liu, Y.; Li, M.; Zhang, Y.; Wu, C.; Yang, K.; Gao, S.; Zheng, M.; Li, X.; Li, H.; Chen, L. Structure Based Discovery of Novel Hexokinase 2 Inhibitors. Bioorg. Chem. 2020, 96, 103609. [Google Scholar] [CrossRef]
- Zheng, M.; Wu, C.; Yang, K.; Yang, Y.; Liu, Y.; Gao, S.; Wang, Q.; Li, C.; Chen, L.; Li, H. Novel Selective Hexokinase 2 Inhibitor Benitrobenrazide Blocks Cancer Cells Growth by Targeting Glycolysis. Pharmacol. Res. 2021, 164, 105367. [Google Scholar] [CrossRef]
- Kim, J.; Dang, C.V. Multifaceted Roles of Glycolytic Enzymes. Trends Biochem. Sci. 2005, 30, 142–150. [Google Scholar] [CrossRef]
- Xu, W.; Seiter, K.; Feldman, E.; Ahmed, T.; Chiao, J. The Differentiation and Maturation Mediator for Human Myeloid Leukemia Cells Shares. Homology with Neuroleukin or Phosphoglucose Isomerase. Blood 1996, 87, 4502–4506. [Google Scholar] [CrossRef]
- Sun, Y.-J.; Chou, C.-C.; Chen, W.-S.; Wu, R.-T.; Meng, M.; Hsiao, C.-D. The Crystal Structure of a Multifunctional Protein: Phosphoglucose Isomerase/Autocrine Motility Factor/Neuroleukin. Proc. Natl. Acad. Sci. USA 1999, 96, 5412–5417. [Google Scholar] [CrossRef]
- Nowak, N.; Kulma, A.; Gutowicz, J. Up-Regulation of Key Glycolysis Proteins in Cancer Development. Open Life Sci. 2018, 13, 569–581. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Yang, Y.-C.; Tien, C.-P.; Yang, C.-J.; Hsiao, M. Roles of Aldolase Family Genes in Human Cancers and Diseases. Trends Endocrinol. Metab. 2018, 29, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-C.; Chan, Y.-C.; Chang, W.-M.; Lin, Y.-F.; Yang, C.-J.; Su, C.-Y.; Huang, M.-S.; Wu, A.T.H.; Hsiao, M. Feedback Regulation of ALDOA Activates the HIF-1α/MMP9 Axis to Promote Lung Cancer Progression. Cancer Lett. 2017, 403, 28–36. [Google Scholar] [CrossRef]
- Li, X.; Jiang, F.; Ge, Z.; Chen, B.; Yu, J.; Xin, M.; Wang, J.; An, L.; Wei, J.; Wu, L. Fructose-Bisphosphate Aldolase A Regulates Hypoxic Adaptation in Hepatocellular Carcinoma and Involved with Tumor Malignancy. Dig. Dis. Sci. 2019, 64, 3215–3227. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Cai, X.; Luo, W.; Chen, L.; Li, K. Role of Aldolase A in Osteosarcoma Progression and Metastasis: In Vitro and in Vivo Evidence. Oncol. Rep. 2014, 32, 2031–2037. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ye, F.; Chen, Y.; Xia, L.; Lian, J.; Yang, S. Aldolase A Overexpression Is Associated with Poor Prognosis and Promotes Tumor Progression by the Epithelial-Mesenchymal Transition in Colon Cancer. Biochem. Biophys. Res. Commun. 2018, 497, 639–645. [Google Scholar] [CrossRef]
- He, J.; Jin, Y.; Chen, Y.; Yao, H.-B.; Xia, Y.-J.; Ma, Y.-Y.; Wang, W.; Shao, Q.-S. Downregulation of ALDOB Is Associated with Poor Prognosis of Patients with Gastric Cancer. Onco Targets 2016, 9, 6099–6109. [Google Scholar] [CrossRef] [PubMed]
- Gizak, A.; Wiśniewski, J.; Heron, P.; Mamczur, P.; Sygusch, J.; Rakus, D. Targeting a Moonlighting Function of Aldolase Induces Apoptosis in Cancer Cells. Cell Death Dis. 2019, 10, 712. [Google Scholar] [CrossRef] [PubMed]
- Heron, P.W.; Abellán-Flos, M.; Salmon, L.; Sygusch, J. Bisphosphonate Inhibitors of Mammalian Glycolytic Aldolase. J. Med. Chem. 2018, 61, 10558–10572. [Google Scholar] [CrossRef]
- Endo, A.; Hasumi, K.; Sakai, K.; Kanbe, T. Specific Inhibition of Glyceraldehyde-3-Phosphate Dehydrogenase by Koningic Acid (Heptelidic Acid). J. Antibiot. 1985, 38, 920–925. [Google Scholar] [CrossRef]
- Sakai, K.; Hasumi, K.; Endo, A. Identification of Koningic Acid (Heptelidic Acid)-Modified Site in Rabbit Muscle Glyceraldehyde-3-Phosphate Dehydrogenase. Biochim. Biophys. Acta 1991, 1077, 192–196. [Google Scholar] [CrossRef]
- Liberti, M.V.; Dai, Z.; Wardell, S.E.; Baccile, J.A.; Liu, X.; Gao, X.; Baldi, R.; Mehrmohamadi, M.; Johnson, M.O.; Madhukar, N.S.; et al. A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab. 2017, 26, 648–659.e8. [Google Scholar] [CrossRef]
- Rahier, N.J.; Molinier, N.; Long, C.; Deshmukh, S.K.; Kate, A.S.; Ranadive, P.; Verekar, S.A.; Jiotode, M.; Lavhale, R.R.; Tokdar, P.; et al. Anticancer Activity of Koningic Acid and Semisynthetic Derivatives. Bioorg. Med. Chem. 2015, 23, 3712–3721. [Google Scholar] [CrossRef]
- Lee, H.J.; Howell, S.K.; Sanford, R.J.; Beisswenger, P.J. Methylglyoxal Can Modify GAPDH Activity and Structure. Ann. N. Y. Acad. Sci. 2005, 1043, 135–145. [Google Scholar] [CrossRef]
- Li, T.; Tan, X.; Yang, R.; Miao, Y.; Zhang, M.; Xi, Y.; Guo, R.; Zheng, M.; Li, B. Discovery of Novel Glyceraldehyde-3-Phosphate Dehydrogenase Inhibitor via Docking-Based Virtual Screening. Bioorg. Chem. 2020, 96, 103620. [Google Scholar] [CrossRef]
- Wang, W.; Jiang, Z.; Hu, C.; Chen, C.; Hu, Z.; Wang, A.; Wang, L.; Liu, J.; Wang, W.; Liu, Q. Pharmacologically Inhibiting Phosphoglycerate Kinase 1 for Glioma with NG52. Acta Pharm. Sin. 2021, 42, 633–640. [Google Scholar] [CrossRef]
- Fothergill-Gilmore, L.A.; Watson, H.C. The Phosphoglycerate Mutases. Adv. Enzym. Relat. Areas Mol. Biol. 1989, 62, 227–313. [Google Scholar] [CrossRef]
- Hitosugi, T.; Zhou, L.; Elf, S.; Fan, J.; Kang, H.-B.; Seo, J.H.; Shan, C.; Dai, Q.; Zhang, L.; Xie, J.; et al. Phosphoglycerate Mutase 1 Coordinates Glycolysis and Biosynthesis to Promote Tumor Growth. Cancer Cell 2012, 22, 585–600. [Google Scholar] [CrossRef]
- Huang, K.; Liang, Q.; Zhou, Y.; Jiang, L.; Gu, W.; Luo, M.; Tang, Y.; Wang, Y.; Lu, W.; Huang, M.; et al. A Novel Allosteric Inhibitor of Phosphoglycerate Mutase 1 Suppresses Growth and Metastasis of Non-Small-Cell Lung Cancer. Cell Metab. 2019, 30, 1107–1119.e8. [Google Scholar] [CrossRef]
- Liang, Q.; Gu, W.-M.; Huang, K.; Luo, M.-Y.; Zou, J.-H.; Zhuang, G.-L.; Lei, H.-M.; Chen, H.-Z.; Zhu, L.; Zhou, L.; et al. HKB99, an Allosteric Inhibitor of Phosphoglycerate Mutase 1, Suppresses Invasive Pseudopodia Formation and Upregulates Plasminogen Activator Inhibitor-2 in Erlotinib-Resistant Non-Small Cell Lung Cancer Cells. Acta Pharm. Sin. 2021, 42, 115–119. [Google Scholar] [CrossRef]
- Evans, M.J.; Saghatelian, A.; Sorensen, E.J.; Cravatt, B.F. Target Discovery in Small-Molecule Cell-Based Screens by in Situ Proteome Reactivity Profiling. Nat. Biotechnol. 2005, 23, 1303–1307. [Google Scholar] [CrossRef]
- Wang, P.; Jiang, L.; Cao, Y.; Zhang, X.; Chen, B.; Zhang, S.; Huang, K.; Ye, D.; Zhou, L. Xanthone Derivatives as Phosphoglycerate Mutase 1 Inhibitors: Design, Synthesis, and Biological Evaluation. Bioorg. Med. Chem. 2018, 26, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.-L.; Huang, K.; Jiang, L.-L.; Lu, X.-X.; Dai, Y.-T.; Shi, M.-M.; Tang, X.-M.; Wang, Q.-B.; Zhang, X.-D.; Wang, P.-H.; et al. An Allosteric PGAM1 Inhibitor Effectively Suppresses Pancreatic Ductal Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 23264–23273. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tang, S.; Wang, Q.-Q.; Leung, E.L.-H.; Jin, H.; Huang, Y.; Liu, J.; Geng, M.; Huang, M.; Yuan, S.; et al. Identification of Epigallocatechin-3- Gallate as an Inhibitor of Phosphoglycerate Mutase 1. Front. Pharmacol. 2017, 8, 325. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, N.; Rasul, A.; Shah, M.A.; Jabeen, F.; Sultana, T. In Silico-Based Identification of Phytochemicals as Novel Human Phosphoglycerate Mutase 1 (PGAM1) Inhibitors for Cancer Therapy. Pak. J. Pharm. Sci. 2021, 34, 665–670. [Google Scholar]
- Didiasova, M.; Schaefer, L.; Wygrecka, M. When Place Matters: Shuttling of Enolase-1 across Cellular Compartments. Front. Cell Dev. Biol. 2019, 7, 61. [Google Scholar] [CrossRef]
- Altenberg, B.; Greulich, K.O. Genes of Glycolysis Are Ubiquitously Overexpressed in 24 Cancer Classes. Genomics 2004, 84, 1014–1020. [Google Scholar] [CrossRef]
- Muller, F.L.; Colla, S.; Aquilanti, E.; Manzo, V.E.; Genovese, G.; Lee, J.; Eisenson, D.; Narurkar, R.; Deng, P.; Nezi, L.; et al. Passenger Deletions Generate Therapeutic Vulnerabilities in Cancer. Nature 2012, 488, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Leonard, P.G.; Satani, N.; Maxwell, D.; Lin, Y.-H.; Hammoudi, N.; Peng, Z.; Pisaneschi, F.; Link, T.M.; Lee, G.R.; Sun, D.; et al. SF2312 Is a Natural Phosphonate Inhibitor of Enolase. Nat. Chem. Biol. 2016, 12, 1053–1058. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Satani, N.; Hammoudi, N.; Yan, V.C.; Barekatain, Y.; Khadka, S.; Ackroyd, J.J.; Georgiou, D.K.; Pham, C.-D.; Arthur, K.; et al. An Enolase Inhibitor for the Targeted Treatment of ENO1-Deleted Cancers. Nat. Metab. 2020, 2, 1413–1426. [Google Scholar] [CrossRef]
- Liu, V.M.; Howell, A.J.; Hosios, A.M.; Li, Z.; Israelsen, W.J.; Vander Heiden, M.G. Cancer-associated Mutations in Human Pyruvate Kinase M2 Impair Enzyme Activity. FEBS Lett. 2020, 594, 646–664. [Google Scholar] [CrossRef]
- Hosios, A.M.; Fiske, B.P.; Gui, D.Y.; Van der Heiden, M.G. Lack of Evidence for PKM2 Protein Kinase Activity. Mol. Cell 2015, 59, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Chen, J.; Zhang, X.; Wang, Z.; Chen, J.; Lin, X.; Huang, H.; Fu, W.; Liang, J.; Wu, W.; et al. The HIF-1α Antisense Long Non-Coding RNA Drives a Positive Feedback Loop of HIF-1α Mediated Transactivation and Glycolysis. Nat. Commun. 2021, 12, 1341. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Gao, Y.; Wang, C.; Yu, X.; Zhang, W.; Guan, H.; Shan, Z.; Teng, W. Aberrant Overexpression of Pyruvate Kinase M2 Is Associated with Aggressive Tumor Features and the BRAF Mutation in Papillary Thyroid Cancer. J. Clin. Endocrinol. Metab. 2013, 98, E1524–E1533. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.-L.; Au, S.L.-K.; Tse, A.P.-W.; Xu, I.M.-J.; Lai, R.K.-H.; Chiu, D.K.-C.; Wei, L.L.; Fan, D.N.-Y.; Tsang, F.H.-C.; Lo, R.C.-L.; et al. Switching of Pyruvate Kinase Isoform L to M2 Promotes Metabolic Reprogramming in Hepatocarcinogenesis. PLoS ONE 2014, 9, e115036. [Google Scholar] [CrossRef]
- Lu, W.; Cao, Y.; Zhang, Y.; Li, S.; Gao, J.; Wang, X.-A.; Mu, J.; Hu, Y.-P.; Jiang, L.; Dong, P.; et al. Up-Regulation of PKM2 Promote Malignancy and Related to Adverse Prognostic Risk Factor in Human Gallbladder Cancer. Sci. Rep. 2016, 6, 26351. [Google Scholar] [CrossRef]
- Azoitei, N.; Becher, A.; Steinestel, K.; Rouhi, A.; Diepold, K.; Genze, F.; Simmet, T.; Seufferlein, T. PKM2 Promotes Tumor Angiogenesis by Regulating HIF-1α through NF-ΚB Activation. Mol. Cancer 2016, 15, 3. [Google Scholar] [CrossRef]
- Israelsen, W.J.; Dayton, T.L.; Davidson, S.M.; Fiske, B.P.; Hosios, A.M.; Bellinger, G.; Li, J.; Yu, Y.; Sasaki, M.; Horner, J.W.; et al. PKM2 Isoform-Specific Deletion Reveals a Differential Requirement for Pyruvate Kinase in Tumor Cells. Cell 2013, 155, 397–409. [Google Scholar] [CrossRef]
- Dayton, T.L.; Gocheva, V.; Miller, K.M.; Israelsen, W.J.; Bhutkar, A.; Clish, C.B.; Davidson, S.M.; Luengo, A.; Bronson, R.T.; Jacks, T.; et al. Germline Loss of PKM2 Promotes Metabolic Distress and Hepatocellular Carcinoma. Genes Dev. 2016, 30, 1020–1033. [Google Scholar] [CrossRef]
- Hillis, A.L.; Lau, A.N.; Devoe, C.X.; Dayton, T.L.; Danai, L.V.; Di Vizio, D.; Vander Heiden, M.G. PKM2 Is Not Required for Pancreatic Ductal Adenocarcinoma. Cancer Metab. 2018, 6, 17. [Google Scholar] [CrossRef]
- Papadaki, C.; Sfakianaki, M.; Lagoudaki, E.; Giagkas, G.; Ioannidis, G.; Trypaki, M.; Tsakalaki, E.; Voutsina, A.; Koutsopoulos, A.; Mavroudis, D.; et al. PKM2 as a Biomarker for Chemosensitivity to Front-Line Platinum-Based Chemotherapy in Patients with Metastatic Non-Small-Cell Lung Cancer. Br. J. Cancer 2014, 111, 1757–1764. [Google Scholar] [CrossRef]
- He, X.; Du, S.; Lei, T.; Li, X.; Liu, Y.; Wang, H.; Tong, R.; Wang, Y. PKM2 in Carcinogenesis and Oncotherapy. Oncotarget 2017, 8, 110656–110670. [Google Scholar] [CrossRef]
