
Complex Mutation Pattern of Omicron BA.2: Evading Antibodies without Losing Receptor Interactions

 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Prevalence of BA.2 Signature Mutations
2.2. Analysis of Structural Data and Impact of Mutations on Overall S-RBD Structure
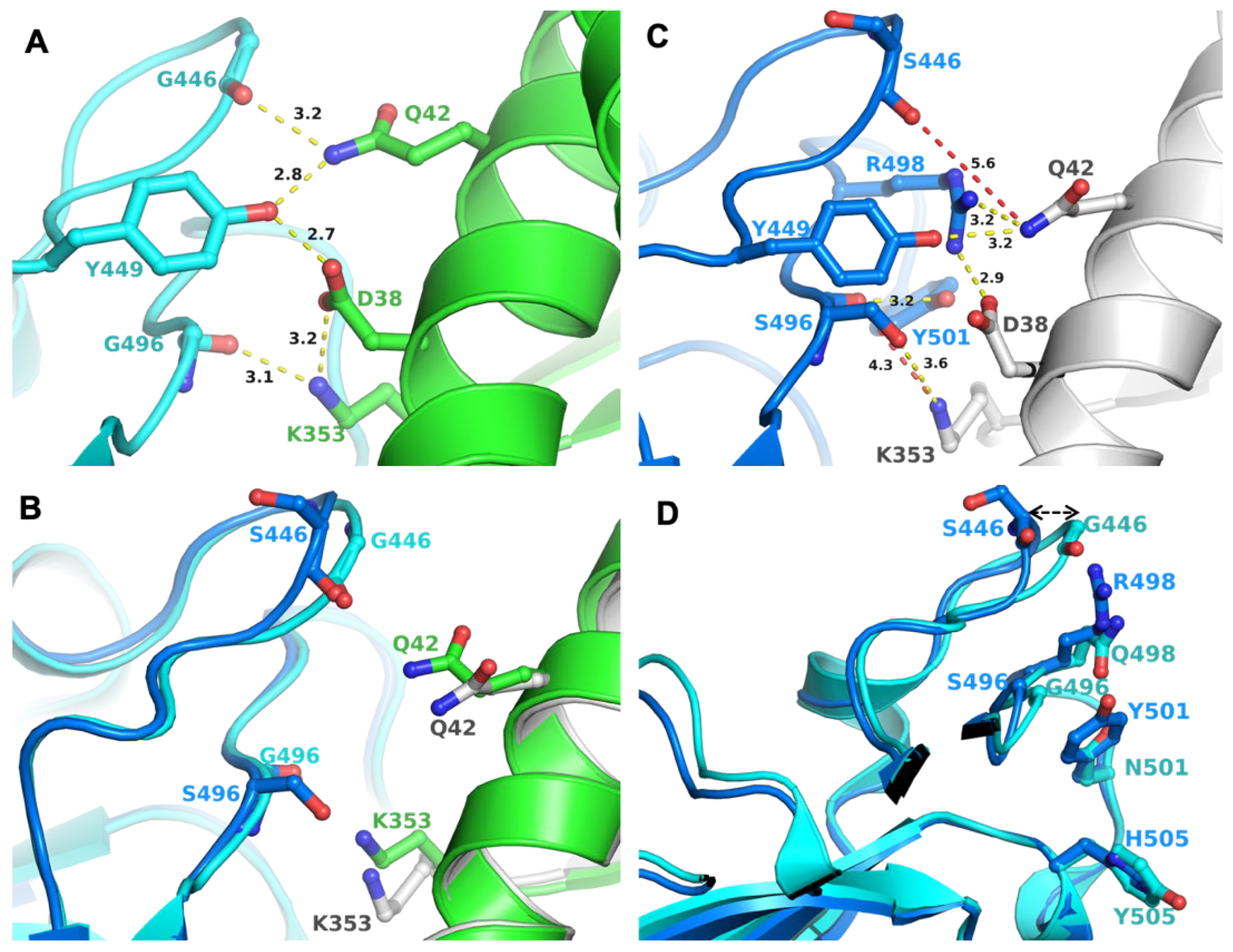
2.3. Impact of Mutations on S-RBD/ACE2 Interaction
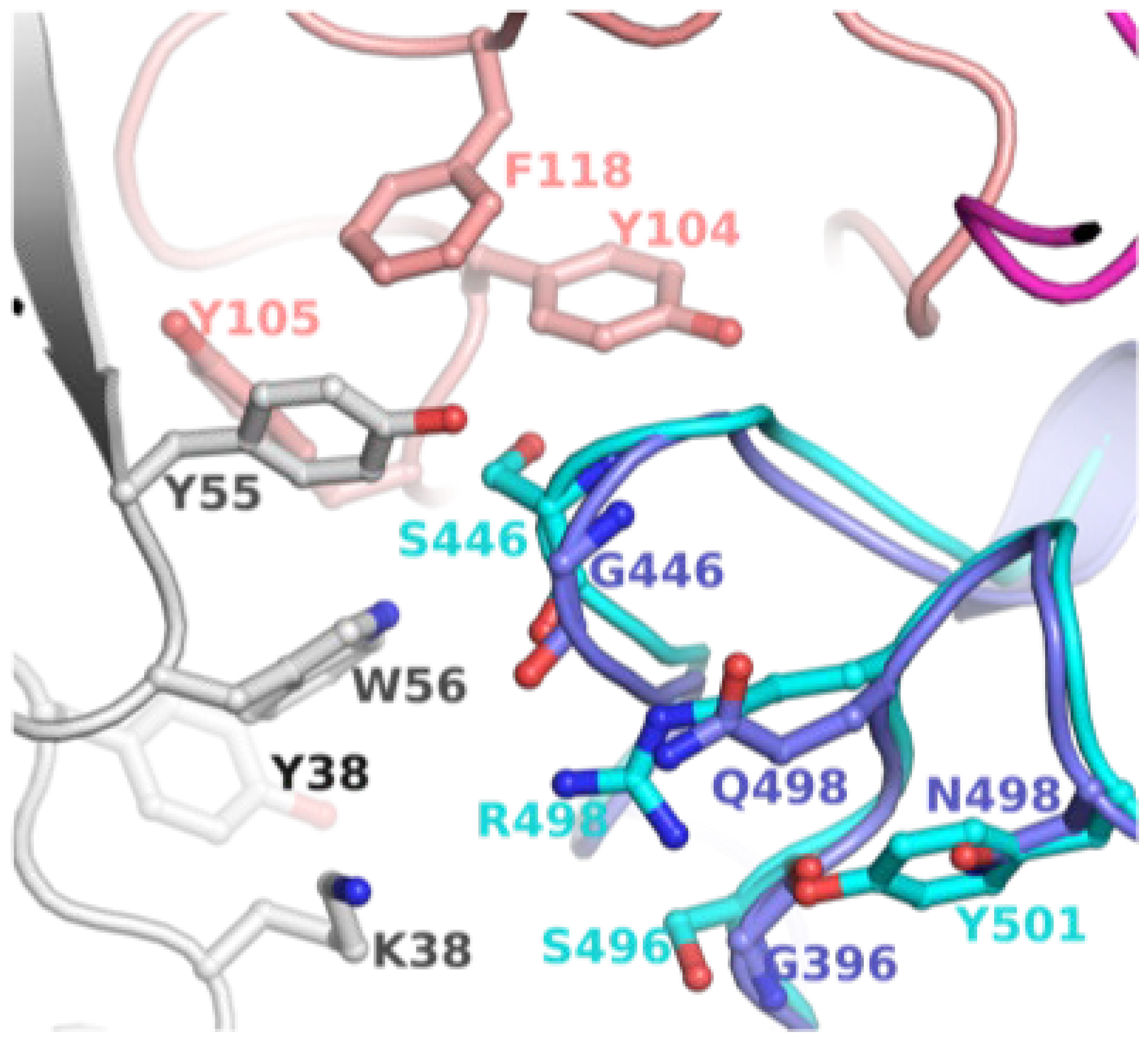
2.4. BA2 Unique Mutations in Relation to mAb Binding
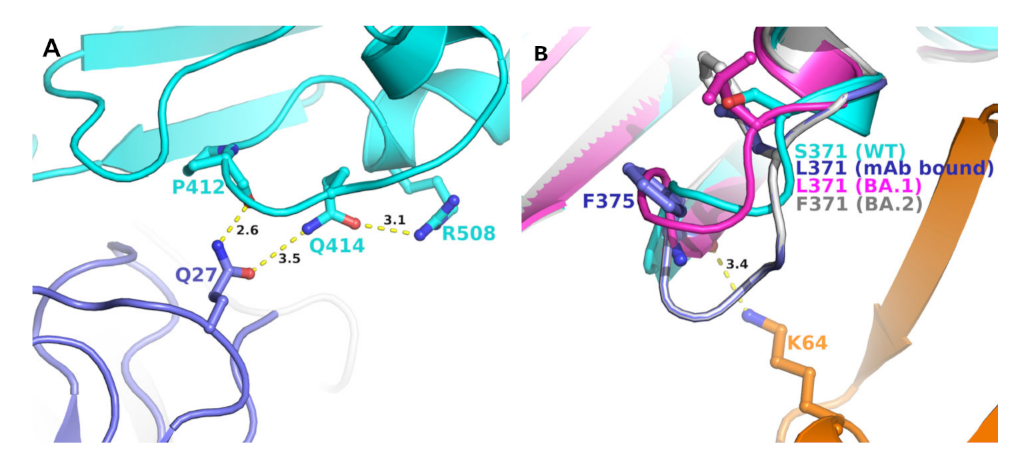
2.5. BA2 Unique Mutations in Relation to N-Terminal Directed mAb Binding
2.6. BA2 Unique Mutations in ORFs Other than the S-Protein
3. Discussion
4. Materials and Methods
4.1. Sequence Acquisition and Analysis
4.2. Structural Analysis
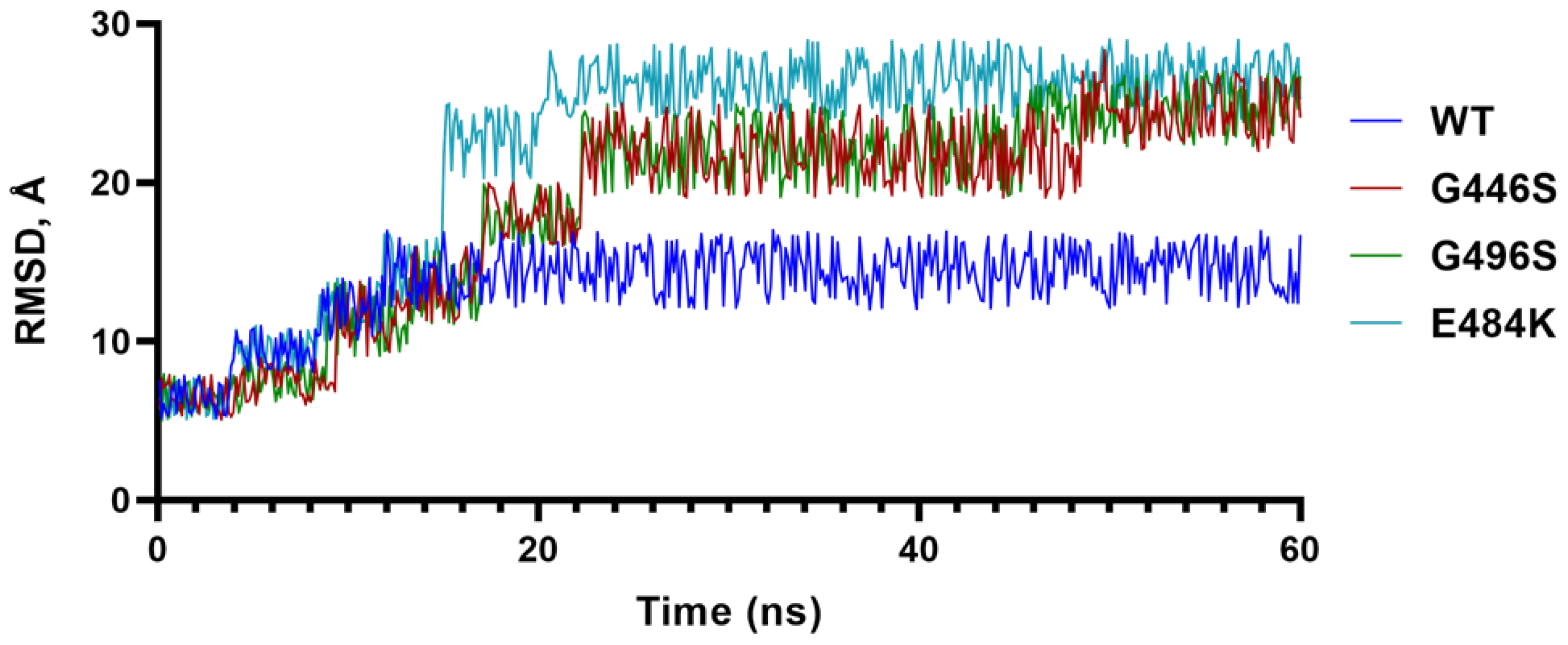
4.3. Molecular Dynamics (MD) Simulation
5. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in china. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauring, A.S.; Malani, P.N. Variants of SARS-CoV-2. JAMA 2021, 326, 880. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.R.; Spratt, A.N.; Cohen, A.R.; Naqvi, S.H.; Chand, H.S.; Quinn, T.P.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Evolutionary analysis of the delta and delta plus variants of the SARS-CoV-2 viruses. J. Autoimmun. 2021, 124, 102715. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Mittal, A.; Khattri, A.; Verma, V. Structural and antigenic variations in the spike protein of emerging SARS-CoV-2 variants. PLoS Pathog. 2022, 18, e1010260. [Google Scholar] [CrossRef]
