
Role of Biliverdin Reductase A in the Regulation of Insulin Signaling in Metabolic and Neurodegenerative Diseases: An Update


 , and
, and 
Abstract
:1. Introduction
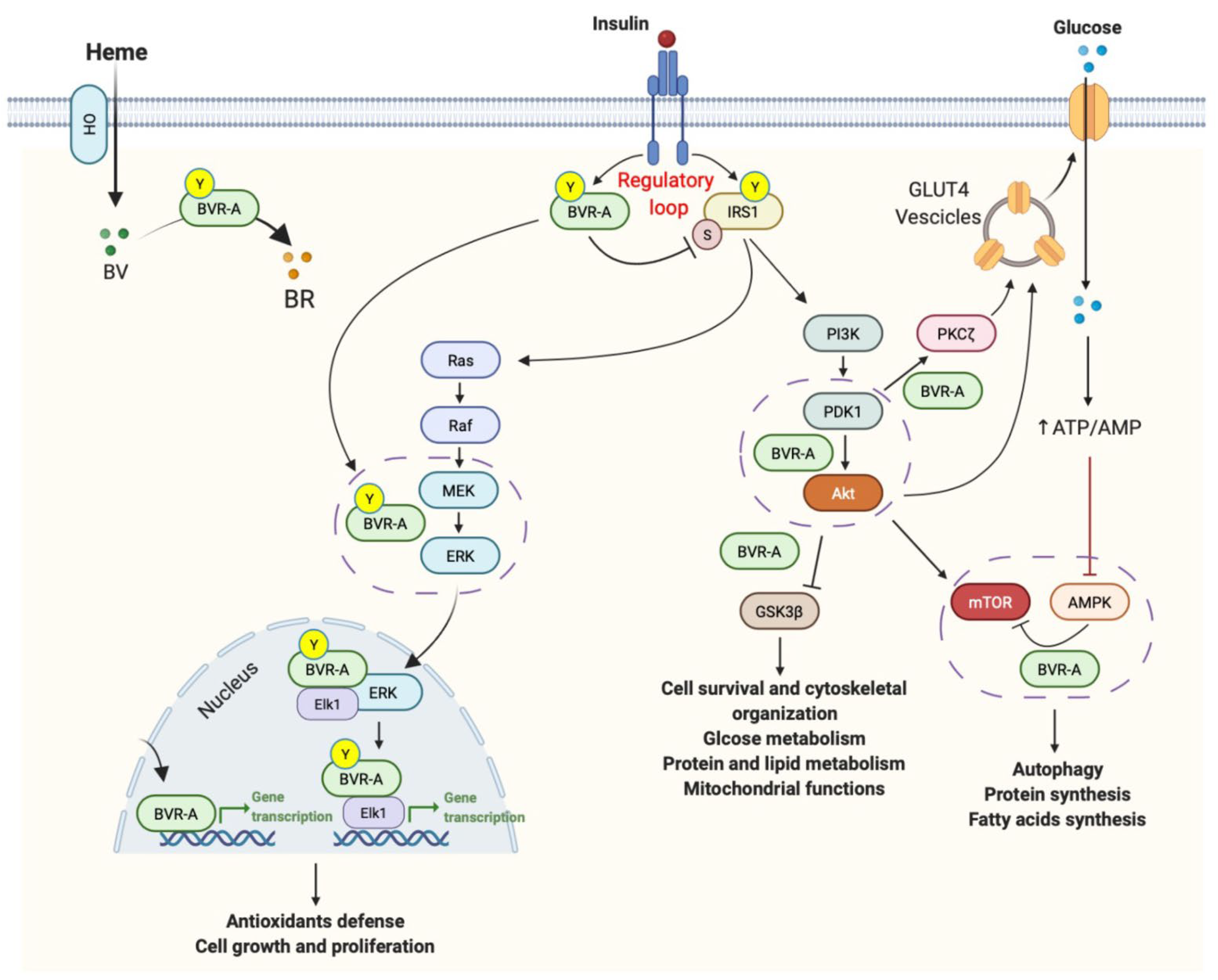
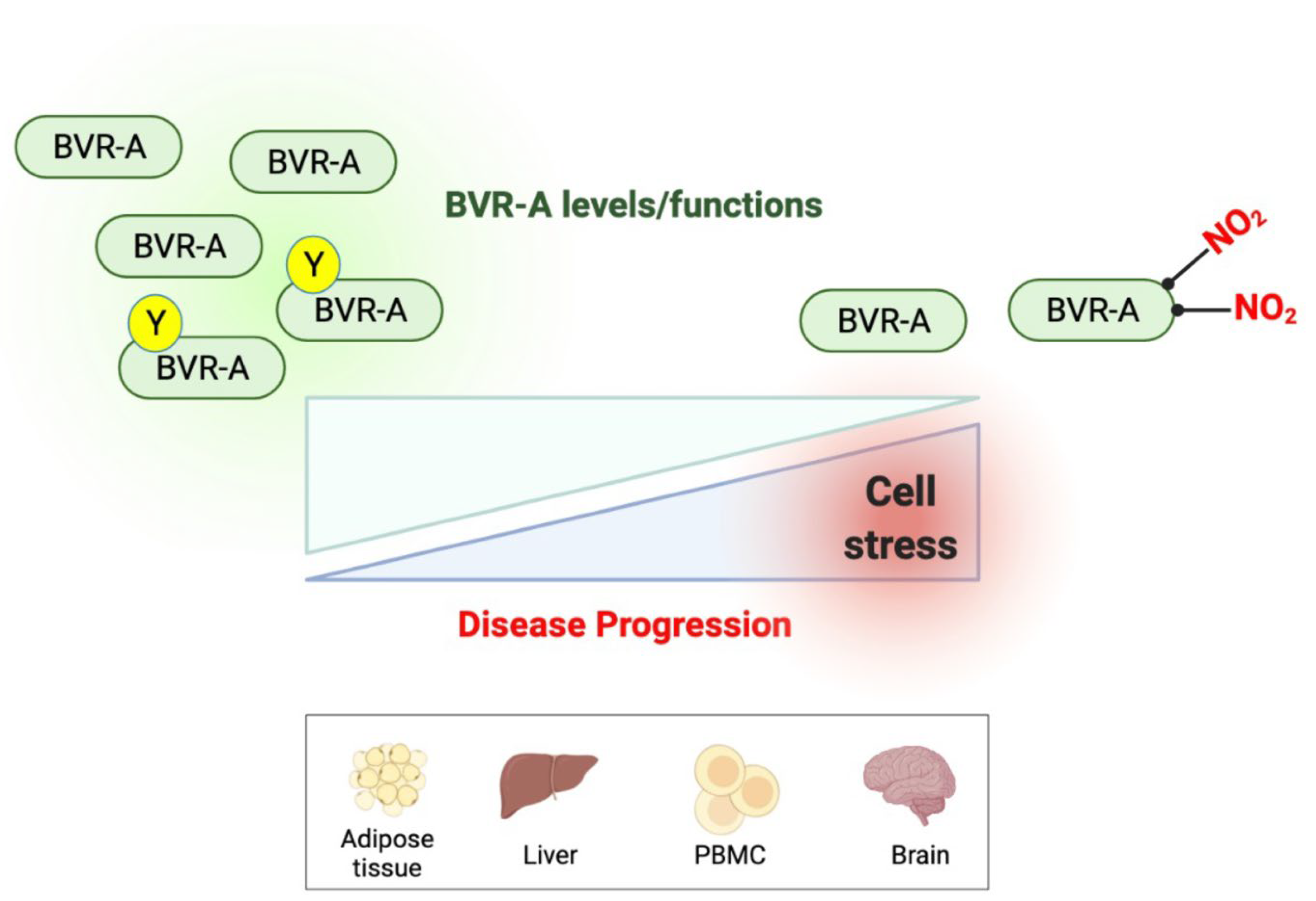
2. BVR-A Is a Protein with Pleiotropic Nature
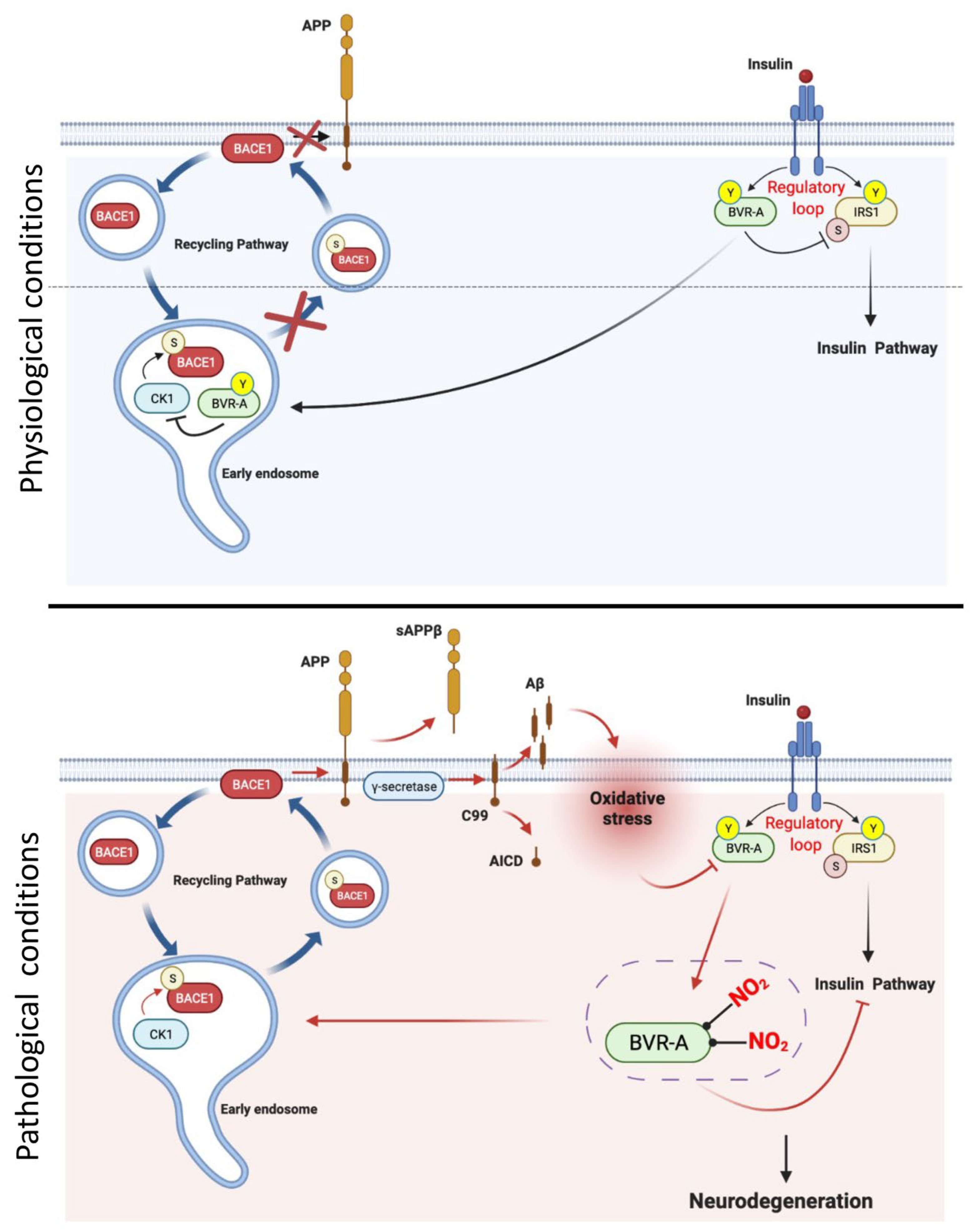
3. BVR-A in Insulin Signaling Pathway
4. Alteration of BVR-A in Metabolic Impairment: Data from Animal and Human Studies
4.1. Animal Studies
4.2. Human Studies
5. BVR-A in Brain Insulin Resistance and Neurodegenerative Disorders
5.1. Animal Studies
5.2. Human Studies

{kind=link}
{kind=link}
{kind=link}
| Model | BVR-A Alteration(s) | Insulin Signaling Alteration(s) | Observed Effect(s) | Ref. |
|---|---|---|---|---|
| 3xTg-AD mice | Reduced BVR-A levels and activation (6–18 months) and increased 3-NT on BVR-A (12–18 months) in the hippocampus | IRS1 hyper-activation (6 months) followed by increased IRS1 inhibition and mTOR hyper-activation (12 months) | Increased Aβ and Tau phosphorylation in the hippocampus | [81] |
| 3xTg-AD mice | Reduced BVR-A levels and Tyr-phosphorylation (6–12 months) in the hippocampus and cortex | IRS1 hyper-activation (6 months) followed by increased IRS1 inhibition (12 months); reduced Akt activation and reduced GSK3β inhibition on Ser9 (6–12 months); reduced Akt-GSK3β physical interaction (6–12 months); ERK1/2 hyperactivation (6 months); mTOR hyper-activation (12 months) | Impairment of cognitive and learning functions (6–12 months); increased Aβ levels and Tau phosphorylation in the hippocampus and cortex (6–12 months) | [73,75] |
| 3xTg-AD mice treated with intranasal insulin | Increased BVR-A Tyr-phosphorylation in the hippocampus and conrtex (6 and 12 months) | Reduced IRS1 hyper-activation (6 months) and reduced IRS1 inhibition (12 months); increased Akt activation (6 and 12 months); block of mTOR hyper-activation (12 months) | Improvement of cognitive and learning functions (6 and 12 months); reduced Aβ levels and Tau phosphorylation in the hippocampus and cortex (6 and 12 months) | [75] |
| C57Bl6 mice | Reduced BVR-A levels and phosphorylation (12 months) and increased 3-NT on BVR-A (18 months) in the hippocampus | Increased IRS1 inhibition (18 months) | - | [81] |
| Canine (beagle) | Reduced BVR-A Tyr-phosphorylation (4–12 months) and increased 3-NT on BVR-A (10–12 months) in the parietal cortex | Reduced Akt activation (4–12 months) | Increased Aβ levels in the cortex (10–12 months) | [82] |
| BVR-A KO mice | Global BVR-A deficiency in the cerebral cortex | mTOR hyper-activation and reduced AMPK levels | Impairment of autophagic flux in the cortex | [98] |
| Ts65dn mice | Reduced BVR-A Tyr-phosphorylation in the frontal cortex (9 months) | Increased IRS1 inhibition; mTOR hyper-activation | Loss of proteins regulating synaptic plasticity; accumulation of APP-C99 | [78] |
| aMCI and AD subjects | Reduced BVR-A Tyr-phosphorylation and increased 3-NT on BVR-A in the hippocampus | Increased IRS1 inhibition; mTOR hyper-activation; decreased interaction with ERK2 | - | [94,117,118] |
| aMCI and AD subjects | Reduced BVR-A levels in the parietal cortex | Reduced GSK3β inhibition; reduced Akt-GSK3β physical interaction | - | [54] |
| DS subjects | Increased 3-NT on BVR-A and reduced BVR-A activation in the frontal cortex | Increased IRS1 inhibition; mTOR hyper-activation | - | [72,125] |
| Centenarians | Increased BVR-A gene expression in blood samples | - | - | [126] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO; World Health Organization. Obesity: Preventing and Managing the Global Epidemic—Report of a WHO Consultation; World Health Organ Technical Report Series; WHO: Geneva, Switzerland, 2000; Volume 894, pp. 1–253.
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deuschl, G.; Beghi, E.; Fazekas, F.; Varga, T.; Christoforidi, K.A.; Sipido, E.; Bassetti, C.L.; Vos, T.; Feigin, V.L. The burden of neurological diseases in Europe: An analysis for the Global Burden of Disease Study 2017. Lancet Public Health 2020, 5, e551–e567. [Google Scholar] [CrossRef]
- Tahami Monfared, A.A.; Byrnes, M.J.; White, L.A.; Zhang, Q. The Humanistic and Economic Burden of Alzheimer’s Disease. Neurol. Ther. 2022, 11, 525–551. [Google Scholar] [CrossRef] [PubMed]