- Su, Q.; Luo, S.; Tan, Q.; Deng, J.; Zhou, S.; Peng, M.; Tao, T.; Yang, X. The Role of Pyruvate Kinase M2 in Anticancer Therapeutic Treatments (Review). Oncol. Lett. 2019, 18, 5663–5672. [Google Scholar] [CrossRef]
- Zhu, S.; Guo, Y.; Zhang, X.; Liu, H.; Yin, M.; Chen, X.; Peng, C. Pyruvate Kinase M2 (PKM2) in Cancer and Cancer Therapeutics. Cancer Lett. 2021, 503, 240–248. [Google Scholar] [CrossRef]
- Shang, D.; Wu, J.; Guo, L.; Xu, Y.; Liu, L.; Lu, J. Metformin Increases Sensitivity of Osteosarcoma Stem Cells to Cisplatin by Inhibiting Expression of PKM2. Int. J. Oncol. 2017, 50, 1848–1856. [Google Scholar] [CrossRef]
- Su, Q.; Tao, T.; Tang, L.; Deng, J.; Darko, K.O.; Zhou, S.; Peng, M.; He, S.; Zeng, Q.; Chen, A.F.; et al. Down-regulation of PKM2 Enhances Anticancer Efficiency of THP on Bladder Cancer. J. Cell. Mol. Med. 2018, 22, 2774–2790. [Google Scholar] [CrossRef]
- Cheng, K.; Hao, M. Metformin Inhibits TGF-Β1-Induced Epithelial-to-Mesenchymal Transition via PKM2 Relative-MTOR/P70s6k Signaling Pathway in Cervical Carcinoma Cells. Int. J. Mol. Sci. 2016, 17, 2000. [Google Scholar] [CrossRef]
- Yin, W.; Liu, Y.; Liu, X.; Ma, X.; Sun, B.; Yu, Z. Metformin Inhibits Epithelial-Mesenchymal Transition of Oral Squamous Cell Carcinoma via the MTOR/HIF-1α/PKM2/STAT3 Pathway. Oncol. Lett. 2021, 21, 31. [Google Scholar] [CrossRef]
- Puckett, D.L.; Alquraishi, M.; Chowanadisai, W.; Bettaieb, A. The Role of PKM2 in Metabolic Reprogramming: Insights into the Regulatory Roles of Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 1171. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, Z.; Su, J.; Li, J.; Zhao, S.; Wu, L.; Zhang, J.; He, Y.; Zhang, G.; Tao, J.; et al. Benserazide Is a Novel Inhibitor Targeting PKM2 for Melanoma Treatment. Int. J. Cancer 2020, 147, 139–151. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, F.; Wu, X.-R. Inhibition of Pyruvate Kinase M2 Markedly Reduces Chemoresistance of Advanced Bladder Cancer to Cisplatin. Sci. Rep. 2017, 7, 45983. [Google Scholar] [CrossRef]
- Thonsri, U.; Seubwai, W.; Waraasawapati, S.; Wongkham, S.; Boonmars, T.; Cha’on, U.; Wongkham, C. Antitumor Effect of Shikonin, a PKM2 Inhibitor, in Cholangiocarcinoma Cell Lines. Anticancer Res. 2020, 40, 5115–5124. [Google Scholar] [CrossRef]
- Zhao, X.; Zhu, Y.; Hu, J.; Jiang, L.; Li, L.; Jia, S.; Zen, K. Shikonin Inhibits Tumor Growth in Mice by Suppressing Pyruvate Kinase M2-Mediated Aerobic Glycolysis. Sci. Rep. 2018, 8, 14517. [Google Scholar] [CrossRef]
- Tang, J.-C.; Zhao, J.; Long, F.; Chen, J.-Y.; Mu, B.; Jiang, Z.; Ren, Y.; Yang, J. Efficacy of Shikonin against Esophageal Cancer Cells and Its Possible Mechanisms in Vitro and in Vivo. J. Cancer 2018, 9, 32–40. [Google Scholar] [CrossRef]
- Wu, H.; Wang, Y.; Wu, C.; Yang, P.; Li, H.; Li, Z. Resveratrol Induces Cancer Cell Apoptosis through MiR-326/PKM2-Mediated ER Stress and Mitochondrial Fission. J. Agric. Food Chem. 2016, 64, 9356–9367. [Google Scholar] [CrossRef]
- Zhao, H.; Han, L.; Jian, Y.; Ma, Y.; Yan, W.; Chen, X.; Xu, H.; Li, L. Resveratrol Induces Apoptosis in Human Melanoma Cell through Negatively Regulating Erk/PKM2/Bcl-2 Axis. OTT 2018, 11, 8995–9006. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Vitamin K(3) and K(5) Are Inhibitors of Tumor Pyruvate Kinase M2. Cancer Lett. 2012, 316, 204–210. [Google Scholar] [CrossRef]
- Verrax, J.; Cadrobbi, J.; Delvaux, M.; Jamison, J.M.; Gilloteaux, J.; Summers, J.L.; Taper, H.S.; Buc Calderon, P. The Association of Vitamins C and K3 Kills Cancer Cells Mainly by Autoschizis, a Novel Form of Cell Death. Basis for Their Potential Use as Coadjuvants in Anticancer Therapy. Eur. J. Med. Chem. 2003, 38, 451–457. [Google Scholar] [CrossRef]
- Hitomi, M.; Yokoyama, F.; Kita, Y.; Nonomura, T.; Masaki, T.; Yoshiji, H.; Inoue, H.; Kinekawa, F.; Kurokohchi, K.; Uchida, N.; et al. Antitumor Effects of Vitamins K1, K2 and K3 on Hepatocellular Carcinoma in Vitro and in Vivo. Int. J. Oncol. 2005, 26, 713–720. [Google Scholar] [CrossRef]
- Ogawa, M.; Nakai, S.; Deguchi, A.; Nonomura, T.; Masaki, T.; Uchida, N.; Yoshiji, H.; Kuriyama, S. Vitamins K2, K3 and K5 Exert Antitumor Effects on Established Colorectal Cancer in Mice by Inducing Apoptotic Death of Tumor Cells. Int. J. Oncol. 2007, 31, 323–331. [Google Scholar] [CrossRef][Green Version]
- Tang, W.; Liu, Z.; Mai, X.; Qi, X.; Li, D.; Gu, Q.; Li, J. Identification of Gliotoxin Isolated from Marine Fungus as a New Pyruvate Kinase M2 Inhibitor. Biochem. Biophys. Res. Commun. 2020, 528, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Qi, H.; Li, R.; Li, Y.; Jin, Y.; McNutt, M.A.; Liu, J.; Yin, Y. Discovery of Novel Naphthoquinone Derivatives as Inhibitors of the Tumor Cell Specific M2 Isoform of Pyruvate Kinase. Eur. J. Med. Chem. 2017, 138, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Chun, J.; Pan, C.; Alesi, G.N.; Li, D.; Magliocca, K.R.; Kang, Y.; Chen, Z.G.; Shin, D.M.; Khuri, F.R.; et al. Phosphorylation-Mediated Activation of LDHA Promotes Cancer Cell Invasion and Tumour Metastasis. Oncogene 2017, 36, 3797–3806. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Hitosugi, T.; Chung, T.-W.; Xie, J.; Ge, Q.; Gu, T.-L.; Polakiewicz, R.D.; Chen, G.Z.; Boggon, T.J.; Lonial, S.; et al. Tyrosine Phosphorylation of Lactate Dehydrogenase A Is Important for NADH/NAD(+) Redox Homeostasis in Cancer Cells. Mol. Cell. Biol. 2011, 31, 4938–4950. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of Lactate Dehydrogenase A Induces Oxidative Stress and Inhibits Tumor Progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef]
- Zhao, Z.; Han, F.; Yang, S.; Wu, J.; Zhan, W. Oxamate-Mediated Inhibition of Lactate Dehydrogenase Induces Protective Autophagy in Gastric Cancer Cells: Involvement of the Akt-MTOR Signaling Pathway. Cancer Lett. 2015, 358, 17–26. [Google Scholar] [CrossRef]
- Valvona, C.J.; Fillmore, H.L. Oxamate, but Not Selective Targeting of LDH-A, Inhibits Medulloblastoma Cell Glycolysis, Growth and Motility. Brain Sci. 2018, 8, 56. [Google Scholar] [CrossRef]
- Stone, S.C.; Rossetti, R.A.M.; Alvarez, K.L.F.; Carvalho, J.P.; Margarido, P.F.R.; Baracat, E.C.; Tacla, M.; Boccardo, E.; Yokochi, K.; Lorenzi, N.P.; et al. Lactate Secreted by Cervical Cancer Cells Modulates Macrophage Phenotype. J. Leukoc. Biol. 2019, 105, 1041–1054. [Google Scholar] [CrossRef]
- Cassim, S.; Raymond, V.-A.; Dehbidi-Assadzadeh, L.; Lapierre, P.; Bilodeau, M. Metabolic Reprogramming Enables Hepatocarcinoma Cells to Efficiently Adapt and Survive to a Nutrient-Restricted Microenvironment. Cell Cycle 2018, 17, 903–916. [Google Scholar] [CrossRef]
- Qiao, T.; Xiong, Y.; Feng, Y.; Guo, W.; Zhou, Y.; Zhao, J.; Jiang, T.; Shi, C.; Han, Y. Inhibition of LDH-A by Oxamate Enhances the Efficacy of Anti-PD-1 Treatment in an NSCLC Humanized Mouse Model. Front. Oncol. 2021, 11, 632364. [Google Scholar] [CrossRef]
- Granchi, C.; Paterni, I.; Rani, R.; Minutolo, F. Small-Molecule Inhibitors of Human LDH5. Future Med. Chem. 2013, 5, 1967–1991. [Google Scholar] [CrossRef]
- Lee, C.-Y.G.; Moon, Y.S.; Yuan, J.H.; Chen, A.F. Enzyme Inactivation and Inhibition by Gossypol. Mol. Cell. Biochem. 1982, 47, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Bertini, S.; Macchia, M.; Minutolo, F. Inhibitors of Lactate Dehydrogenase Isoforms and Their Therapeutic Potentials. CMC 2010, 17, 672–697. [Google Scholar] [CrossRef]
- Zeng, Y.; Ma, J.; Xu, L.; Wu, D. Natural Product Gossypol and Its Derivatives in Precision Cancer Medicine. CMC 2019, 26, 1849–1873. [Google Scholar] [CrossRef]
- Shelley, M.D.; Hartley, L.; Fish, R.G.; Groundwater, P.; Morgan, J.J.; Mort, D.; Mason, M.; Evans, A. Stereo-Specific Cytotoxic Effects of Gossypol Enantiomers and Gossypolone in Tumour Cell Lines. Cancer Lett. 1999, 135, 171–180. [Google Scholar] [CrossRef]
- Coyle, T.; Levante, S.; Shetler, M.; Winfield, J. In Vitro and in Vivo Cytotoxicity of Gossypol against Central Nervous System Tumor Cell Lines. J. Neurooncol. 1994, 19, 25–35. [Google Scholar] [CrossRef]
- Gilbert, N.E.; O’Reilly, J.E.; Chang, C.J.G.; Lin, Y.C.; Brueggemeier, R.W. Antiproliferative Activity of Gossypol and Gossypolone on Human Breast Cancer Cells. Life Sci. 1995, 57, 61–67. [Google Scholar] [CrossRef]
- Volate, S.R.; Kawasaki, B.T.; Hurt, E.M.; Milner, J.A.; Kim, Y.S.; White, J.; Farrar, W.L. Gossypol Induces Apoptosis by Activating P53 in Prostate Cancer Cells and Prostate Tumor–Initiating Cells. Mol. Cancer 2010, 9, 461–470. [Google Scholar] [CrossRef]
- Lan, L.; Appelman, C.; Smith, A.R.; Yu, J.; Larsen, S.; Marquez, R.T.; Liu, H.; Wu, X.; Gao, P.; Roy, A.; et al. Natural Product (−)-Gossypol Inhibits Colon Cancer Cell Growth by Targeting RNA-Binding Protein Musashi-1. Mol. Oncol. 2015, 9, 1406–1420. [Google Scholar] [CrossRef]
- Wolter, K.G.; Wang, S.J.; Henson, B.S.; Wang, S.; Griffith, K.A.; Kumar, B.; Chen, J.; Carey, T.E.; Bradford, C.R.; D’Silva, N.J. (−)-Gossypol Inhibits Growth and Promotes Apoptosis of Human Head and Neck Squamous Cell Carcinoma In Vivo. Neoplasia 2006, 8, 163–172. [Google Scholar] [CrossRef]
- Jaroszewski, J.W.; Kaplan, O.; Cohen, J.S. Action of Gossypol and Rhodamine 123 on Wild Type and Multidrug-Resistant MCF-7 Human Breast Cancer Cells: 31P Nuclear Magnetic Resonance and Toxicity Studies. Cancer Res. 1990, 50, 6936–6943. [Google Scholar]
- Yu, Z.-H.; Chan, H.C. Gossypol as a Male Antifertility Agent—Why Studies Should Have Been Continued: Gossypol as a Male Infertility Agent. Int. J. Androl. 1998, 21, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.; Banerjee, D. Lactate Dehydrogenases as Metabolic Links between Tumor and Stroma in the Tumor Microenvironment. Cancers 2019, 11, 750. [Google Scholar] [CrossRef] [PubMed]
- Xian, Z.-Y.; Liu, J.-M.; Chen, Q.-K.; Chen, H.-Z.; Ye, C.-J.; Xue, J.; Yang, H.-Q.; Li, J.-L.; Liu, X.-F.; Kuang, S.-J. Inhibition of LDHA Suppresses Tumor Progression in Prostate Cancer. Tumor Biol. 2015, 36, 8093–8100. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, X.; Yu, Y.; Li, J.; Hu, Q.; Xue, C.; Chen, J.; Shen, S.; Luo, Y.; Ren, F.; et al. LDHA Is a Direct Target of MiR-30d-5p and Contributes to Aggressive Progression of Gallbladder Carcinoma. Mol. Carcinog. 2018, 57, 772–783. [Google Scholar] [CrossRef]
- Rellinger, E.J.; Craig, B.T.; Alvarez, A.L.; Dusek, H.L.; Kim, K.W.; Qiao, J.; Chung, D.H. FX11 Inhibits Aerobic Glycolysis and Growth of Neuroblastoma Cells. Surgery 2017, 161, 747–752. [Google Scholar] [CrossRef]
- Rani, R.; Kumar, V. Recent Update on Human Lactate Dehydrogenase Enzyme 5 (h LDH5) Inhibitors: A Promising Approach for Cancer Chemotherapy: Miniperspective. J. Med. Chem. 2016, 59, 487–496. [Google Scholar] [CrossRef]
- Billiard, J.; Dennison, J.B.; Briand, J.; Annan, R.S.; Chai, D.; Colón, M.; Dodson, C.S.; Gilbert, S.A.; Greshock, J.; Jing, J.; et al. Quinoline 3-Sulfonamides Inhibit Lactate Dehydrogenase A and Reverse Aerobic Glycolysis in Cancer Cells. Cancer Metab. 2013, 1, 19. [Google Scholar] [CrossRef]
- Granchi, C.; Roy, S.; De Simone, A.; Salvetti, I.; Tuccinardi, T.; Martinelli, A.; Macchia, M.; Lanza, M.; Betti, L.; Giannaccini, G.; et al. N-Hydroxyindole-Based Inhibitors of Lactate Dehydrogenase against Cancer Cell Proliferation. Eur. J. Med. Chem. 2011, 46, 5398–5407. [Google Scholar] [CrossRef]
- Granchi, C.; Roy, S.; Giacomelli, C.; Macchia, M.; Tuccinardi, T.; Martinelli, A.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; et al. Discovery of N-Hydroxyindole-Based Inhibitors of Human Lactate Dehydrogenase Isoform A (LDH-A) as Starvation Agents against Cancer Cells. J. Med. Chem. 2011, 54, 1599–1612. [Google Scholar] [CrossRef]