- Mahase, E. COVID-19: What do we know about omicron sublineages? BMJ 2022, 376, o358. [Google Scholar] [CrossRef]
- Schmidt, F.; Weisblum, Y.; Rutkowska, M.; Poston, D.; DaSilva, J.; Zhang, F.; Bednarski, E.; Cho, A.; Schaefer-Babajew, D.J.; Gaebler, C.; et al. High genetic barrier to SARS-CoV-2 polyclonal neutralizing antibody escape. Nature 2021, 600, 512. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: Gisaid’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Kannan, S.R.; Spratt, A.N.; Sharma, K.; Chand, H.S.; Byrareddy, S.N.; Singh, K. Omicron SARS-CoV-2 variant: Unique features and their impact on pre-existing antibodies. J. Autoimmun. 2022, 126, 102779. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 omicron variant: Antibody evasion and cryo-em structure of spike protein-ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Cameroni, E.; Bowen, J.E.; Rosen, L.E.; Saliba, C.; Zepeda, S.K.; Culap, K.; Pinto, D.; VanBlargan, L.A.; De Marco, A.; di Iulio, J.; et al. Broadly neutralizing antibodies overcome SARS-CoV-2 omicron antigenic shift. Nature 2022, 602, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Dong, J.; Zost, S.J.; Greaney, A.J.; Starr, T.N.; Dingens, A.S.; Chen, E.C.; Chen, R.E.; Case, J.B.; Sutton, R.E.; Gilchuk, P.; et al. Genetic and structural basis for SARS-CoV-2 variant neutralization by a two-antibody cocktail. Nat. Microbiol. 2021, 6, 1233–1244. [Google Scholar] [CrossRef]