- Terzo, S.; Amato, A.; Mulè, F. From obesity to Alzheimer’s disease through insulin resistance. J. Diabetes Complicat. 2021, 35, 108026. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Chávez-Castillo, M.; Bautista, J.; Ortega, Á.; Nava, M.; Salazar, J.; Díaz-Camargo, E.; Medina, O.; Rojas-Quintero, J.; Bermúdez, V. Alzheimer’s disease and type 2 diabetes mellitus: Pathophysiologic and pharmacotherapeutics links. World J. Diabetes 2021, 12, 745–766. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Perluigi, M.; Butterfield, D.A. The interplay among oxidative stress, brain insulin resistance and AMPK dysfunction contribute to neurodegeneration in type 2 diabetes and Alzheimer disease. Free Radic. Biol. Med. 2021, 176, 16–33. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Di Domenico, F.; Barone, E. Elevated risk of type 2 diabetes for development of Alzheimer disease: A key role for oxidative stress in brain. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2014, 1842, 1693–1706. [Google Scholar] [CrossRef] [Green Version]
- Lynn, J.; Park, M.; Ogunwale, C.; Acquaah-Mensah, G.K. A Tale of Two Diseases: Exploring Mechanisms Linking Diabetes Mellitus with Alzheimer’s Disease. J. Alzheimers Dis. 2022, 85, 485–501. [Google Scholar] [CrossRef]
- Femminella, G.D.; Bencivenga, L.; Petraglia, L.; Visaggi, L.; Gioia, L.; Grieco, F.V.; de Lucia, C.; Komici, K.; Corbi, G.; Edison, P.; et al. Antidiabetic Drugs in Alzheimer’s Disease: Mechanisms of Action and Future Perspectives. J. Diabetes Res. 2017, 2017, 7420796. [Google Scholar] [CrossRef] [PubMed]
- Bendlin, B.B. Antidiabetic therapies and Alzheimer disease. Dialogues Clin. Neurosci. 2019, 21, 83–91. [Google Scholar] [PubMed]
- Yan, X.; Hu, Y.; Wang, B.; Wang, S.; Zhang, X. Metabolic Dysregulation Contributes to the Progression of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 530219. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Häring, H.-U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- De Felice, F.G.; Gonçalves, R.A.; Ferreira, S.T. Impaired insulin signalling and allostatic load in Alzheimer disease. Nat. Rev. Neurosci. 2022, 23, 215–230. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Lerner-Marmarosh, N.; Shen, J.; Torno, M.D.; Kravets, A.; Hu, Z.; Maines, M.D. Human biliverdin reductase: A member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 7109–7114. [Google Scholar] [CrossRef] [Green Version]
- Singleton, J.W.; Laster, L. Biliverdin reductase of guinea pig liver. J. Biol. Chem. 1965, 240, 4780–4789. [Google Scholar] [CrossRef]
- Kutty, R.K.; Maines, M.D. Purification and characterization of biliverdin reductase from rat liver. J. Biol. Chem. 1981, 256, 3956–3962. [Google Scholar] [CrossRef]
- Maines, M.D.; Trakshel, G.M. Purification and characterization of human biliverdin reductase. Arch. Biochem. Biophys. 1993, 300, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Otterbein, L.E. Go green: The anti-inflammatory effects of biliverdin reductase. Front. Pharmacol. 2012, 3, 47. [Google Scholar] [CrossRef] [Green Version]
- Barone, E.; Di Domenico, F.; Mancuso, C.; Butterfield, D.A. The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: It’s time for reconciliation. Neurobiol. Dis. 2014, 62, 144–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapitulnik, J.; Maines, M.D. Pleiotropic functions of biliverdin reductase: Cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol. Sci. 2009, 30, 129–137. [Google Scholar] [CrossRef]
- Canesin, G.; Hejazi, S.M.; Swanson, K.D.; Wegiel, B. Heme-Derived Metabolic Signals Dictate Immune Responses. Front. Immunol. 2020, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Maines, M.D. Overview of heme degradation pathway. Curr. Protoc. Toxicol. 2001. [Google Scholar] [CrossRef]
- Barone, E.; Trombino, S.; Cassano, R.; Sgambato, A.; De Paola, B.; Di Stasio, E.; Picci, N.; Preziosi, P.; Mancuso, C. Characterization of the S-denitrosylating activity of bilirubin. J. Cell. Mol. Med. 2009, 13, 2365–2375. [Google Scholar] [CrossRef]