- Maftouh, M.; Avan, A.; Sciarrillo, R.; Granchi, C.; Leon, L.G.; Rani, R.; Funel, N.; Smid, K.; Honeywell, R.; Boggi, U.; et al. Synergistic Interaction of Novel Lactate Dehydrogenase Inhibitors with Gemcitabine against Pancreatic Cancer Cells in Hypoxia. Br. J. Cancer 2014, 110, 172–182. [Google Scholar] [CrossRef]
- Di Bussolo, V.; Calvaresi, E.C.; Granchi, C.; Del Bino, L.; Frau, I.; Dasso Lang, M.C.; Tuccinardi, T.; Macchia, M.; Martinelli, A.; Hergenrother, P.J.; et al. Synthesis and Biological Evaluation of Non-Glucose Glycoconjugated N-Hydroyxindole Class LDH Inhibitors as Anticancer Agents. RSC Adv. 2015, 5, 19944–19954. [Google Scholar] [CrossRef]
- Calvaresi, E.C.; Granchi, C.; Tuccinardi, T.; Di Bussolo, V.; Huigens, R.W.; Lee, H.Y.; Palchaudhuri, R.; Macchia, M.; Martinelli, A.; Minutolo, F.; et al. Dual Targeting of the Warburg Effect with a Glucose-Conjugated Lactate Dehydrogenase Inhibitor. ChemBioChem 2013, 14, 2263–2267. [Google Scholar] [CrossRef]
- Manerba, M.; Vettraino, M.; Fiume, L.; Di Stefano, G.; Sartini, A.; Giacomini, E.; Buonfiglio, R.; Roberti, M.; Recanatini, M. Galloflavin (CAS 568-80-9): A Novel Inhibitor of Lactate Dehydrogenase. ChemMedChem 2012, 7, 311–317. [Google Scholar] [CrossRef]
- Han, X.; Sheng, X.; Jones, H.M.; Jackson, A.L.; Kilgore, J.; Stine, J.E.; Schointuch, M.N.; Zhou, C.; Bae-Jump, V.L. Evaluation of the Anti-Tumor Effects of Lactate Dehydrogenase Inhibitor Galloflavin in Endometrial Cancer Cells. J. Hematol. Oncol. 2015, 8, 2. [Google Scholar] [CrossRef]
- Vettraino, M.; Manerba, M.; Govoni, M.; Di Stefano, G. Galloflavin Suppresses Lactate Dehydrogenase Activity and Causes MYC Downregulation in Burkitt Lymphoma Cells through NAD/NADH-Dependent Inhibition of Sirtuin-1. Anticancer Drugs 2013, 24, 862–870. [Google Scholar] [CrossRef]
- Farabegoli, F.; Vettraino, M.; Manerba, M.; Fiume, L.; Roberti, M.; Di Stefano, G. Galloflavin, a New Lactate Dehydrogenase Inhibitor, Induces the Death of Human Breast Cancer Cells with Different Glycolytic Attitude by Affecting Distinct Signaling Pathways. Eur. J. Pharm. Sci. 2012, 47, 729–738. [Google Scholar] [CrossRef]
- Manerba, M.; Di Ianni, L.; Govoni, M.; Roberti, M.; Recanatini, M.; Di Stefano, G. LDH Inhibition Impacts on Heat Shock Response and Induces Senescence of Hepatocellular Carcinoma Cells. Eur. J. Pharm. Sci. 2017, 105, 91–98. [Google Scholar] [CrossRef]
- Wendt, E.H.U.; Schoenrogge, M.; Vollmar, B.; Zechner, D. Galloflavin Plus Metformin Treatment Impairs Pancreatic Cancer Cells. Anticancer Res. 2020, 40, 153–160. [Google Scholar] [CrossRef]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour Hypoxia Induces a Metabolic Shift Causing Acidosis: A Common Feature in Cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Y.; Dong, D.; Wang, F.; Ma, X.; Guan, F.; Sun, L. MCT1 Regulates Aggressive and Metabolic Phenotypes in Bladder Cancer. J. Cancer 2018, 9, 2492–2501. [Google Scholar] [CrossRef]
- Choi, S.Y.C.; Ettinger, S.L.; Lin, D.; Xue, H.; Ci, X.; Nabavi, N.; Bell, R.H.; Mo, F.; Gout, P.W.; Fleshner, N.E.; et al. Targeting MCT 4 to Reduce Lactic Acid Secretion and Glycolysis for Treatment of Neuroendocrine Prostate Cancer. Cancer Med. 2018, 7, 3385–3392. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Santos, C.R.; Spencer-Dene, B.; Martinez, P.; Endesfelder, D.; Burrell, R.A.; Vetter, M.; Jiang, M.; Saunders, R.E.; Kelly, G.; et al. Genome-wide RNA Interference Analysis of Renal Carcinoma Survival Regulators Identifies MCT4 as a Warburg Effect Metabolic Target. J. Pathol. 2012, 227, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Curtis, N.J.; Mooney, L.; Hopcroft, L.; Michopoulos, F.; Whalley, N.; Zhong, H.; Murray, C.; Logie, A.; Revill, M.; Byth, K.F.; et al. Pre-Clinical Pharmacology of AZD3965, a Selective Inhibitor of MCT1: DLBCL, NHL and Burkitt’s Lymphoma Anti-Tumor Activity. Oncotarget 2017, 8, 69219–69236. [Google Scholar] [CrossRef] [PubMed]
- Quanz, M.; Bender, E.; Kopitz, C.; Grünewald, S.; Schlicker, A.; Schwede, W.; Eheim, A.; Toschi, L.; Neuhaus, R.; Richter, C.; et al. Preclinical Efficacy of the Novel Monocarboxylate Transporter 1 Inhibitor BAY-8002 and Associated Markers of Resistance. Mol. Cancer 2018, 17, 2285–2296. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Li, G.; Bao, Z.; Zhou, Z.; Li, L. Mitochondrial Pyruvate Carrier 1: A Novel Prognostic Biomarker That Predicts Favourable Patient Survival in Cancer. Cancer Cell Int. 2021, 21, 288. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Raemy, E.; Montessuit, S.; Veuthey, J.-L.; Zamboni, N.; Westermann, B.; Kunji, E.R.S.; Martinou, J.-C. Identification and Functional Expression of the Mitochondrial Pyruvate Carrier. Science 2012, 337, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Yonashiro, R.; Eguchi, K.; Wake, M.; Takeda, N.; Nakayama, K. Pyruvate Dehydrogenase PDH-E1β Controls Tumor Progression by Altering the Metabolic Status of Cancer Cells. Cancer Res. 2018, 78, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Shan, C.; Kang, H.-B.; Elf, S.; Xie, J.; Tucker, M.; Gu, T.-L.; Aguiar, M.; Lonning, S.; Chen, H.; et al. Tyr Phosphorylation of PDP1 Toggles Recruitment between ACAT1 and SIRT3 to Regulate the Pyruvate Dehydrogenase Complex. Mol. Cell 2014, 53, 534–548. [Google Scholar] [CrossRef]
- Jin, L.; Kim, E.-Y.; Chung, T.-W.; Han, C.W.; Park, S.Y.; Han, J.H.; Bae, S.-J.; Lee, J.R.; Kim, Y.W.; Jang, S.B.; et al. Hemistepsin A Suppresses Colorectal Cancer Growth through Inhibiting Pyruvate Dehydrogenase Kinase Activity. Sci. Rep. 2020, 10, 21940. [Google Scholar] [CrossRef]
- Korga, A.; Ostrowska, M.; Iwan, M.; Herbet, M.; Dudka, J. Inhibition of Glycolysis Disrupts Cellular Antioxidant Defense and Sensitizes HepG2 Cells to Doxorubicin Treatment. FEBS Open Bio 2019, 9, 959–972. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, A.; Zhou, X.; Wang, F. Suppression of Pyruvate Dehydrogenase Kinase-2 Re-Sensitizes Paclitaxel-Resistant Human Lung Cancer Cells to Paclitaxel. Oncotarget 2017, 8, 52642–52650. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Choudhary, D.; Mikhalyuk, A.; Trammel, C.; Shanmugam, S.; Abbott, E.; Pilbeam, C.C.; Taylor, J.A. The Role of Pyruvate Dehydrogenase Kinase-4 (PDK4) in Bladder Cancer and Chemoresistance. Mol. Cancer 2018, 17, 2004–2012. [Google Scholar] [CrossRef]
- Tataranni, T.; Piccoli, C. Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications. Oxid. Med. Cell. Longev. 2019, 2019, 8201079. [Google Scholar] [CrossRef]
- Kissling, G.E.; Malarkey, D.E.; Vallant, M.K.; Johnson, J.D.; Hejtmancik, M.R.; Herbert, R.A.; Boorman, G.A. Evaluation of Dichloroacetic Acid for Carcinogenicity in Genetically Modified Tg.AC Hemizygous and P53 Haploinsufficient Mice. Toxicol. Sci. 2009, 107, 19–26. [Google Scholar] [CrossRef]
- Xu, C.; Yang, H.; Xiao, Z.; Zhang, T.; Guan, Z.; Chen, J.; Lai, H.; Xu, X.; Huang, Y.; Huang, Z.; et al. Reduction-Responsive Dehydroepiandrosterone Prodrug Nanoparticles Loaded with Camptothecin for Cancer Therapy by Enhancing Oxidation Therapy and Cell Replication Inhibition. Int. J. Pharm. 2021, 603, 120671. [Google Scholar] [CrossRef]
- Lucarelli, G.; Galleggiante, V.; Rutigliano, M.; Sanguedolce, F.; Cagiano, S.; Bufo, P.; Lastilla, G.; Maiorano, E.; Ribatti, D.; Giglio, A.; et al. Metabolomic Profile of Glycolysis and the Pentose Phosphate Pathway Identifies the Central Role of Glucose-6-Phosphate Dehydrogenase in Clear Cell-Renal Cell Carcinoma. Oncotarget 2015, 6, 13371–13386. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Amelio, I.; Berkers, C.R.; Antonov, A.; Vousden, K.H.; Melino, G.; Zolla, L. Metabolic Effect of TAp63α: Enhanced Glycolysis and Pentose Phosphate Pathway, Resulting in Increased Antioxidant Defense. Oncotarget 2014, 5, 7722–7733. [Google Scholar] [CrossRef][Green Version]
- Jiang, P.; Du, W.; Wu, M. Regulation of the Pentose Phosphate Pathway in Cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef]
- Mele, L.; Paino, F.; Papaccio, F.; Regad, T.; Boocock, D.; Stiuso, P.; Lombardi, A.; Liccardo, D.; Aquino, G.; Barbieri, A.; et al. A New Inhibitor of Glucose-6-Phosphate Dehydrogenase Blocks Pentose Phosphate Pathway and Suppresses Malignant Proliferation and Metastasis in Vivo. Cell Death Dis. 2018, 9, 572. [Google Scholar] [CrossRef]
- Gordon, G.; Mackow, M.C.; Levy, H.R. On the Mechanism of Interaction of Steroids with Human Glucose 6-Phosphate Dehydrogenase. Arch. Biochem. Biophys. 1995, 318, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Jiang, C.; Feng, Y.; Chen, R.; Lin, X.; Zhang, Z.; Han, L.; Chen, X.; Li, H.; Guo, Y.; et al. Effects of G6PD Activity Inhibition on the Viability, ROS Generation and Mechanical Properties of Cervical Cancer Cells. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 2245–2254. [Google Scholar] [CrossRef]
- Ho, H.-Y.; Cheng, M.-L.; Chiu, H.-Y.; Weng, S.-F.; Chiu, D.T.-Y. Dehydroepiandrosterone Induces Growth Arrest of Hepatoma Cells via Alteration of Mitochondrial Gene Expression and Function. Int. J. Oncol. 2008, 33, 969–977. [Google Scholar] [PubMed]
- Di Monaco, M.; Pizzini, A.; Gatto, V.; Leonardi, L.; Gallo, M.; Brignardello, E.; Boccuzzi, G. Role of Glucose-6-Phosphate Dehydrogenase Inhibition in the Antiproliferative Effects of Dehydroepiandrosterone on Human Breast Cancer Cells. Br. J. Cancer 1997, 75, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.; Dwarakanath, B.; Jain, V. Radiosensitization by 6-Aminonicotinamide and 2-Deoxy-D-Glucose in Human Cancer Cells. Int. J. Radiat. Biol. 2005, 81, 397–408. [Google Scholar] [CrossRef]
- Catanzaro, D.; Gaude, E.; Orso, G.; Giordano, C.; Guzzo, G.; Rasola, A.; Ragazzi, E.; Caparrotta, L.; Frezza, C.; Montopoli, M. Inhibition of Glucose-6-Phosphate Dehydrogenase Sensitizes Cisplatin-Resistant Cells to Death. Oncotarget 2015, 6, 30102–30114. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, W.; Zhang, Y.; Wu, S.; Liu, Y.; Deng, X.; Xie, L.; Yang, J.; Yu, H.; Su, J.; et al. ABT737 Reverses Cisplatin Resistance by Targeting Glucose Metabolism of Human Ovarian Cancer Cells. Int. J. Oncol. 2018, 53, 1055–1068. [Google Scholar] [CrossRef]
- Hong, W.; Cai, P.; Xu, C.; Cao, D.; Yu, W.; Zhao, Z.; Huang, M.; Jin, J. Inhibition of Glucose-6-Phosphate Dehydrogenase Reverses Cisplatin Resistance in Lung Cancer Cells via the Redox System. Front. Pharmacol. 2018, 9, 43. [Google Scholar] [CrossRef]
- Chen, X.; Xu, Z.; Zhu, Z.; Chen, A.; Fu, G.; Wang, Y.; Pan, H.; Jin, B. Modulation of G6PD Affects Bladder Cancer via ROS Accumulation and the AKT Pathway in Vitro. Int. J. Oncol. 2018, 53, 1703–1712. [Google Scholar] [CrossRef]
- Arbe, M.F.; Agnetti, L.; Breininger, E.; Glikin, G.C.; Finocchiaro, L.M.E.; Villaverde, M.S. Glucose 6-Phosphate Dehydrogenase Inhibition Sensitizes Melanoma Cells to Metformin Treatment. Transl. Oncol. 2020, 13, 100842. [Google Scholar] [CrossRef]
- Giacomini, I.; Ragazzi, E.; Pasut, G.; Montopoli, M. The Pentose Phosphate Pathway and Its Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 937. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, S.; Zhang, Y.; Wu, J.; Peng, H.; Fan, J.; Liao, J. Reactive Oxygen Species-Mediated Endoplasmic Reticulum Stress and Mitochondrial Dysfunction Contribute to Polydatin-Induced Apoptosis in Human Nasopharyngeal Carcinoma CNE Cells. J. Cell. Biochem. 2011, 112, 3695–3703. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Wu, Y.; Du, D. Polydatin Inhibits Cell Proliferation, Invasion and Migration, and Induces Cell Apoptosis in Hepatocellular Carcinoma. Braz. J. Med. Biol. Res. 2018, 51, e6867. [Google Scholar] [CrossRef]