- Zost, S.J.; Gilchuk, P.; Case, J.B.; Binshtein, E.; Chen, R.E.; Nkolola, J.P.; Schafer, A.; Reidy, J.X.; Trivette, A.; Nargi, R.S.; et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature 2020, 584, 443–449. [Google Scholar] [CrossRef]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 delta p681r mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef]
- Yan, L.; Yang, Y.; Li, M.; Zhang, Y.; Zheng, L.; Ge, J.; Huang, Y.C.; Liu, Z.; Wang, T.; Gao, S.; et al. Coupling of n7-methyltransferase and 3′-5′ exoribonuclease with SARS-CoV-2 polymerase reveals mechanisms for capping and proofreading. Cell 2021, 184, 3474–3485.e11. [Google Scholar] [CrossRef]
- Zhao, Y.; Fang, C.; Zhang, Q.; Zhang, R.; Zhao, X.; Duan, Y.; Wang, H.; Zhu, Y.; Feng, L.; Zhao, J.; et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor pf-07321332. Protein Cell 2021. [Google Scholar] [CrossRef]
- Rockett, R.; Basile, K.; Maddocks, S.; Fong, W.; Agius, J.E.; Johnson-Mackinnon, J.; Arnott, A.; Chandra, S.; Gall, M.; Draper, J.; et al. Resistance mutations in SARS-CoV-2 delta variant after sotrovimab use. N. Engl. J. Med. 2022, 386, 1477–1479. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA x: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Troshin, P.V.; Procter, J.B.; Barton, G.J. Java bioinformatics analysis web services for multiple sequence alignment—Jabaws: MSA. Bioinformatics 2011, 27, 2001–2002. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. An open-source molecular graphics tool. CCP4 newsletter on protein crystallography. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VOC | Country and Date of Origin | Mutations in S-Protein |