- Vasavda, C.; Kothari, R.; Malla, A.P.; Tokhunts, R.; Lin, A.; Ji, M.; Ricco, C.; Xu, R.; Saavedra, H.G.; Sbodio, J.I.; et al. Bilirubin Links Heme Metabolism to Neuroprotection by Scavenging Superoxide. Cell Chem. Biol. 2019, 26, 1450–1460.e7. [Google Scholar] [CrossRef]
- Gibbs, P.E.; Miralem, T.; Maines, M.D. Biliverdin reductase: A target for cancer therapy? Front. Pharmacol. 2015, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, L.; Hosick, P.A.; John, K.; Stec, D.E.; Hinds, T.D., Jr. Biliverdin reductase isozymes in metabolism. Trends Endocrinol. Metab. 2015, 26, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, T.; Cooper, J.A. Protein-tyrosine kinases. Annu. Rev. Biochem. 1985, 54, 897–930. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T.; Scott, J.D. Protein phosphorylation in signaling—50 years and counting. Trends Biochem. Sci. 2005, 30, 286–290. [Google Scholar] [CrossRef]
- Gibbs, P.E.; Lerner-Marmarosh, N.; Poulin, A.; Farah, E.; Maines, M.D. Human biliverdin reductase-based peptides activate and inhibit glucose uptake through direct interaction with the kinase domain of insulin receptor. FASEB J. 2014, 28, 2478–2491. [Google Scholar] [CrossRef] [Green Version]
- Miralem, T.; Lerner-Marmarosh, N.; Gibbs, P.E.; Jenkins, J.L.; Heimiller, C.; Maines, M.D. Interaction of human biliverdin reductase with Akt/protein kinase B and phosphatidylinositol-dependent kinase 1 regulates glycogen synthase kinase 3 activity: A novel mechanism of Akt activation. FASEB J. 2016, 30, 2926–2944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maines, M.D. Biliverdin reductase: PKC interaction at the cross-talk of MAPK and PI3K signaling pathways. Antioxid. Redox Signal. 2007, 9, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- Lerner-Marmarosh, N.; Miralem, T.; Gibbs, P.E.; Maines, M.D. Regulation of TNF-alpha-activated PKC-zeta signaling by the human biliverdin reductase: Identification of activating and inhibitory domains of the reductase. FASEB J. 2007, 21, 3949–3962. [Google Scholar] [CrossRef]
- Lerner-Marmarosh, N.; Miralem, T.; Gibbs, P.E.; Maines, M.D. Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 6870–6875. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, P.E.; Miralem, T.; Lerner-Marmarosh, N.; Tudor, C.; Maines, M.D. Formation of ternary complex of human biliverdin reductase-protein kinase Cδ-ERK2 protein is essential for ERK2-mediated activation of Elk1 protein, nuclear factor-κB, and inducible nitric-oxidase synthase (iNOS). J. Biol. Chem. 2012, 287, 1066–1079. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, P.E.; Tudor, C.; Maines, M.D. Biliverdin reductase: More than a namesake—The reductase, its Peptide fragments, and biliverdin regulate activity of the three classes of protein kinase C. Front. Pharmacol. 2012, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Lucke-Wold, R.C.; Turner, A.F.; Logsdon, J.W.; Simpkins, D.L.; Alkon, K.E.; Smith, Y.W.; Chen, Z.; Tan, J.D.; Huber, C.L. Common mechanisms of Alzheimer’s disease and ischemic stroke: The role of protein kinase C in the progression of age-related neurodegeneration. J. Alzheimers Dis. 2015, 43, 711–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, Z.; Salim, M.; Maines, M.D. Human biliverdin reductase is a leucine zipper-like DNA-binding protein and functions in transcriptional activation of heme oxygenase-1 by oxidative stress. J. Biol. Chem. 2002, 277, 9226–9232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kravets, A.; Hu, Z.; Miralem, T.; Torno, M.D.; Maines, M.D. Biliverdin reductase, a novel regulator for induction of activating transcription factor-2 and heme oxygenase-1. J. Biol. Chem. 2004, 279, 19916–19923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miralem, T.; Hu, Z.; Torno, M.D.; Lelli, K.M.; Maines, M.D. Small interference RNA-mediated gene silencing of human biliverdin reductase, but not that of heme oxygenase-1, attenuates arsenite-mediated induction of the oxygenase and increases apoptosis in 293A kidney cells. J. Biol. Chem. 2005, 280, 17084–17092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, P.E.; Maines, M.D. Biliverdin inhibits activation of NF-kappaB: Reversal of inhibition by human biliverdin reductase. Int. J. Cancer 2007, 121, 2567–2574. [Google Scholar] [CrossRef] [PubMed]