- Wang, C.; Luo, Y.; Lu, J.; Wang, Y.; Sheng, G. Polydatin Induces Apoptosis and Inhibits Growth of Acute Monocytic Leukemia Cells: EFFECT OF PD ON THP-1 CELLS. J. Biochem. Mol. Toxicol. 2016, 30, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhuang, Z.; Meng, Q.; Jiao, Y.; Xu, J.; Fan, S. Polydatin Inhibits Growth of Lung Cancer Cells by Inducing Apoptosis and Causing Cell Cycle Arrest. Oncol. Lett. 2014, 7, 295–301. [Google Scholar] [CrossRef]
- Cremon, C.; Stanghellini, V.; Barbaro, M.R.; Cogliandro, R.F.; Bellacosa, L.; Santos, J.; Vicario, M.; Pigrau, M.; Alonso Cotoner, C.; Lobo, B.; et al. Randomised Clinical Trial: The Analgesic Properties of Dietary Supplementation with Palmitoylethanolamide and Polydatin in Irritable Bowel Syndrome. Aliment. Pharmacol. Ther. 2017, 45, 909–922. [Google Scholar] [CrossRef]
- Indraccolo, U.; Indraccolo, S.R.; Mignini, F. Micronized Palmitoylethanolamide/Trans-Polydatin Treatment of Endometriosis-Related Pain: A Meta-Analysis. Ann. Ist. Super. Sanita 2017, 53, 125–134. [Google Scholar] [CrossRef]
- Wang, X.; Wu, G.; Cao, G.; Yang, L.; Xu, H.; Huang, J.; Hou, J. Zoledronic Acid Inhibits the Pentose Phosphate Pathway through Attenuating the Ras-TAp73-G6PD Axis in Bladder Cancer Cells. Mol. Med. Rep. 2015, 12, 4620–4625. [Google Scholar] [CrossRef]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The Emerging Role and Targetability of the TCA Cycle in Cancer Metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Kang, W.; Suzuki, M.; Saito, T.; Miyado, K. Emerging Role of TCA Cycle-Related Enzymes in Human Diseases. Int. J. Mol. Sci. 2021, 22, 13057. [Google Scholar] [CrossRef]
- Cardaci, S.; Ciriolo, M.R. TCA Cycle Defects and Cancer: When Metabolism Tunes Redox State. Int. J. Cell Biol. 2012, 2012, 1–9. [Google Scholar] [CrossRef]
- Griss, T.; Vincent, E.E.; Egnatchik, R.; Chen, J.; Ma, E.H.; Faubert, B.; Viollet, B.; DeBerardinis, R.J.; Jones, R.G. Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biol. 2015, 13, e1002309. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.-M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin Suppresses Gluconeogenesis by Inhibiting Mitochondrial Glycerophosphate Dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef]
- Andrzejewski, S.; Gravel, S.-P.; Pollak, M.; St-Pierre, J. Metformin Directly Acts on Mitochondria to Alter Cellular Bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef]
- Gasmi, A.; Peana, M.; Arshad, M.; Butnariu, M.; Menzel, A.; Bjørklund, G. Krebs Cycle: Activators, Inhibitors and Their Roles in the Modulation of Carcinogenesis. Arch. Toxicol. 2021, 95, 1161–1178. [Google Scholar] [CrossRef]
- Strydom, C.; Robinson, C.; Pretorius, E.; Whitcutt, J.M.; Marx, J.; Bornman, M.S. The Effect of Selected Metals on the Central Metabolic Pathways in Biology: A Review. Water SA 2006, 32, 543–554. [Google Scholar] [CrossRef]
- Dejure, F.R.; Royla, N.; Herold, S.; Kalb, J.; Walz, S.; Ade, C.P.; Mastrobuoni, G.; Vanselow, J.T.; Schlosser, A.; Wolf, E.; et al. The MYC MRNA 3′-UTR Couples RNA Polymerase II Function to Glutamine and Ribonucleotide Levels. EMBO J. 2017, 36, 1854–1868. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and Cancer: Cell Biology, Physiology, and Clinical Opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Brunengraber, H.; Roe, C.R. Anaplerotic Molecules: Current and Future. J. Inherit. Metab. Dis. 2006, 29, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Bröer, A.; Rahimi, F.; Bröer, S. Deletion of Amino Acid Transporter ASCT2 (SLC1A5) Reveals an Essential Role for Transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to Sustain Glutaminolysis in Cancer Cells. J. Biol. Chem. 2016, 291, 13194–13205. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. C-Myc Suppression of MiR-23a/b Enhances Mitochondrial Glutaminase Expression and Glutamine Metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, G.; Mao, Q.; Li, S.; Xiong, W.; Lin, Y.; Ge, J. Glutamate Dehydrogenase (GDH) Regulates Bioenergetics and Redox Homeostasis in Human Glioma. Oncotarget 2016, 5, 1–12. [Google Scholar] [CrossRef]
- Li, C.; Li, M.; Chen, P.; Narayan, S.; Matschinsky, F.M.; Bennett, M.J.; Stanley, C.A.; Smith, T.J. Green Tea Polyphenols Control Dysregulated Glutamate Dehydrogenase in Transgenic Mice by Hijacking the ADP Activation Site. J. Biol. Chem. 2011, 286, 34164–34174. [Google Scholar] [CrossRef]
- Jin, L.; Li, D.; Alesi, G.N.; Fan, J.; Kang, H.-B.; Lu, Z.; Boggon, T.J.; Jin, P.; Yi, H.; Wright, E.R.; et al. Glutamate Dehydrogenase 1 Signals through Antioxidant Glutathione Peroxidase 1 to Regulate Redox Homeostasis and Tumor Growth. Cancer Cell 2015, 27, 257–270. [Google Scholar] [CrossRef]
- Ren, J.-G.; Seth, P.; Ye, H.; Guo, K.; Hanai, J.; Husain, Z.; Sukhatme, V.P. Citrate Suppresses Tumor Growth in Multiple Models through Inhibition of Glycolysis, the Tricarboxylic Acid Cycle and the IGF-1R Pathway. Sci. Rep. 2017, 7, 4537. [Google Scholar] [CrossRef]
- Esslinger, C.S.; Cybulski, K.A.; Rhoderick, J.F. Nγ-Aryl Glutamine Analogues as Probes of the ASCT2 Neutral Amino Acid Transporter Binding Site. Bioorg. Med. Chem. 2005, 13, 1111–1118. [Google Scholar] [CrossRef]
- Fuchs, B.C.; Bode, B.P. Amino Acid Transporters ASCT2 and LAT1 in Cancer: Partners in Crime? Semin. Cancer Biol. 2005, 15, 254–266. [Google Scholar] [CrossRef]
- Hassanein, M.; Hoeksema, M.D.; Shiota, M.; Qian, J.; Harris, B.K.; Chen, H.; Clark, J.E.; Alborn, W.E.; Eisenberg, R.; Massion, P.P. SLC1A5 Mediates Glutamine Transport Required for Lung Cancer Cell Growth and Survival. Clin. Cancer Res. 2013, 19, 560–570. [Google Scholar] [CrossRef]
- Hassanein, M.; Qian, J.; Hoeksema, M.D.; Wang, J.; Jacobovitz, M.; Ji, X.; Harris, F.T.; Harris, B.K.; Boyd, K.L.; Chen, H.; et al. Targeting SLC1a5-Mediated Glutamine Dependence in Non-Small Cell Lung Cancer: Targeting SLC1A5 in Lung Cancer. Int. J. Cancer 2015, 137, 1587–1597. [Google Scholar] [CrossRef]
- Ren, P.; Yue, M.; Xiao, D.; Xiu, R.; Gan, L.; Liu, H.; Qing, G. ATF4 and N-Myc Coordinate Glutamine Metabolism in MYCN-Amplified Neuroblastoma Cells through ASCT2 Activation. J. Pathol. 2015, 235, 90–100. [Google Scholar] [CrossRef]
- Wang, Q.; Hardie, R.; Hoy, A.J.; van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated Glutamine Uptake Blocks Prostate Cancer Growth and Tumour Development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef]
- Bolzoni, M.; Chiu, M.; Accardi, F.; Vescovini, R.; Airoldi, I.; Storti, P.; Todoerti, K.; Agnelli, L.; Missale, G.; Andreoli, R.; et al. Dependence on Glutamine Uptake and Glutamine Addiction Characterize Myeloma Cells: A New Attractive Target. Blood 2016, 128, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 Controls Glutamine Uptake and Tumour Growth in Triple-Negative Basal-like Breast Cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.D.; van Geldermalsen, M.; Otte, N.J.; Lum, T.; Vellozzi, M.; Thoeng, A.; Pang, A.; Nagarajah, R.; Zhang, B.; Wang, Q.; et al. ASCT2 Regulates Glutamine Uptake and Cell Growth in Endometrial Carcinoma. Oncogenesis 2017, 6, e367. [Google Scholar] [CrossRef] [PubMed]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological Blockade of ASCT2-Dependent Glutamine Transport Leads to Antitumor Efficacy in Preclinical Models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Bröer, A.; Fairweather, S.; Bröer, S. Disruption of Amino Acid Homeostasis by Novel ASCT2 Inhibitors Involves Multiple Targets. Front. Pharmacol. 2018, 9, 785. [Google Scholar] [CrossRef]
- Grewer, C.; Grabsch, E. New Inhibitors for the Neutral Amino Acid Transporter ASCT2 Reveal Its Na+-Dependent Anion Leak: Na+-Dependent ASCT2 Leak Anion Conductance. J. Physiol. 2004, 557, 747–759. [Google Scholar] [CrossRef]
- Van Geldermalsen, M.; Quek, L.-E.; Turner, N.; Freidman, N.; Pang, A.; Guan, Y.F.; Krycer, J.R.; Ryan, R.; Wang, Q.; Holst, J. Benzylserine Inhibits Breast Cancer Cell Growth by Disrupting Intracellular Amino Acid Homeostasis and Triggering Amino Acid Response Pathways. BMC Cancer 2018, 18, 689. [Google Scholar] [CrossRef]
- Sun, H.-J.; Meng, L.-Y.; Shen, Y.; Zhu, Y.-Z.; Liu, H.-R. S-Benzyl-Cysteine-Mediated Cell Cycle Arrest and Apoptosis Involving Activation of Mitochondrial-Dependent Caspase Cascade through the P53 Pathway in Human Gastric Cancer SGC-7901 Cells. Asian Pac. J. Cancer Prev. 2013, 14, 6379–6384. [Google Scholar] [CrossRef]
- Lemberg, K.M.; Vornov, J.J.; Rais, R.; Slusher, B.S. We’re Not “DON” Yet: Optimal Dosing and Prodrug Delivery of 6-Diazo-5-Oxo-L-Norleucine. Mol. Cancer 2018, 17, 1824–1832. [Google Scholar] [CrossRef]
- Thomas, A.G.; Rojas, C.; Tanega, C.; Shen, M.; Simeonov, A.; Boxer, M.B.; Auld, D.S.; Ferraris, D.V.; Tsukamoto, T.; Slusher, B.S. Kinetic Characterization of Ebselen, Chelerythrine and Apomorphine as Glutaminase Inhibitors. Biochem. Biophys. Res. Commun. 2013, 438, 243–248. [Google Scholar] [CrossRef]
- Rubin, J.; Sorensen, S.; Schutt, A.J.; van Hazel, G.A.; O’Connell, M.J.; Moertel, C.G. A Phase II Study of 6-Diazo-5-Oxo-L-Norleucine (DON, NSC-7365) in Advanced Large Bowel Carcinoma. Am. J. Clin. Oncol. 1983, 6, 325–326. [Google Scholar] [CrossRef]
- Coffey, G.L.; Ehrlich, J.; Fisher, M.W.; Hillegas, A.B.; Kohberger, D.L.; Machamer, H.E.; Rightsel, W.A.; Roegner, F.R. 6-Diazo-5-Oxo-L-Norleucine, a New Tumor-Inhibitory Substance. I. Biologic Studies. Antibiot. Chemother. 1956, 6, 487–497. [Google Scholar]
- Eagan, R.T.; Frytak, S.; Nichols, W.C.; Creagan, E.T.; Ingle, J.N. Phase II Study of DON in Patients with Previously Treated Advanced Lung Cancer. J. Natl. Cancer Inst. 1982, 66, 1665–1666. [Google Scholar]
- Tanaka, K.; Sasayama, T.; Irino, Y.; Takata, K.; Nagashima, H.; Satoh, N.; Kyotani, K.; Mizowaki, T.; Imahori, T.; Ejima, Y.; et al. Compensatory Glutamine Metabolism Promotes Glioblastoma Resistance to MTOR Inhibitor Treatment. J. Clin. Investig. 2015, 125, 1591–1602. [Google Scholar] [CrossRef]
- Hanaford, A.R.; Alt, J.; Rais, R.; Wang, S.Z.; Kaur, H.; Thorek, D.L.J.; Eberhart, C.G.; Slusher, B.S.; Martin, A.M.; Raabe, E.H. Orally Bioavailable Glutamine Antagonist Prodrug JHU-083 Penetrates Mouse Brain and Suppresses the Growth of MYC-Driven Medulloblastoma. Transl. Oncol. 2019, 12, 1314–1322. [Google Scholar] [CrossRef]
- Nedelcovych, M.T.; Tenora, L.; Kim, B.-H.; Kelschenbach, J.; Chao, W.; Hadas, E.; Jančařík, A.; Prchalová, E.; Zimmermann, S.C.; Dash, R.P.; et al. N-(Pivaloyloxy)Alkoxy-Carbonyl Prodrugs of the Glutamine Antagonist 6-Diazo-5-Oxo-l-Norleucine (DON) as a Potential Treatment for HIV Associated Neurocognitive Disorders. J. Med. Chem. 2017, 60, 7186–7198. [Google Scholar] [CrossRef]
- Ramachandran, S.; Pan, C.Q.; Zimmermann, S.C.; Duvall, B.; Tsukamoto, T.; Low, B.C.; Sivaraman, J. Structural Basis for Exploring the Allosteric Inhibition of Human Kidney Type Glutaminase. Oncotarget 2016, 7, 57943–57954. [Google Scholar] [CrossRef]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of Glutaminase Preferentially Slows Growth of Glioma Cells with Mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B-Cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine Supports Pancreatic Cancer Growth through a KRAS-Regulated Metabolic Pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Qie, S.; Chu, C.; Li, W.; Wang, C.; Sang, N. ErbB2 Activation Upregulates Glutaminase 1 Expression Which Promotes Breast Cancer Cell Proliferation. J. Cell. Biochem. 2014, 115, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel, A.P.J.; Jing, J.; Wooster, R.F.; Bachman, K.E. Analysis of Glutamine Dependency in Non-Small Cell Lung Cancer: GLS1 Splice Variant GAC Is Essential for Cancer Cell Growth. Cancer Biol. Ther. 2012, 13, 1185–1194. [Google Scholar] [CrossRef]