|---|---|---|
| Alpha | The United Kingdom, September-2020 | ∆H69, ∆V70, ∆Y144, E484K, N501Y, A570D, D614G, P681H, T716I, S982A, and D1118H |
| Beta | South Africa, May-2020 | L18F, D80A, D215G, ∆L242, ∆A243, ∆L244, R246I, K417N, E484K, N501Y, D614G, and A701V |
| Gamma | Brazil, November-2020 | L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, and T1027I |
| Delta | India, October-2020 | T19R, V70F, T95I, G142D, DelE156-, F157-, R158G, A222V, W258L, K417N, L452R, T478K, D614G, P681R, and D950N |
| Omicron (BA.1) | South Africa, November-2021 | A76V, T95I, Y145del, L212I, G339D, S371L, S373P, S375F, K417N N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, and L981F |
| BA.1 (Original) | BA.2 | WT | Delta |
|---|---|---|---|
| T19I | T19R | ||
| PPA25-27Del | |||
| A67V | |||
| T95I | T95 | T95I | |
| G142D | G142D | ||
| VYY143-145Del | |||
| N211Del | |||
| L212I | |||
| V213G | |||
| R214EPEins | |||
| S371L | S371F | ||
| T376A | |||
| D405N | |||
| R408S | |||
| G446S | G446 | ||
| L452 | L452R | ||
| G496S | G496 | ||
| T547K | |||
| N856K | |||
| D950 | D950N | ||
| L981F |
| BA.1 (Original) | BA.2 |
|---|---|
| M:D3G | |
| N:S413R | |
| nsp1:S135R | |
| nsp3:T24I | |
| nsp3:K38R | |
| nsp3:L1266I | |
| nsp3:G489S | |
| nsp3:A1892T | |
| nsp4:L264F | |
| nsp4:T327I | |
| nsp4:L438F | |