- Miralem, T.; Gibbs, P.E.; Revert, F.; Saus, J.; Maines, M.D. Human biliverdin reductase suppresses Goodpasture antigen-binding protein (GPBP) kinase activity: The reductase regulates tumor necrosis factor-alpha-NF-kappaB-dependent GPBP expression. J. Biol. Chem. 2010, 285, 12551–12558. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, P.E.; Miralem, T.; Maines, M.D. Characterization of the human biliverdin reductase gene structure and regulatory elements: Promoter activity is enhanced by hypoxia and suppressed by TNF-alpha-activated NF-kappaB. FASEB J. 2010, 24, 3239–3254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegiel, B.; Baty, C.J.; Gallo, D.; Csizmadia, E.; Scott, J.R.; Akhavan, A. Cell surface biliverdin reductase mediates biliverdin-induced anti-inflammatory effects via phosphatidylinositol 3-kinase and Akt. J. Biol. Chem. 2009, 284, 21369–21378. [Google Scholar] [CrossRef] [Green Version]
- Achori, A.S.; Smith, A.; McDonald, P.; Zhang, L.; Dzau, V.J.; Melo, L.G. Heme-oxygenase-1-induced protection against hypoxia/reoxygenation is dependent on biliverdin reductase and its interaction with PI3K/Akt pathway. J. Mol. Cell. Cardiol. 2007, 43, 580–592. [Google Scholar] [CrossRef] [Green Version]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Pomytkin, I.; Pinelis, V. Brain Insulin Resistance: Focus on Insulin Receptor-Mitochondria Interactions. Life 2021, 11, 262. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Kotani, K.; Ciaraldi, T.P.; Henry, R.R.; Kahn, B.B. Insulin-stimulated protein kinase C lambda/zeta activity is reduced in skeletal muscle of humans with obesity and type 2 diabetes: Reversal with weight reduction. Diabetes 2003, 52, 1935–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Tramutola, A.; Lanzillotta, C.; Arena, A.; Blarzino, C.; Cassano, T.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer’s disease. Neurobiol. Dis. 2019, 125, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, P.E.; Miralem, T.; Lerner-Marmarosh, N.; Maines, M.D. Nanoparticle Delivered Human Biliverdin Reductase-Based Peptide Increases Glucose Uptake by Activating IRK/Akt/GSK3 Axis: The Peptide Is Effective in the Cell and Wild-Type and Diabetic Ob/Ob Mice. J. Diabetes Res. 2016, 2016, 4712053. [Google Scholar] [CrossRef] [Green Version]
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; Vanden Heuvel, J.P.; Stec, D.E. Biliverdin Reductase a Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3β Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) α. J. Biol. Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef] [Green Version]
- Stec, D.E.; Gordon, D.M.; Nestor-Kalinoski, A.L.; Donald, M.C.; Mitchell, Z.L.; Creeden, J.F.; Hinds, T.D., Jr. Biliverdin Reductase A (BVRA) Knockout in Adipocytes Induces Hypertrophy and Reduces Mitochondria in White Fat of Obese Mice. Biomolecules 2020, 10, 387. [Google Scholar] [CrossRef] [Green Version]
- Hinds, T.D., Jr.; Creeden, J.F.; Gordon, D.M.; Spegele, A.C.; Britton, S.L.; Koch, L.G.; Stec, D.E. Rats Genetically Selected for High Aerobic Exercise Capacity Have Elevated Plasma Bilirubin by Upregulation of Hepatic Biliverdin Reductase-A (BVRA) and Suppression of UGT1A1. Antioxidants 2020, 9, 889. [Google Scholar] [CrossRef]
- Chen, W.; Tumanov, S.; Fazakerley, D.J.; Cantley, J.; James, D.E.; Dunn, L.L.; Shaik, T.; Suarna, C.; Stocker, R. Bilirubin deficiency renders mice susceptible to hepatic steatosis in the absence of insulin resistance. Redox Biol. 2021, 47, 102152. [Google Scholar] [CrossRef]
- Cimini, F.A.; Arena, A.; Barchetta, I.; Tramutola, A.; Ceccarelli, V.; Lanzillotta, C.; Fontana, M.; Bertoccini, L.; Leonetti, F.; Capoccia, D.; et al. Reduced biliverdin reductase-A levels are associated with early alterations of insulin signaling in obesity. BiochimBiophys. Acta BBA-Mol. Basis Dis. 2019, 1865, 1490–1501. [Google Scholar] [CrossRef]
- Ceccarelli, V.; Barchetta, I.; Cimini, F.A.; Bertoccini, L.; Chiappetta, C.; Capoccia, D.; Carletti, R.; Di Cristofano, C.; Silecchia, G.; Fontana, M.; et al. Reduced Biliverdin Reductase-A Expression in Visceral Adipose Tissue is Associated with Adipocyte Dysfunction and NAFLD in Human Obesity. Int. J. Mol. Sci. 2020, 21, 9091. [Google Scholar] [CrossRef]