- Robinson, M.M.; McBryant, S.J.; Tsukamoto, T.; Rojas, C.; Ferraris, D.V.; Hamilton, S.K.; Hansen, J.C.; Curthoys, N.P. Novel Mechanism of Inhibition of Rat Kidney-Type Glutaminase by Bis-2-(5-Phenylacetamido-1,2,4-Thiadiazol-2-Yl)Ethyl Sulfide (BPTES). Biochem. J. 2007, 406, 407–414. [Google Scholar] [CrossRef]
- Xu, X.; Meng, Y.; Li, L.; Xu, P.; Wang, J.; Li, Z.; Bian, J. Overview of the Development of Glutaminase Inhibitors: Achievements and Future Directions. J. Med. Chem. 2019, 62, 1096–1115. [Google Scholar] [CrossRef]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a Target for Cancer Therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef]
- Varghese, S.; Pramanik, S.; Williams, L.J.; Hodges, H.R.; Hudgens, C.W.; Fischer, G.M.; Luo, C.K.; Knighton, B.; Tan, L.; Lorenzi, P.L.; et al. The Glutaminase Inhibitor CB-839 (Telaglenastat) Enhances the Antimelanoma Activity of T-Cell-Mediated Immunotherapies. Mol. Cancer 2021, 20, 500–511. [Google Scholar] [CrossRef]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer 2014, 13, 890–901. [Google Scholar] [CrossRef]
- Ruan, J.J.; Yu, Y.; Hou, W.; Chen, Z.; Fang, J.; Zhang, J.; Ni, M.; Li, D.; Lu, S.; Rui, J.; et al. Kidney-Type Glutaminase Inhibitor Hexylselen Selectively Kills Cancer Cells via a Three-Pronged Mechanism. ACS Pharmacol. Transl. Sci. 2019, 2, 18–30. [Google Scholar] [CrossRef]
- Gregory, M.A.; Nemkov, T.; Park, H.J.; Zaberezhnyy, V.; Gehrke, S.; Adane, B.; Jordan, C.T.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin. Cancer Res. 2019, 25, 4079–4090. [Google Scholar] [CrossRef]
- Matre, P.; Shariati, M.; Velez, J.; Qi, Y.; Konoplev, S.; Su, X.; DiNardo, C.D.; Daver, N.; Majeti, R.; Andreeff, M.; et al. Efficacy of Novel Glutaminase Inhibitor CB-839 in Acute Myeloid Leukemia. Blood 2014, 124, 3763. [Google Scholar] [CrossRef]
- Shah, R.; Chen, S. Metabolic Signaling Cascades Prompted by Glutaminolysis in Cancer. Cancers 2020, 12, 2624. [Google Scholar] [CrossRef]
- Xie, C.; Jin, J.; Bao, X.; Zhan, W.-H.; Han, T.-Y.; Gan, M.; Zhang, C.; Wang, J. Inhibition of Mitochondrial Glutaminase Activity Reverses Acquired Erlotinib Resistance in Non-Small Cell Lung Cancer. Oncotarget 2015, 7, 610–621. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Cheng, M.; Koch, K.; Marchionni, L.; Fan, X.; Raabe, E.H.; Maciaczyk, J.; Glunde, K.; Eberhart, C.G. Alterations in Cellular Metabolome after Pharmacological Inhibition of Notch in Glioblastoma Cells. Int. J. Cancer 2016, 138, 1246–1255. [Google Scholar] [CrossRef]
- Simpson, N.E.; Tryndyak, V.P.; Beland, F.A.; Pogribny, I.P. An in Vitro Investigation of Metabolically Sensitive Biomarkers in Breast Cancer Progression. Breast Cancer Res. Treat. 2012, 133, 959–968. [Google Scholar] [CrossRef]
- Yuan, L.; Sheng, X.; Clark, L.H.; Zhang, L.; Guo, H.; Jones, H.M.; Willson, A.K.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. Glutaminase Inhibitor Compound 968 Inhibits Cell Proliferation and Sensitizes Paclitaxel in Ovarian Cancer. Am. J. Transl. Res. 2016, 8, 4265–4277. [Google Scholar]
- Koundouros, N.; Poulogiannis, G. Reprogramming of Fatty Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty Acid Oxidation: An Emerging Facet of Metabolic Transformation in Cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef]
- Mozolewska, P.; Duzowska, K.; Pakiet, A.; Mika, A.; Śledziński, T. Inhibitors of Fatty Acid Synthesis and Oxidation as Potential Anticancer Agents in Colorectal Cancer Treatment. Anticancer Res. 2020, 40, 4843–4856. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer Metabolism: Fatty Acid Oxidation in the Limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijón, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glodé, L.M.; Eckel, R.H.; et al. Lipid Catabolism via CPT1 as a Therapeutic Target for Prostate Cancer. Mol. Cancer 2014, 13, 2361–2371. [Google Scholar] [CrossRef]
- Fritz, I.B.; Schultz, S.K.; Srere, P.A. Properties of Partially Purified Carnitine Acetyltransferase. J. Biol. Chem. 1963, 238, 2509–2517. [Google Scholar] [CrossRef]
- Murthy, M.S.; Pande, S.V. Malonyl-CoA Binding Site and the Overt Carnitine Palmitoyltransferase Activity Reside on the Opposite Sides of the Outer Mitochondrial Membrane. Proc. Natl. Acad. Sci. USA 1987, 84, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.W. The Role of the Carnitine System in Human Metabolism. Ann. N. Y. Acad. Sci. 2004, 1033, 1–16. [Google Scholar] [CrossRef]
- Bristow, M. Etomoxir: A New Approach to Treatment of Chronic Heart Failure. Lancet 2000, 356, 1621–1622. [Google Scholar] [CrossRef]
- Ratheiser, K.; Schneeweiß, B.; Waldhäusl, W.; Fasching, P.; Korn, A.; Nowotny, P.; Rohac, M.; Wolf, H.P.O. Inhibition by Etomoxir of Carnitine Palmitoyltransferase I Reduces Hepatic Glucose Production and Plasma Lipids in Non-Insulin-Dependent Diabetes Mellitus. Metab. Clin. Exp. 1991, 40, 1185–1190. [Google Scholar] [CrossRef]
- Holubarsch, C.J.F.; Rohrbach, M.; Karrasch, M.; Boehm, E.; Polonski, L.; Ponikowski, P.; Rhein, S. A Double-Blind Randomized Multicentre Clinical Trial to Evaluate the Efficacy and Safety of Two Doses of Etomoxir in Comparison with Placebo in Patients with Moderate Congestive Heart Failure: The ERGO (Etomoxir for the Recovery of Glucose Oxidation) Study. Clin. Sci. 2007, 113, 205–212. [Google Scholar] [CrossRef]
- Tan, Z.; Xiao, L.; Tang, M.; Bai, F.; Li, J.; Li, L.; Shi, F.; Li, N.; Li, Y.; Du, Q.; et al. Targeting CPT1A-Mediated Fatty Acid Oxidation Sensitizes Nasopharyngeal Carcinoma to Radiation Therapy. Theranostics 2018, 8, 2329–2347. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic Inhibition of Fatty Acid Oxidation Sensitizes Human Leukemia Cells to Apoptosis Induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef]
- Jariwala, N.; Mehta, G.A.; Bhatt, V.; Hussein, S.; Parker, K.A.; Yunus, N.; Parker, J.S.; Guo, J.Y.; Gatza, M.L. CPT1A and Fatty Acid β-Oxidation Are Essential for Tumor Cell Growth and Survival in Hormone Receptor-Positive Breast Cancer. NAR Cancer 2021, 3, zcab035. [Google Scholar] [CrossRef] [PubMed]
- Giannessi, F.; Pessotto, P.; Tassoni, E.; Chiodi, P.; Conti, R.; De Angelis, F.; Dell’Uomo, N.; Catini, R.; Deias, R.; Tinti, M.O.; et al. Discovery of a Long-Chain Carbamoyl Aminocarnitine Derivative, a Reversible Carnitine Palmitoyltransferase Inhibitor with Antiketotic and Antidiabetic Activity. J. Med. Chem. 2003, 46, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Rufer, A.C.; Thoma, R.; Benz, J.; Stihle, M.; Gsell, B.; De Roo, E.; Banner, D.W.; Mueller, F.; Chomienne, O.; Hennig, M. The Crystal Structure of Carnitine Palmitoyltransferase 2 and Implications for Diabetes Treatment. Structure 2006, 14, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Pacilli, A.; Calienni, M.; Margarucci, S.; D’Apolito, M.; Petillo, O.; Rocchi, L.; Pasquinelli, G.; Nicolai, R.; Koverech, A.; Calvani, M.; et al. Carnitine-Acyltransferase System Inhibition, Cancer Cell Death, and Prevention of Myc-Induced Lymphomagenesis. J. Natl. Cancer Inst. 2013, 105, 489–498. [Google Scholar] [CrossRef]
- Ricciardi, M.R.; Mirabilii, S.; Allegretti, M.; Licchetta, R.; Calarco, A.; Torrisi, M.R.; Foà, R.; Nicolai, R.; Peluso, G.; Tafuri, A. Targeting the Leukemia Cell Metabolism by the CPT1a Inhibition: Functional Preclinical Effects in Leukemias. Blood 2015, 126, 1925–1929. [Google Scholar] [CrossRef]
- Gugiatti, E.; Tenca, C.; Ravera, S.; Fabbi, M.; Ghiotto, F.; Mazzarello, A.N.; Bagnara, D.; Reverberi, D.; Zarcone, D.; Cutrona, G.; et al. A Reversible Carnitine Palmitoyltransferase (CPT1) Inhibitor Offsets the Proliferation of Chronic Lymphocytic Leukemia Cells. Haematologica 2018, 103, e531–e536. [Google Scholar] [CrossRef]
- Mao, S.; Ling, Q.; Pan, J.; Li, F.; Huang, S.; Ye, W.; Wei, W.; Lin, X.; Qian, Y.; Wang, Y.; et al. Inhibition of CPT1a as a Prognostic Marker Can Synergistically Enhance the Antileukemic Activity of ABT199. J. Transl. Med. 2021, 19, 181. [Google Scholar] [CrossRef]
- Keung, W.; Ussher, J.R.; Jaswal, J.S.; Raubenheimer, M.; Lam, V.H.M.; Wagg, C.S.; Lopaschuk, G.D. Inhibition of Carnitine Palmitoyltransferase-1 Activity Alleviates Insulin Resistance in Diet-Induced Obese Mice. Diabetes 2013, 62, 711–720. [Google Scholar] [CrossRef]
- Mascagna, D.; Ghanem, G.; Morandini, R.; d’Ischia, M.; Misuraca, G.; Lejeune, F.; Prota, G. Synthesis and Cytotoxic Properties of New N-Substituted 4-Aminophenol Derivatives with a Potential as Antimelanoma Agents. Melanoma Res. 1992, 2, 25–32. [Google Scholar] [CrossRef]
- Lee, E.A.; Angka, L.; Rota, S.-G.; Hanlon, T.; Mitchell, A.; Hurren, R.; Wang, X.M.; Gronda, M.; Boyaci, E.; Bojko, B.; et al. Targeting Mitochondria with Avocatin B Induces Selective Leukemia Cell Death. Cancer Res. 2015, 75, 2478–2488. [Google Scholar] [CrossRef]
- Liu, P.; Liu, J.; Jiang, W.; Carew, J.S.; Ogasawara, M.A.; Pelicano, H.; Croce, C.M.; Estrov, Z.; Xu, R.; Keating, M.J.; et al. Elimination of Chronic Lymphocytic Leukemia Cells in Stromal Microenvironment by Targeting CPT with an Anti-Angina Drug Perhexiline. Oncogene 2016, 35, 5663–5673. [Google Scholar] [CrossRef]
- Ashrafian, H.; Horowitz, J.D.; Frenneaux, M.P. Perhexiline. Cardiovasc. Drug Rev. 2007, 25, 76–97. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Gandour, R.D.; van der Leij, F.R. Molecular Enzymology of Carnitine Transfer and Transport. Biochim. Biophys. Acta 2001, 1546, 21–43. [Google Scholar] [CrossRef]
- Kim, W.T.; Yun, S.J.; Yan, C.; Jeong, P.; Kim, Y.H.; Lee, I.-S.; Kang, H.-W.; Park, S.; Moon, S.-K.; Choi, Y.-H.; et al. Metabolic Pathway Signatures Associated with Urinary Metabolite Biomarkers Differentiate Bladder Cancer Patients from Healthy Controls. Yonsei Med. J. 2016, 57, 865–871. [Google Scholar] [CrossRef]
- Valentino, A.; Calarco, A.; Di Salle, A.; Finicelli, M.; Crispi, S.; Calogero, R.A.; Riccardo, F.; Sciarra, A.; Gentilucci, A.; Galderisi, U.; et al. Deregulation of MicroRNAs Mediated Control of Carnitine Cycle in Prostate Cancer: Molecular Basis and Pathophysiological Consequences. Oncogene 2017, 36, 6030–6040. [Google Scholar] [CrossRef]
- Chegary, M.; te Brinke, H.; Doolaard, M.; IJlst, L.; Wijburg, F.A.; Wanders, R.J.A.; Houten, S.M. Characterization of L-Aminocarnitine, an Inhibitor of Fatty Acid Oxidation. Mol. Genet. Metab. 2008, 93, 403–410. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial Fatty Acid Metabolism in Health and Disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, W.; Devarakonda, T.; Zhou, H.; Wang, X.-Y.; Salloum, F.N.; Spiegel, S.; Fang, X. Functional Analysis of Molecular and Pharmacological Modulators of Mitochondrial Fatty Acid Oxidation. Sci. Rep. 2020, 10, 1450. [Google Scholar] [CrossRef]
- Chen, M.; Huang, J. The Expanded Role of Fatty Acid Metabolism in Cancer: New Aspects and Targets. Precis. Clin. Med. 2019, 2, 183–191. [Google Scholar] [CrossRef]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- Browne, C.D.; Hindmarsh, E.J.; Smith, J.W. Inhibition of Endothelial Cell Proliferation and Angiogenesis by Orlistat, a Fatty Acid Synthase Inhibitor. FASEB J. 2006, 20, 2027–2035. [Google Scholar] [CrossRef] [PubMed]
- Orlistat (marked as Alli and Xenical) Information FDA. Available online: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/orlistat-marketed-alli-and-xenical-information (accessed on 15 January 2022).