| nsp6:I189V | |
| nsp14:R391C | |
| nsp15:T112I | |
| ORF3a:T223I | |
| ORF6:D61L |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kannan, S.R.; Spratt, A.N.; Sharma, K.; Goyal, R.; Sönnerborg, A.; Apparsundaram, S.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Complex Mutation Pattern of Omicron BA.2: Evading Antibodies without Losing Receptor Interactions. Int. J. Mol. Sci. 2022, 23, 5534. https://doi.org/10.3390/ijms23105534
Kannan SR, Spratt AN, Sharma K, Goyal R, Sönnerborg A, Apparsundaram S, Lorson CL, Byrareddy SN, Singh K. Complex Mutation Pattern of Omicron BA.2: Evading Antibodies without Losing Receptor Interactions. International Journal of Molecular Sciences. 2022; 23(10):5534. https://doi.org/10.3390/ijms23105534
Chicago/Turabian StyleKannan, Saathvik R., Austin N. Spratt, Kalicharan Sharma, Ramesh Goyal, Anders Sönnerborg, Subbu Apparsundaram, Christian L. Lorson, Siddappa N. Byrareddy, and Kamal Singh. 2022. "Complex Mutation Pattern of Omicron BA.2: Evading Antibodies without Losing Receptor Interactions" International Journal of Molecular Sciences 23, no. 10: 5534. https://doi.org/10.3390/ijms23105534
APA StyleKannan, S. R., Spratt, A. N., Sharma, K., Goyal, R., Sönnerborg, A., Apparsundaram, S., Lorson, C. L., Byrareddy, S. N., & Singh, K. (2022). Complex Mutation Pattern of Omicron BA.2: Evading Antibodies without Losing Receptor Interactions. International Journal of Molecular Sciences, 23(10), 5534. https://doi.org/10.3390/ijms23105534