- Takata, T.; Okada, Y. Effects of deprivation of oxygen or glucose on the neural activity in the guinea pig hippocampal slice—Intracellular recording study of pyramidal neurons. Brain Res. 1995, 683, 109–116. [Google Scholar] [CrossRef]
- Cimini, F.A.; Barchetta, I.; Zuliani, I.; Pagnotta, S.; Bertoccini, L.; Dule, S.; Zampieri, M.; Reale, A.; Baroni, M.G.; Cavallo, M.G.; et al. Biliverdin reductase-A protein levels are reduced in type 2 diabetes and are associated with poor glycometabolic control. Life Sci. 2021, 284, 119913. [Google Scholar] [CrossRef] [PubMed]
- Howarth, C.; Gleeson, P.; Attwell, D. Updated energy budgets for neural computation in the neocortex and cerebellum. J. Cereb. Blood Flow Metab. 2012, 32, 1222–1232. [Google Scholar] [CrossRef]
- Han, R.; Liang, J.; Zhou, B. Glucose Metabolic Dysfunction in Neurodegenerative Diseases-New Mechanistic Insights and the Potential of Hypoxia as a Prospective Therapy Targeting Metabolic Reprogramming. Int. J. Mol. Sci. 2021, 22, 5887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Alshakhshir, N.; Zhao, L. Glycolytic Metabolism, Brain Resilience, and Alzheimer’s Disease. Front. Neurosci. 2021, 15, 662242. [Google Scholar] [CrossRef]
- Yamane, K.; Yokono, K.; Okada, Y. Anaerobic glycolysis is crucial for the maintenance of neural activity in guinea pig hippocampal slices. J. Neurosci. Methods 2000, 103, 163–171. [Google Scholar] [CrossRef]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflügers Arch. Eur. J. Physiol. 2020, 472, 1299–1343. [Google Scholar] [CrossRef]
- White, M.F.; Copps, K.D. The mechanisms of insulin action. In Endocrinology: Adult and Pediatric, 7th ed.; Saunders, W.B., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 556–585. [Google Scholar]
- Kapogiannis, D.; Mustapic, M.; Shardell, M.D.; Berkowitz, S.T.; Diehl, T.C.; Spangler, R.D.; Tran, J.; Lazaropoulos, M.P.; Chawla, S.; Gulyani, S.; et al. Association of Extracellular Vesicle Biomarkers with Alzheimer Disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol. 2019, 76, 1340–1351. [Google Scholar] [CrossRef]
- Tramutola, A.; Lanzillotta, C.; Di Domenico, F.; Head, E.; Butterfield, D.A.; Perluigi, M.; Barone, E. Brain insulin resistance triggers early onset Alzheimer disease in Down syndrome. Neurobiol. Dis. 2020, 137, 104772. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinelli, M.; Fusco, S.; Mainardi, M.; Scala, F.; Natale, F.; Lapenta, R.; Mattera, A.; Rinaudo, M.; Li Puma, D.D.; Ripoli, C.; et al. Brain insulin resistance impairs hippocampal synaptic plasticity and memory by increasing GluA1 palmitoylation through FoxO3a. Nat. Commun. 2017, 8, 2009. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Tramutola, A.; Triani, F.; Calcagnini, S.; Di Domenico, F.; Ripoli, C.; Gaetani, S.; Grassi, C.; Butterfield, D.A.; Cassano, T.; et al. Biliverdin reductase-a mediates the beneficial effects of intranasal insulin in Alzheimer disease. Mol. Neurobiol. 2019, 56, 2922–2943. [Google Scholar] [CrossRef]
- Franklin, W.; Krishnan, B.; Taglialatela, G. Chronic synaptic insulin resistance after traumatic brain injury abolishes insulin protection from amyloid beta and tau oligomer-induced synaptic dysfunction. Sci. Rep. 2019, 9, 8228. [Google Scholar] [CrossRef] [PubMed]
- Melo, H.M.; Seixas da Silva GD, S.; Sant’Ana, M.R.; Teixeira CV, L.; Clarke, J.R.; Coreixas, V.S.; de Melo, B.C.; Fortuna, J.T.; Forny-Germano, L.; Ledo, J.H.; et al. Palmitate is increased in the cerebrospinal fluid of humans with obesity and induces memory impairment in mice via pro-inflammatory TNF-alpha. Cell Rep. 2020, 30, 2180–2194.e8. [Google Scholar] [CrossRef] [Green Version]
- Lanzillotta, C.; Tramutola, A.; Di Giacomo, G.; Marini, F.; Butterfield, D.A.; Di Domenico, F.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Insulin resistance, oxidative stress and mitochondrial defects in Ts65dn mice brain: A harmful synergistic path in down syndrome. Free Radic. Biol. Med. 2021, 165, 152–170. [Google Scholar] [CrossRef]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Abeta oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Barone, E.; Di Domenico, F.; Cassano, T.; Arena, A.; Tramutola, A.; Lavecchia, M.A.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic. Biol. Med. 2016, 91, 127–142. [Google Scholar] [CrossRef]