- Sternby, B.; Hartmann, D.; Borgstroom, B.; Nilsson, P. Degree of in Vivo Inhibition of Human Gastric and Pancreatic Lipases by Orlistat (Tetrahydrolipstatin, THL) in the Stomach and Small Intestine. Clin. Nutr. 2002, 21, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Kridel, S.J.; Axelrod, F.; Rozenkrantz, N.; Smith, J.W. Orlistat Is a Novel Inhibitor of Fatty Acid Synthase with Antitumor Activity. Cancer Res. 2004, 64, 2070–2075. [Google Scholar] [CrossRef]
- Papaevangelou, E.; Almeida, G.S.; Box, C.; deSouza, N.M.; Chung, Y.-L. The Effect of FASN Inhibition on the Growth and Metabolism of a Cisplatin-Resistant Ovarian Carcinoma Model: FASN Inhibition in Cisplatin-Resistant Cancer. Int. J. Cancer 2018, 143, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Grube, S.; Dünisch, P.; Freitag, D.; Klausnitzer, M.; Sakr, Y.; Walter, J.; Kalff, R.; Ewald, C. Overexpression of Fatty Acid Synthase in Human Gliomas Correlates with the WHO Tumor Grade and Inhibition with Orlistat Reduces Cell Viability and Triggers Apoptosis. J. Neurooncol. 2014, 118, 277–287. [Google Scholar] [CrossRef]
- Mims, J.; Bansal, N.; Bharadwaj, M.S.; Chen, X.; Molina, A.J.; Tsang, A.W.; Furdui, C.M. Energy Metabolism in a Matched Model of Radiation Resistance for Head and Neck Squamous Cell Cancer. Radiat. Res. 2015, 183, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-F.; Fang, F.-M.; Chen, Y.-Y.; Liu, T.-T.; Chan, T.-C.; Yu, S.-C.; Chen, L.-T.; Huang, H.-Y. Overexpressed Fatty Acid Synthase in Gastrointestinal Stromal Tumors: Targeting a Progression-Associated Metabolic Driver Enhances the Antitumor Effect of Imatinib. Clin. Cancer Res. 2017, 23, 4908–4918. [Google Scholar] [CrossRef]
- Kant, S.; Kumar, A.; Singh, S.M. Fatty Acid Synthase Inhibitor Orlistat Induces Apoptosis in T Cell Lymphoma: Role of Cell Survival Regulatory Molecules. Biochim. Biophys. Acta 2012, 1820, 1764–1773. [Google Scholar] [CrossRef]
- Czumaj, A.; Zabielska, J.; Pakiet, A.; Mika, A.; Rostkowska, O.; Makarewicz, W.; Kobiela, J.; Sledzinski, T.; Stelmanska, E. In Vivo Effectiveness of Orlistat in the Suppression of Human Colorectal Cancer Cell Proliferation. Anticancer Res. 2019, 39, 3815–3822. [Google Scholar] [CrossRef]
- Carvalho, M.A.; Zecchin, K.G.; Seguin, F.; Bastos, D.C.; Agostini, M.; Rangel, A.L.C.A.; Veiga, S.S.; Raposo, H.F.; Oliveira, H.C.F.; Loda, M.; et al. Fatty Acid Synthase Inhibition with Orlistat Promotes Apoptosis and Reduces Cell Growth and Lymph Node Metastasis in a Mouse Melanoma Model. Int. J. Cancer 2008, 123, 2557–2565. [Google Scholar] [CrossRef]
- Chuang, H.-Y.; Lee, Y.-P.; Lin, W.-C.; Lin, Y.-H.; Hwang, J.-J. Fatty Acid Inhibition Sensitizes Androgen-Dependent and -Independent Prostate Cancer to Radiotherapy via FASN/NF-ΚB Pathway. Sci. Rep. 2019, 9, 13284. [Google Scholar] [CrossRef]
- Sokolowska, E.; Presler, M.; Goyke, E.; Milczarek, R.; Swierczynski, J.; Sledzinski, T. Orlistat Reduces Proliferation and Enhances Apoptosis in Human Pancreatic Cancer Cells (PANC-1). Anticancer Res. 2017, 37, 6321–6327. [Google Scholar] [CrossRef]
- You, B.-J.; Chen, L.-Y.; Hsu, P.-H.; Sung, P.-H.; Hung, Y.-C.; Lee, H.-Z. Orlistat Displays Antitumor Activity and Enhances the Efficacy of Paclitaxel in Human Hepatoma Hep3B Cells. Chem. Res. Toxicol. 2019, 32, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhang, J.; Yan, M.; Wu, J.; Lian, S.; Sun, K.; Li, B.; Ma, J.; Xia, J.; Lian, C. Orlistat Induces Ferroptosis-like Cell Death of Lung Cancer Cells. Front. Med. 2021, 15, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Zhi, J.; Melia, A.T.; Eggers, H.; Joly, R.; Patel, I.H. Review of Limited Systemic Absorption of Orlistat, a Lipase Inhibitor, in Healthy Human Volunteers. J. Clin. Pharm. 1995, 35, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Funabashi, H.; Kawaguchi, A.; Tomoda, H.; Ömura, S.; Okuda, S.; Iwasaki, S. Binding Site of Cerulenin in Fatty Acid Synthetase1. J. Biochem. 1989, 105, 751–755. [Google Scholar] [CrossRef]
- Pizer, E.S.; Jackisch, C.; Wood, F.D.; Pasternack, G.R.; Davidson, N.E.; Kuhajda, F.P. Inhibition of Fatty Acid Synthesis Induces Programmed Cell Death in Human Breast Cancer Cells. Cancer Res. 1996, 56, 2745–2747. [Google Scholar] [PubMed]
- Pizer, E.S.; Wood, F.D.; Heine, H.S.; Romantsev, F.E.; Pasternack, G.R.; Kuhajda, F.P. Inhibition of Fatty Acid Synthesis Delays Disease Progression in a Xenograft Model of Ovarian Cancer. Cancer Res. 1996, 56, 1189–1193. [Google Scholar]
- Shiragami, R.; Murata, S.; Kosugi, C.; Tezuka, T.; Yamazaki, M.; Hirano, A.; Yoshimura, Y.; Suzuki, M.; Shuto, K.; Koda, K. Enhanced Antitumor Activity of Cerulenin Combined with Oxaliplatin in Human Colon Cancer Cells. Int. J. Oncol. 2013, 43, 431–438. [Google Scholar] [CrossRef]
- Chang, L.; Wu, P.; Senthilkumar, R.; Tian, X.; Liu, H.; Shen, X.; Tao, Z.; Huang, P. Loss of Fatty Acid Synthase Suppresses the Malignant Phenotype of Colorectal Cancer Cells by Down-Regulating Energy Metabolism and MTOR Signaling Pathway. J. Cancer Res. Clin. Oncol. 2016, 142, 59–72. [Google Scholar] [CrossRef]
- Deepa, P.R.; Vandhana, S.; Jayanthi, U.; Krishnakumar, S. Therapeutic and Toxicologic Evaluation of Anti-Lipogenic Agents in Cancer Cells Compared with Non-Neoplastic Cells. Basic Clin. Pharm. Toxicol. 2012, 110, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Li, E.-H.; Lu, Y.-Y.; Jiang, Q.; Cui, D.; Jing, Y.-F.; Xia, S.-J. Inhibition of Fatty-Acid Synthase Suppresses P-AKT and Induces Apoptosis in Bladder Cancer. Urology 2012, 80, 484. [Google Scholar] [CrossRef]
- Gouw, A.M.; Eberlin, L.S.; Margulis, K.; Sullivan, D.K.; Toal, G.G.; Tong, L.; Zare, R.N.; Felsher, D.W. Oncogene KRAS Activates Fatty Acid Synthase, Resulting in Specific ERK and Lipid Signatures Associated with Lung Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 4300–4305. [Google Scholar] [CrossRef]
- Kuhajda, F.P.; Pizer, E.S.; Li, J.N.; Mani, N.S.; Frehywot, G.L.; Townsend, C.A. Synthesis and Antitumor Activity of an Inhibitor of Fatty Acid Synthase. Proc. Natl. Acad. Sci. USA 2000, 97, 3450–3454. [Google Scholar] [CrossRef] [PubMed]
- Angeles, T.S.; Hudkins, R.L. Recent Advances in Targeting the Fatty Acid Biosynthetic Pathway Using Fatty Acid Synthase Inhibitors. Expert Opin. Drug Discov. 2016, 11, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Rendina, A.R.; Cheng, D. Characterization of the Inactivation of Rat Fatty Acid Synthase by C75: Inhibition of Partial Reactions and Protection by Substrates. Biochem. J. 2005, 388, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Loftus, T.M.; Jaworsky, D.E.; Frehywot, G.L.; Townsend, C.A.; Ronnett, G.V.; Lane, M.D.; Kuhajda, F.P. Reduced Food Intake and Body Weight in Mice Treated with Fatty Acid Synthase Inhibitors. Science 2000, 288, 2379–2381. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.; Fragkoulis, G.I.; Chalmers, A.J. Cytotoxicity and Radiosensitizing Activity of the Fatty Acid Synthase Inhibitor C75 Is Enhanced by Blocking Fatty Acid Uptake in Prostate Cancer Cells. Adv. Radiat. Oncol. 2020, 5, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Lin, L.-P.; Zhu, C.-H.; Chen, Y.; Hou, Y.-T.; Ding, J. Growth Arrest Induced by C75, A Fatty Acid Synthase Inhibitor, Was Partially Modulated by P38 MAPK but Not by P53 in Human Hepatocellular Carcinoma. Cancer Biol. Ther. 2006, 5, 978–985. [Google Scholar] [CrossRef]
- Brenner, A.J.; Von Hoff, D.D.; Infante, J.R.; Patel, M.R.; Jones, S.F.; Burris, H.A.; Rubino, C.; McCulloch, W.; Zhukova-Harrill, V.; Kemble, G. First-in-Human Investigation of the Oral First-in-Class Fatty Acid Synthase (FASN) Inhibitor, TVB-2640. JCO 2015, 33, TPS2615. [Google Scholar] [CrossRef]
- Ventura, R.; Mordec, K.; Waszczuk, J.; Wang, Z.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G.; Heuer, T.S. Inhibition of de Novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression. EBioMedicine 2015, 2, 808–824. [Google Scholar] [CrossRef]
- Zaytseva, Y.Y.; Rychahou, P.G.; Le, A.-T.; Scott, T.L.; Flight, R.M.; Kim, J.T.; Harris, J.; Liu, J.; Wang, C.; Morris, A.J.; et al. Preclinical Evaluation of Novel Fatty Acid Synthase Inhibitors in Primary Colorectal Cancer Cells and a Patient-Derived Xenograft Model of Colorectal Cancer. Oncotarget 2018, 9, 24787–24800. [Google Scholar] [CrossRef]
- Tao, T.; Su, Q.; Xu, S.; Deng, J.; Zhou, S.; Zhuang, Y.; Huang, Y.; He, C.; He, S.; Peng, M.; et al. Down-regulation of PKM2 Decreases FASN Expression in Bladder Cancer Cells through AKT/MTOR/SREBP-1c Axis. J. Cell. Physiol. 2019, 234, 3088–3104. [Google Scholar] [CrossRef]
- Stoddard, B.L.; Dean, A.; Koshland, D.E. Structure of Isocitrate Dehydrogenase with Isocitrate, Nicotinamide Adenine Dinucleotide Phosphate, and Calcium at 2.5-.ANG. Resolution: A Pseudo-Michaelis Ternary Complex. Biochemistry 1993, 32, 9310–9316. [Google Scholar] [CrossRef]
- Han, C.H.; Batchelor, T.T. Isocitrate Dehydrogenase Mutation as a Therapeutic Target in Gliomas. Chin. Clin. Oncol. 2017, 6, 33. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; van Noorden, C.J.F. Wild-Type and Mutated IDH1/2 Enzymes and Therapy Responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef]
- Stein, E.M. Molecular Pathways: IDH2 Mutations—Co-Opting Cellular Metabolism for Malignant Transformation. Clin. Cancer Res. 2016, 22, 16–19. [Google Scholar] [CrossRef][Green Version]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Krönke, J.; Bullinger, L.; Späth, D.; Kayser, S.; Zucknick, M.; Götze, K.; et al. IDH1 and IDH2 Mutations Are Frequent Genetic Alterations in Acute Myeloid Leukemia and Confer Adverse Prognosis in Cytogenetically Normal Acute Myeloid Leukemia with NPM1 Mutation without FLT3 Internal Tandem Duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Seo, S.I.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutational Analysis of IDH1 Codon 132 in Glioblastomas and Other Common Cancers. Int. J. Cancer 2009, 125, 353–355. [Google Scholar] [CrossRef]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent Mutation of Isocitrate Dehydrogenase (IDH)1 and IDH2 in Cholangiocarcinoma Identified through Broad-Based Tumor Genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Bojdani, E.; Xing, M. Identification and Functional Characterization of Isocitrate Dehydrogenase 1 (IDH1) Mutations in Thyroid Cancer. Biochem. Biophys. Res. Commun. 2010, 393, 555–559. [Google Scholar] [CrossRef]
- Ghiam, A.F.; Cairns, R.A.; Thoms, J.; Dal Pra, A.; Ahmed, O.; Meng, A.; Mak, T.W.; Bristow, R.G. IDH Mutation Status in Prostate Cancer. Oncogene 2012, 31, 3826. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Ivosidenib: First Global Approval. Drugs 2018, 78, 1509–1516. [Google Scholar] [CrossRef]
- Fan, B.; Dai, D.; DiNardo, C.D.; Stein, E.; de Botton, S.; Attar, E.C.; Liu, H.; Liu, G.; Lemieux, I.; Agresta, S.V.; et al. Clinical Pharmacokinetics and Pharmacodynamics of Ivosidenib in Patients with Advanced Hematologic Malignancies with an IDH1 Mutation. Cancer Chemother. Pharm. 2020, 85, 959–968. [Google Scholar] [CrossRef]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Commissioner, O. FDA Approves New Targeted Treatment for Relapsed or Refractory Acute Myeloid Leukemia. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-targeted-treatment-relapsed-or-refractory-acute-myeloid-leukemia (accessed on 13 March 2022).
- Liu, X.; Gong, Y. Isocitrate Dehydrogenase Inhibitors in Acute Myeloid Leukemia. Biomark. Res. 2019, 7, 22. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Herbst, L.; Pusch, S.; Klett, L.; Goparaju, R.; Stichel, D.; Kaulfuss, S.; Panknin, O.; Zimmermann, K.; Toschi, L.; et al. Pan-Mutant-IDH1 Inhibitor BAY1436032 Is Highly Effective against Human IDH1 Mutant Acute Myeloid Leukemia in Vivo. Leukemia 2017, 31, 2020–2028. [Google Scholar] [CrossRef]
- Heuser, M.; Palmisiano, N.; Mantzaris, I.; Mims, A.; DiNardo, C.; Silverman, L.R.; Wang, E.S.; Fiedler, W.; Baldus, C.; Schwind, S.; et al. Safety and Efficacy of BAY1436032 in IDH1-Mutant AML: Phase I Study Results. Leukemia 2020, 34, 2903–2913. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Gupta, C.; Gabdoulline, R.; Nora, M.B.; Goparaju, R.; Kaulfuss, S.; Görlich, K.; Schottmann, R.; Othman, B.; Welzenbach, J.; et al. Synergistic Activity of IDH1 Inhibitor BAY1436032 with Azacitidine in IDH1 Mutant Acute Myeloid Leukemia. Haematologica 2020, 106, 565–573. [Google Scholar] [CrossRef]
- Caravella, J.A.; Lin, J.; Diebold, R.B.; Campbell, A.-M.; Ericsson, A.; Gustafson, G.; Wang, Z.; Castro, J.; Clarke, A.; Gotur, D.; et al. Structure-Based Design and Identification of FT-2102 (Olutasidenib), a Potent Mutant-Selective IDH1 Inhibitor. J. Med. Chem. 2020, 63, 1612–1623. [Google Scholar] [CrossRef]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef] [PubMed]
- Celgene A Phase 1/2, Multicenter, Open-Label, Dose-Escalation and Expansion, Safety, Pharmacokinetic, Pharmacodynamic, and Clinical Activity Study of Orally Administered AG-221 in Subjects with Advanced Hematologic Malignancies with an IDH2 Mutation. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT01915498 (accessed on 5 February 2022).
- Kim, E.S. Enasidenib: First Global Approval. Drugs 2017, 77, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted Inhibition of Mutant IDH2 in Leukemia Cells Induces Cellular Differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef]
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and Downsides of Reactive Oxygen Species for Cancer: The Roles of Reactive Oxygen Species in Tumorigenesis, Prevention, and Therapy. Antioxid. Redox Signal. 2012, 16, 1295–1322. [Google Scholar] [CrossRef]
- Pacheco-Velázquez, S.C.; Robledo-Cadena, D.X.; Hernández-Reséndiz, I.; Gallardo-Pérez, J.C.; Moreno-Sánchez, R.; Rodríguez-Enríquez, S. Energy Metabolism Drugs Block Triple Negative Breast Metastatic Cancer Cell Phenotype. Mol. Pharm. 2018, 15, 2151–2164. [Google Scholar] [CrossRef]
- Nayak, A.; Kapur, A.; Barroilhet, L.; Patankar, M. Oxidative Phosphorylation: A Target for Novel Therapeutic Strategies Against Ovarian Cancer. Cancers 2018, 10, 337. [Google Scholar] [CrossRef]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene Ablation-Resistant Pancreatic Cancer Cells Depend on Mitochondrial Function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef]
- Ralph, S.J.; Low, P.; Dong, L.; Lawen, A.; Neuzil, J. Mitocans: Mitochondrial Targeted Anti-Cancer Drugs as Improved Therapies and Related Patent Documents. Recent Pat. Anticancer Drug Discov. 2006, 1, 327–346. [Google Scholar] [CrossRef]
- Neuzil, J.; Dong, L.-F.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of Mitocans, Anti-Cancer Drugs Acting on Mitochondria. Mitochondrion 2013, 13, 199–208. [Google Scholar] [CrossRef]
- Hd, U. Bcl-2 Family Proteins as Regulators of Cancer Cell Invasion and Metastasis: A Review Focusing on Mitochondrial Respiration and Reactive Oxygen Species. Oncotarget 2016, 7, 5193–5203. [Google Scholar] [CrossRef]
- Hafezi, S.; Rahmani, M. Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives. Cancers 2021, 13, 1292. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An Inhibitor of Bcl-2 Family Proteins Induces Regression of Solid Tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Cleary, J.M.; Lima, C.M.S.R.; Hurwitz, H.I.; Montero, A.J.; Franklin, C.; Yang, J.; Graham, A.; Busman, T.; Mabry, M.; Holen, K.; et al. A Phase I Clinical Trial of Navitoclax, a Targeted High-Affinity Bcl-2 Family Inhibitor, in Combination with Gemcitabine in Patients with Solid Tumors. Investig. New Drugs 2014, 32, 937–945. [Google Scholar] [CrossRef]
- Tolcher, A.W.; LoRusso, P.; Arzt, J.; Busman, T.A.; Lian, G.; Rudersdorf, N.S.; Vanderwal, C.A.; Kirschbrown, W.; Holen, K.D.; Rosen, L.S. Safety, Efficacy, and Pharmacokinetics of Navitoclax (ABT-263) in Combination with Erlotinib in Patients with Advanced Solid Tumors. Cancer Chemother. Pharm. 2015, 76, 1025–1032. [Google Scholar] [CrossRef]
- Vlahovic, G.; Karantza, V.; Wang, D.; Cosgrove, D.; Rudersdorf, N.; Yang, J.; Xiong, H.; Busman, T.; Mabry, M. A Phase I Safety and Pharmacokinetic Study of ABT-263 in Combination with Carboplatin/Paclitaxel in the Treatment of Patients with Solid Tumors. Investig. New Drugs 2014, 32, 976–984. [Google Scholar] [CrossRef]
- Richard-Carpentier, G.; DiNardo, C.D. Venetoclax for the Treatment of Newly Diagnosed Acute Myeloid Leukemia in Patients Who Are Ineligible for Intensive Chemotherapy. Adv. Hematol. 2019, 10, 2040620719882822. [Google Scholar] [CrossRef]
- Deeks, E.D. Venetoclax: First Global Approval. Drugs 2016, 76, 979–987. [Google Scholar] [CrossRef]
- Stilgenbauer, S.; Eichhorst, B.; Schetelig, J.; Hillmen, P.; Seymour, J.F.; Coutre, S.; Jurczak, W.; Mulligan, S.P.; Schuh, A.; Assouline, S.; et al. Venetoclax for Patients with Chronic Lymphocytic Leukemia With 17p Deletion: Results from the Full Population of a Phase II Pivotal Trial. J. Clin. Oncol. 2018, 36, 1973–1980. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting Cancer Cells by ROS-Mediated Mechanisms: A Radical Therapeutic Approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting Mitochondria for Cancer Therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Wright, S.E.; Kim, S.-H.; Srivastava, S.K. Phenethyl Isothiocyanate: A Comprehensive Review of Anti-Cancer Mechanisms. Biochim. Biophys. Acta 2014, 1846, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.E.; Dave, R.A. Pharmacokinetics and Pharmacodynamics of Phenethyl Isothiocyanate: Implications in Breast Cancer Prevention. AAPS J. 2014, 16, 705–713. [Google Scholar] [CrossRef]
- Trachootham, D.; Zhang, H.; Zhang, W.; Feng, L.; Du, M.; Zhou, Y.; Chen, Z.; Pelicano, H.; Plunkett, W.; Wierda, W.G.; et al. Effective Elimination of Fludarabine-Resistant CLL Cells by PEITC through a Redox-Mediated Mechanism. Blood 2008, 112, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Xiao, Y.; Fu, B.; Qin, Z. TPP-Based Mitocans: A Potent Strategy for Anticancer Drug Design. RSC Med. Chem. 2020, 11, 858–875. [Google Scholar] [CrossRef]
- Don, A.S.; Kisker, O.; Dilda, P.; Donoghue, N.; Zhao, X.; Decollogne, S.; Creighton, B.; Flynn, E.; Folkman, J.; Hogg, P.J. A Peptide Trivalent Arsenical Inhibits Tumor Angiogenesis by Perturbing Mitochondrial Function in Angiogenic Endothelial Cells. Cancer Cell 2003, 3, 497–509. [Google Scholar] [CrossRef]
- Zhivotovsky, B.; Galluzzi, L.; Kepp, O.; Kroemer, G. Adenine Nucleotide Translocase: A Component of the Phylogenetically Conserved Cell Death Machinery. Cell Death Differ. 2009, 16, 1419–1425. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Golan, M. Mitochondrial VDAC1: Function in Cell Life and Death and a Target for Cancer Therapy. CMC 2012, 19, 714–735. [Google Scholar] [CrossRef]
- Simamura, E.; Shimada, H.; Ishigaki, Y.; Hatta, T.; Higashi, N.; Hirai, K.-I. Bioreductive Activation of Quinone Antitumor Drugs by Mitochondrial Voltage-Dependent Anion Channel 1. Anat. Sci. Int. 2008, 83, 261–266. [Google Scholar] [CrossRef]
- Jóźwiak, P.; Ciesielski, P.; Forma, E.; Kozal, K.; Wójcik-Krowiranda, K.; Cwonda, Ł.; Bieńkiewicz, A.; Bryś, M.; Krześlak, A. Expression of Voltage-Dependent Anion Channels in Endometrial Cancer and Its Potential Prognostic Significance. Tumour Biol. 2020, 42, 1010428320951057. [Google Scholar] [CrossRef]
- Marchetti, P.; Zamzami, N.; Joseph, B.; Schraen-Maschke, S.; Méreau-Richard, C.; Costantini, P.; Métivier, D.; Susin, S.A.; Kroemer, G.; Formstecher, P. The Novel Retinoid 6-[3-(1-Adamantyl)-4-Hydroxyphenyl]-2-Naphtalene Carboxylic Acid Can Trigger Apoptosis through a Mitochondrial Pathway Independent of the Nucleus. Cancer Res. 1999, 59, 6257–6266. [Google Scholar]
- Notario, B.; Zamora, M.; Viñas, O.; Mampel, T. All-Trans-Retinoic Acid Binds to and Inhibits Adenine Nucleotide Translocase and Induces Mitochondrial Permeability Transition. Mol. Pharm. 2003, 63, 224–231. [Google Scholar] [CrossRef]
- Rohlena, J.; Dong, L.; Neuzil, J. Targeting the Mitochondrial Electron Transport Chain Complexes for the Induction of Apoptosis and Cancer Treatment. Curr. Pharm. Biotechnol. 2013, 14, 377–389. [Google Scholar] [CrossRef]
- Urra, F.A.; Weiss-López, B.; Araya-Maturana, R. Determinants of Anti-Cancer Effect of Mitochondrial Electron Transport Chain Inhibitors: Bioenergetic Profile and Metabolic Flexibility of Cancer Cells. Curr. Pharm. Des. 2016, 22, 5998–6008. [Google Scholar] [CrossRef]
- Modica-Napolitano, J.S.; Aprille, J.R. Delocalized Lipophilic Cations Selectively Target the Mitochondria of Carcinoma Cells. Adv. Drug Deliv. Rev. 2001, 49, 63–70. [Google Scholar] [CrossRef]
- Wang, F.; Ogasawara, M.A.; Huang, P. Small Mitochondria-Targeting Molecules as Anti-Cancer Agents. Mol. Asp. Med. 2010, 31, 75–92. [Google Scholar] [CrossRef]
- Modica-Napolitano, J.S.; Aprille, J.R. Basis for the Selective Cytotoxicity of Rhodamine 123. Cancer Res. 1987, 47, 4361–4365. [Google Scholar]
- Lampidis, T.J.; Bernal, S.D.; Summerhayes, I.C.; Chen, L.B. Selective Toxicity of Rhodamine 123 in Carcinoma Cells in Vitro. Cancer Res. 1983, 43, 716–720. [Google Scholar]
- Wang, J.; He, H.; Xiang, C.; Fan, X.-Y.; Yang, L.-Y.; Yuan, L.; Jiang, F.-L.; Liu, Y. Uncoupling Effect of F16 Is Responsible for Its Mitochondrial Toxicity and Anticancer Activity. Toxicol. Sci. 2018, 161, 431–442. [Google Scholar] [CrossRef]