- Triani, F.; Tramutola, A.; Di Domenico, F.; Sharma, N.; Butterfield, D.A.; Head, E.; Perluigi, M.; Barone, E. Biliverdin reductase-A impairment links brain insulin resistance with increased Abeta production in an animal model of aging: Implications for Alzheimer disease. BiochimBiophys. Acta BBA-Mol. Basis Dis. 2018, 1864, 3181–3194. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimers Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef]
- Steen, B.M.; Terry, E.J.; Rivera, J.L.; Cannon, T.R.; Neely, R.; Tavares, X.J.; Xu, J.R.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Monte, S.M. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs 2012, 72, 49–66. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef] [Green Version]
- Pastorino, L.; Ikin, A.F.; Nairn, A.C.; Pursnani, A.; Buxbaum, J.D. The carboxyl-terminus of BACE contains a sorting signal that regulates BACE trafficking but not the formation of total A(beta). Mol. Cell. Neurosci. 2002, 19, 175–185. [Google Scholar] [CrossRef]
- Walter, R.; Fluhrer, B.; Hartung, M.; Willem, C.; Kaether, A.; Capell, S.; Lammich, G.; Multhaup, C.; Haass, C. Phosphorylation regulates intracellular trafficking of beta-secretase. J. Biol. Chem. 2001, 276, 14634–14641. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR Signaling. Cold Spring Harb Perspect. Biol. 2012, 4, a011593. [Google Scholar] [CrossRef]
- O’Neill, C. PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp. Gerontol. 2013, 48, 647–653. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: Effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef] [Green Version]
- Vartak, R.S.; Rodin, A.; Oddo, S. Differential activation of the mTOR/autophagy pathway predicts cognitive performance in APP/PS1 mice. Neurobiol. Aging 2019, 83, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Triplett, J.C.; Di Domenico, F.; Niedowicz, D.M.; Murphy, M.P.; Coccia, R.; Perluigi, M.; Butterfield, D.A. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): Analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J. Neurochem. 2015, 133, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Tramutola, A.; Barone, E.; Lanzillotta, C.; Defever, O.; Arena, A.; Zuliani, I.; Foppoli, C.; Iavarone, F.; Vincenzoni, F.; et al. Restoration of aberrant mTOR signaling by intranasal rapamycin reduces oxidative damage: Focus on HNE-modified proteins in a mouse model of down syndrome. Redox Biol. 2019, 23, 101162. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Lanzillotta, C.; Arena, A.; Barone, E.; Perluigi, M.; Di Domenico, F. Increased Mammalian Target of Rapamycin Signaling Contributes to the Accumulation of Protein Oxidative Damage in a Mouse Model of Down’s Syndrome. Neurodegener. Dis. 2016, 16, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Di Domenico, F.; Barone, E.; Butterfield, D.A. mTOR in Alzheimer disease and its earlier stages: Links to oxidative damage in the progression of this dementing disorder. Free Radic. Biol. Med. 2021, 169, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, C.; Zuliani, I.; Vasavda, C.; Snyder, S.H.; Paul, B.D.; Perluigi, M.; Di Domenico, F.; Barone, E. BVR-A Deficiency Leads to Autophagy Impairment through the Dysregulation of AMPK/mTOR Axis in the Brain—Implications for Neurodegeneration. Antioxidants 2020, 9, 671. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Carling, D. AMPK signalling in health and disease. Curr. Opin. Cell Biol. 2017, 45, 31–37. [Google Scholar] [CrossRef]
- Peixoto, C.A.; Oliveira, W.H.; Araujo, S.; Nunes, A.K.S. AMPK activation: Role in the signaling pathways of neuroinflammation and neurodegeneration. Exp. Neurol. 2017, 298, 31–41. [Google Scholar] [CrossRef]
- De Felice, F.G. Alzheimer’s disease and insulin resistance: Translating basic science into clinical applications. J. Clin. Investig. 2013, 123, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Reger, M.A.; Watson, G.S.; Frey, W.H.; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostolatos, A.; Song, S.; Acosta, S.; Peart, M.; Watson, J.E.; Bickford, P.; Cooper, D.R.; Patel, N.A. Insulin promotes neuronal survival via the alternatively spliced protein kinase CdeltaII isoform. J. Biol. Chem. 2012, 287, 9299–9310. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.F.; Guo, Z.; Zheng, T.; Jiang, Y.; Yan, Y.; Yin, X.; Chen, Y.; Zhang, B. Intranasal insulin alleviates cognitive deficits and amyloid pathology in young adult APPswe/PS1dE9 mice. Aging Cell 2016, 15, 893–902. [Google Scholar] [CrossRef] [Green Version]