- Alves, I.D.; Carré, M.; Montero, M.-P.; Castano, S.; Lecomte, S.; Marquant, R.; Lecorché, P.; Burlina, F.; Schatz, C.; Sagan, S.; et al. A Proapoptotic Peptide Conjugated to Penetratin Selectively Inhibits Tumor Cell Growth. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1838, 2087–2098. [Google Scholar] [CrossRef]
- Rao, V.A.; Klein, S.R.; Bonar, S.J.; Zielonka, J.; Mizuno, N.; Dickey, J.S.; Keller, P.W.; Joseph, J.; Kalyanaraman, B.; Shacter, E. The Antioxidant Transcription Factor Nrf2 Negatively Regulates Autophagy and Growth Arrest Induced by the Anticancer Redox Agent Mitoquinone. J. Biol. Chem. 2010, 285, 34447–34459. [Google Scholar] [CrossRef] [PubMed]
- Capeloa, T.; Krzystyniak, J.; Rodriguez, A.C.; Payen, V.L.; Zampieri, L.X.; Pranzini, E.; Derouane, F.; Vazeille, T.; Bouzin, C.; Duhoux, F.P.; et al. MitoQ Prevents Human Breast Cancer Recurrence and Lung Metastasis in Mice. Cancers 2022, 14, 1488. [Google Scholar] [CrossRef] [PubMed]
- Capeloa, T.; Krzystyniak, J.; d’Hose, D.; Canas Rodriguez, A.; Payen, V.L.; Zampieri, L.X.; Van de Velde, J.A.; Benyahia, Z.; Pranzini, E.; Vazeille, T.; et al. MitoQ Inhibits Human Breast Cancer Cell Migration, Invasion and Clonogenicity. Cancers 2022, 14, 1516. [Google Scholar] [CrossRef] [PubMed]
- Titova, E.; Shagieva, G.; Ivanova, O.; Domnina, L.; Domninskaya, M.; Strelkova, O.; Khromova, N.; Kopnin, P.; Chernyak, B.; Skulachev, V.; et al. Mitochondria-Targeted Antioxidant SkQ1 Suppresses Fibrosarcoma and Rhabdomyosarcoma Tumour Cell Growth. Cell Cycle 2018, 17, 1797–1811. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial Metabolism and ROS Generation Are Essential for Kras-Mediated Tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Starenki, D.; Park, J.-I. Mitochondria-Targeted Nitroxide, Mito-CP, Suppresses Medullary Thyroid Carcinoma Cell Survival In Vitro and In Vivo. J. Clin. Endocrinol. Metab. 2013, 98, 1529–1540. [Google Scholar] [CrossRef]
- Zielonka, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; McAllister, D.M.; Mackinnon, A.C.; Joseph, J.; Dwinell, M.B.; Kalyanaraman, B. Mitochondria-Targeted Vitamin E Analogs Inhibit Breast Cancer Cell Energy Metabolism and Promote Cell Death. BMC Cancer 2013, 13, 285. [Google Scholar] [CrossRef]
- Reddy, C.A.; Somepalli, V.; Golakoti, T.; Kanugula, A.K.; Karnewar, S.; Rajendiran, K.; Vasagiri, N.; Prabhakar, S.; Kuppusamy, P.; Kotamraju, S.; et al. Mitochondrial-Targeted Curcuminoids: A Strategy to Enhance Bioavailability and Anticancer Efficacy of Curcumin. PLoS ONE 2014, 9, e89351. [Google Scholar] [CrossRef]
- Huang, M.; Myers, C.R.; Wang, Y.; You, M. Mitochondria as a Novel Target for Cancer Chemoprevention: Emergence of Mitochondrial Targeting Agents. Cancer Prev. Res. 2021, 14, 285–306. [Google Scholar] [CrossRef]
- Sassi, N.; Mattarei, A.; Azzolini, M.; Bernardi, P.; Szabo’, I.; Paradisi, C.; Zoratti, M.; Biasutto, L. Mitochondria-Targeted Resveratrol Derivatives Act as Cytotoxic pro-Oxidants. Curr. Pharm. Des. 2014, 20, 172–179. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Gubaidullin, R.R.; Dubinin, M.V.; Belosludtsev, K.N. Conjugation of Natural Triterpenic Acids with Delocalized Lipophilic Cations: Selective Targeting Cancer Cell Mitochondria. J. Pers. Med. 2021, 11, 470. [Google Scholar] [CrossRef]
- Tsepaeva, O.V.; Nemtarev, A.V.; Abdullin, T.I.; Grigor’eva, L.R.; Kuznetsova, E.V.; Akhmadishina, R.A.; Ziganshina, L.E.; Cong, H.H.; Mironov, V.F. Design, Synthesis, and Cancer Cell Growth Inhibitory Activity of Triphenylphosphonium Derivatives of the Triterpenoid Betulin. J. Nat. Prod 2017, 80, 2232–2239. [Google Scholar] [CrossRef]
- Nedopekina, D.A.; Gubaidullin, R.R.; Odinokov, V.N.; Maximchik, P.V.; Zhivotovsky, B.; Bel’skii, Y.P.; Khazanov, V.A.; Manuylova, A.V.; Gogvadze, V.; Spivak, A.Y. Mitochondria-Targeted Betulinic and Ursolic Acid Derivatives: Synthesis and Anticancer Activity. MedChemComm 2017, 8, 1934–1945. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Bel’skii, Y.P.; Bel’skaya, N.V.; Khazanov, V.A. Triphenylphosphonium Cations of Betulinic Acid Derivatives: Synthesis and Antitumor Activity. Med. Chem. Res. 2017, 26, 518–531. [Google Scholar] [CrossRef]
- Jin, L.; Dai, L.; Ji, M.; Wang, H. Mitochondria-Targeted Triphenylphosphonium Conjugated Glycyrrhetinic Acid Derivatives as Potent Anticancer Drugs. Bioorg. Chem. 2019, 85, 179–190. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Semenova, A.A.; Nedopekina, D.A.; Davletshin, E.V.; Spivak, A.Y.; Belosludtsev, K.N. Effect of F16-Betulin Conjugate on Mitochondrial Membranes and Its Role in Cell Death Initiation. Membranes 2021, 11, 352. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Semenova, A.A.; Ilzorkina, A.I.; Penkov, N.V.; Nedopekina, D.A.; Sharapov, V.A.; Khoroshavina, E.I.; Davletshin, E.V.; Belosludtseva, N.V.; Spivak, A.Y.; et al. Mitochondria-Targeted Prooxidant Effects of Betulinic Acid Conjugated with Delocalized Lipophilic Cation F16. Free Radic. Biol. Med. 2021, 168, 55–69. [Google Scholar] [CrossRef]
- Wolfram, R.K.; Fischer, L.; Kluge, R.; Ströhl, D.; Al-Harrasi, A.; Csuk, R. Homopiperazine-Rhodamine B Adducts of Triterpenoic Acids Are Strong Mitocans. Eur. J. Med. Chem. 2018, 155, 869–879. [Google Scholar] [CrossRef]
- Hoenke, S.; Serbian, I.; Deigner, H.-P.; Csuk, R. Mitocanic Di- and Triterpenoid Rhodamine B Conjugates. Molecules 2020, 25, 5443. [Google Scholar] [CrossRef]
- Heise, N.V.; Major, D.; Hoenke, S.; Kozubek, M.; Serbian, I.; Csuk, R. Rhodamine 101 Conjugates of Triterpenoic Amides Are of Comparable Cytotoxicity as Their Rhodamine B Analogs. Molecules 2022, 27, 2220. [Google Scholar] [CrossRef] [PubMed]
- Sommerwerk, S.; Heller, L.; Kerzig, C.; Kramell, A.E.; Csuk, R. Rhodamine B Conjugates of Triterpenoic Acids Are Cytotoxic Mitocans Even at Nanomolar Concentrations. Eur. J. Med. Chem. 2017, 127, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Colvin, M. Alkylating Agents in Holland-Frei Cancer Medicine, 6th ed.; Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Bast, R.C., Jr., Gansler, T.S., Holland, J.F., Frei, E., III, Eds.; BC Decker: Hamilton, ON, Canada, 2003. [Google Scholar]
- Goede, V.; Eichhorst, B.; Fischer, K.; Wendtner, C.-M.; Hallek, M. Past, Present and Future Role of Chlorambucil in the Treatment of Chronic Lymphocytic Leukemia. Leuk. Lymphoma 2015, 56, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- PubChem Chlorambucil. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/2708 (accessed on 1 March 2022).
- Millard, M.; Gallagher, J.D.; Olenyuk, B.Z.; Neamati, N. A Selective Mitochondrial-Targeted Chlorambucil with Remarkable Cytotoxicity in Breast and Pancreatic Cancers. J. Med. Chem. 2013, 56, 9170–9179. [Google Scholar] [CrossRef]
- Battogtokh, G.; Cho, Y.-Y.; Lee, J.Y.; Lee, H.S.; Kang, H.C. Mitochondrial-Targeting Anticancer Agent Conjugates and Nanocarrier Systems for Cancer Treatment. Front. Pharmacol. 2018, 9, 922. [Google Scholar] [CrossRef]
- Han, M.; Vakili, M.R.; Soleymani Abyaneh, H.; Molavi, O.; Lai, R.; Lavasanifar, A. Mitochondrial Delivery of Doxorubicin via Triphenylphosphine Modification for Overcoming Drug Resistance in MDA-MB-435/DOX Cells. Mol. Pharm. 2014, 11, 2640–2649. [Google Scholar] [CrossRef]
- Liu, H.-N.; Guo, N.-N.; Wang, T.-T.; Guo, W.-W.; Lin, M.-T.; Huang-Fu, M.-Y.; Vakili, M.R.; Xu, W.-H.; Chen, J.-J.; Wei, Q.-C.; et al. Mitochondrial Targeted Doxorubicin-Triphenylphosphonium Delivered by Hyaluronic Acid Modified and PH Responsive Nanocarriers to Breast Tumor: In Vitro and in Vivo Studies. Mol. Pharm. 2018, 15, 882–891. [Google Scholar] [CrossRef]
- Boukalova, S.; Stursa, J.; Werner, L.; Ezrova, Z.; Cerny, J.; Bezawork-Geleta, A.; Pecinova, A.; Dong, L.; Drahota, Z.; Neuzil, J. Mitochondrial Targeting of Metformin Enhances Its Activity against Pancreatic Cancer. Mol. Cancer Ther. 2016, 15, 2875–2886. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; Ouari, O.; Lopez, M.; McAllister, D.; Boyle, K.; Barrios, C.S.; Weber, J.J.; Johnson, B.D.; Hardy, M.; et al. Mitochondria-Targeted Analogs of Metformin Exhibit Enhanced Antiproliferative and Radiosensitizing Effects in Pancreatic Cancer Cells. Cancer Res. 2016, 76, 3904–3915. [Google Scholar] [CrossRef]
- Pathak, R.K.; Marrache, S.; Harn, D.A.; Dhar, S. Mito-DCA: A Mitochondria Targeted Molecular Scaffold for Efficacious Delivery of Metabolic Modulator Dichloroacetate. ACS Chem. Biol. 2014, 9, 1178–1187. [Google Scholar] [CrossRef]
- Sasaki, R.; Suzuki, Y.; Yonezawa, Y.; Ota, Y.; Okamoto, Y.; Demizu, Y.; Huang, P.; Yoshida, H.; Sugimura, K.; Mizushina, Y. DNA Polymerase Gamma Inhibition by Vitamin K3 Induces Mitochondria-Mediated Cytotoxicity in Human Cancer Cells. Cancer Sci. 2008, 99, 1040–1048. [Google Scholar] [CrossRef]
- Umeda, S.; Muta, T.; Ohsato, T.; Takamatsu, C.; Hamasaki, N.; Kang, D. The D-Loop Structure of Human MtDNA Is Destabilized Directly by 1-Methyl-4-Phenylpyridinium Ion (MPP+), a Parkinsonism-Causing Toxin. Eur. J. Biochem. 2000, 267, 200–206. [Google Scholar] [CrossRef]
- Modica-Napolitano, J.S.; Koya, K.; Weisberg, E.; Brunelli, B.T.; Li, Y.; Chen, L.B. Selective Damage to Carcinoma Mitochondria by the Rhodacyanine MKT-077. Cancer Res. 1996, 56, 544–550. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubik, J.; Humeniuk, E.; Adamczuk, G.; Madej-Czerwonka, B.; Korga-Plewko, A. Targeting Energy Metabolism in Cancer Treatment. Int. J. Mol. Sci. 2022, 23, 5572. https://doi.org/10.3390/ijms23105572
Kubik J, Humeniuk E, Adamczuk G, Madej-Czerwonka B, Korga-Plewko A. Targeting Energy Metabolism in Cancer Treatment. International Journal of Molecular Sciences. 2022; 23(10):5572. https://doi.org/10.3390/ijms23105572
Chicago/Turabian StyleKubik, Joanna, Ewelina Humeniuk, Grzegorz Adamczuk, Barbara Madej-Czerwonka, and Agnieszka Korga-Plewko. 2022. "Targeting Energy Metabolism in Cancer Treatment" International Journal of Molecular Sciences 23, no. 10: 5572. https://doi.org/10.3390/ijms23105572
APA StyleKubik, J., Humeniuk, E., Adamczuk, G., Madej-Czerwonka, B., & Korga-Plewko, A. (2022). Targeting Energy Metabolism in Cancer Treatment. International Journal of Molecular Sciences, 23(10), 5572. https://doi.org/10.3390/ijms23105572