- Salameh, T.S.; Bullock, K.M.; Hujoel, I.A.; Niehoff, M.L.; Wolden-Hanson, T.; Kim, J.; Morley, J.E.; Farr, S.A.; Banks, W.A. Central nervous system delivery of intranasal insulin: Mechanisms of uptake and effects on cognition. J. Alzheimers Dis. 2015, 47, 715–728. [Google Scholar] [CrossRef]
- Zhang, Y.; Dai, C.L.; Chen, Y.; Iqbal, K.; Liu, F.; Gong, C.X. Intranasal insulin prevents anesthesia-induced spatial learning and memory deficit in mice. Sci. Rep. 2016, 6, 21186. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Dai, C.L.; Wu, Z.; Iqbal, K.; Liu, F.; Zhang, B.; Gong, C.X. Intranasal insulin prevents anesthesia-induced cognitive impairment and chronic neurobehavioral changes. Front. Aging Neurosci. 2017, 9, 136. [Google Scholar] [CrossRef] [Green Version]
- Nedelcovych, M.T.; Gadiano, A.J.; Wu, Y.; Manning, A.A.; Thomas, A.G.; Khuder, S.S.; Yoo, S.W.; Xu, J.; McArthur, J.C.; Haughey, N.J.; et al. Pharmacokinetics of intranasal versus subcutaneous insulin in the mouse. ACS Chem. Neurosci. 2018, 9, 809–816. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef]
- Chauhan, M.B.; Chauhan, N.B. Brain uptake of neurotherapeutics after intranasal versus intraperitoneal delivery in mice. J. Neurol. Neurosurg. 2015, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Wiseman, F.K.; Al-Janabi, T.; Hardy, J.; Karmiloff-Smith, A.; Nizetic, D.; Tybulewicz, V.L.; Fisher, E.; Strydom, A. A genetic cause of Alzheimer disease: Mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 2015, 16, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Perluigi, M.; Picca, A.; Montanari, E.; Calvani, R.; Marini, F.; Matassa, R.; Tramutola, A.; Villani, A.; Familiari, G.; Domenico, F.D.; et al. Aberrant crosstalk between insulin signaling and mTOR in young Down syndrome individuals revealed by neuronal-derived extracellular vesicles. Alzheimers Dement. 2021. [Google Scholar] [CrossRef]
- Antonarakis, S.E.; Skotko, B.G.; Rafii, M.S.; Strydom, A.; Pape, S.E.; Bianchi, D.W.; Sherman, S.L.; Reeves, R.H. Down syndrome. Nat. Rev. Dis. Primers 2020, 6, 9. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Cini, C.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Biliverdin reductase-a protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. BiochimBiophys. Acta BBA-Mol. Basis Dis. 2011, 1812, 480–487. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Coccia, R.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer’s disease and amnestic mild cognitive impairment. J. Alzheimers Dis. 2011, 25, 623–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, R.; Roediger, F.; Jordan, B.; Mattson, M.P.; Keller, J.N.; Waeg, G.; Butterfield, D.A. The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortical synaptosomal membrane proteins. J. Neurochem. 1997, 69, 1161–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, D.A.; Di Domenico, F.; Swomley, A.M.; Head, E.; Perluigi, M. Redox proteomics analysis to decipher the neurobiology of Alzheimer-like neurodegeneration: Overlaps in Down’s syndrome and Alzheimer’s disease brain. Biochem. J. 2014, 463, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Stadman, E.R. Protein oxidation processes in aging brain. In Advances in Cell Aging and Gerontology; Paula, S.T., Bittar, E.E., Eds.; Elsevier: Amsterdam, The Netherlands, 1997; pp. 161–191. [Google Scholar]
- Hyman, B.T.; Elvhage, T.E.; Reiter, J. Extracellular signal regulated kinases. Localization of protein and mRNA in the human hippocampal formation in Alzheimer’s disease. Am. J. Pathol. 1994, 144, 565–572. [Google Scholar] [PubMed]
- Perluigi, M.; Barone, E. Aberrant protein networks in Alzheimer disease. Nat. Rev. Neurol. 2022, 18, 255–256. [Google Scholar] [CrossRef]
- Johnson, E.C.; Carter, E.K.; Dammer, E.B.; Duong, D.M.; Gerasimov, E.S.; Liu, Y.; Liu, J.; Betarbet, R.; Ping, L.; Yin, L.; et al. Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat. Neurosci. 2022, 25, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Palozza, P.; Barone, E.; Mancuso, C.; Picci, N. The protective role of carotenoids against 7-keto-cholesterol formation in solution. Mol. Cell. Biochem. 2008, 309, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Pupo, G.; Mancuso, C.; Barone, E.; Paolini, F.; Arena, A.; Blarzino, C.; Schmitt, F.A.; Head, E.; Butterfield, D.A.; et al. Bach1 overexpression in Down syndrome correlates with the alteration of the HO-1/BVR-a system: Insights for transition to Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 1107–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garagnani, P.; Marquis, J.; Delledonne, M.; Pirazzini, C.; Marasco, E.; Kwiatkowska, K.M.; Iannuzzi, V.; Bacalini, M.G.; Valsesia, A.; Carayol, J.; et al. Whole-genome sequencing analysis of semi-supercentenarians. Elife 2021, 10, e57849. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.; Doblhammer, G.; Rau, R.; Vaupel, J.W. Ageing populations: The challenges ahead. Lancet 2009, 374, 1196–1208. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Bonafè, M. Centenarians as a model for healthy aging. Biochem. Soc. Trans. 2003, 31, 457–461. [Google Scholar] [CrossRef]
- Lin, J.R.; Sin-Chan, P.; Napolioni, V.; Torres, G.G.; Mitra, J.; Zhang, Q.; Jabalameli, M.R.; Wang, Z.; Nguyen, N.; Gao, T.; et al. Rare genetic coding variants associated with human longevity and protection against age-related diseases. Nat. Aging 2021, 1, 783–794. [Google Scholar] [CrossRef]
| Model | BVR-A Alteration(s) | Insulin Signaling Alteration(s) | Observed Effect(s) | Ref. |
|---|---|---|---|---|
| Ob/Ob mice | Stimulation of BVR-A kinase activity by KYCCSRK peptide | Increase of IR activation | Rapid glucose clearance from the circulation | [55] |
| Liver-specific BVRA KO mice | Liver deletion of BVR-A | Reduced GSK3β inhibition | Impaired glucose tolerance and development of fatty liver | [56] |
| HFD-treated BVRA KO mice | Adipocyte deletion of BVR-A | Decreased Akt activation and reduced GLUT4 levels | High fasting blood glucose levels; adipocytes hypertrophy and reduction of mitochondrial number in white adipose tissue | [57] |
| BVRA KO mice | Global BVRA deficiency | - | Fatty liver without alteration in glucose metabolism and insulin sensitivity | [58] |
| Obese subjects | Reduced BVR-A levels in PBMC | Aberrant activation of insulin signalling characterized by: reduced IRSSer307/IRS1 ratio; increased pAktSer473/Akt and increased pGSK3βSer9/GSK3β ratio; increased AS160-mediated GLUT4 translocation | Metabolic syndrome, presence and severity of NAFLD and adipose tissue dysfunction | [59] |
| Obese subjects | Reduced BVR-A expression in visceral adipose tissue | - | Larger adipocytes size and greater local expression of inflammatory and hypoxia markers | [60] |
| T2D subjects | Reduced BVR-A levels in PBMC | - | Glyco-metabolic impairment and increased inflammatory condition | [61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cimini, F.A.; Perluigi, M.; Barchetta, I.; Cavallo, M.G.; Barone, E. Role of Biliverdin Reductase A in the Regulation of Insulin Signaling in Metabolic and Neurodegenerative Diseases: An Update. Int. J. Mol. Sci. 2022, 23, 5574. https://doi.org/10.3390/ijms23105574
Cimini FA, Perluigi M, Barchetta I, Cavallo MG, Barone E. Role of Biliverdin Reductase A in the Regulation of Insulin Signaling in Metabolic and Neurodegenerative Diseases: An Update. International Journal of Molecular Sciences. 2022; 23(10):5574. https://doi.org/10.3390/ijms23105574
Chicago/Turabian StyleCimini, Flavia Agata, Marzia Perluigi, Ilaria Barchetta, Maria Gisella Cavallo, and Eugenio Barone. 2022. "Role of Biliverdin Reductase A in the Regulation of Insulin Signaling in Metabolic and Neurodegenerative Diseases: An Update" International Journal of Molecular Sciences 23, no. 10: 5574. https://doi.org/10.3390/ijms23105574
APA StyleCimini, F. A., Perluigi, M., Barchetta, I., Cavallo, M. G., & Barone, E. (2022). Role of Biliverdin Reductase A in the Regulation of Insulin Signaling in Metabolic and Neurodegenerative Diseases: An Update. International Journal of Molecular Sciences, 23(10), 5574. https://doi.org/10.3390/ijms23105574