
Effects of RU486 in Treatment of Traumatic Stress-Induced Glucocorticoid Dysregulation and Fear-Related Abnormalities: Early versus Late Intervention



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
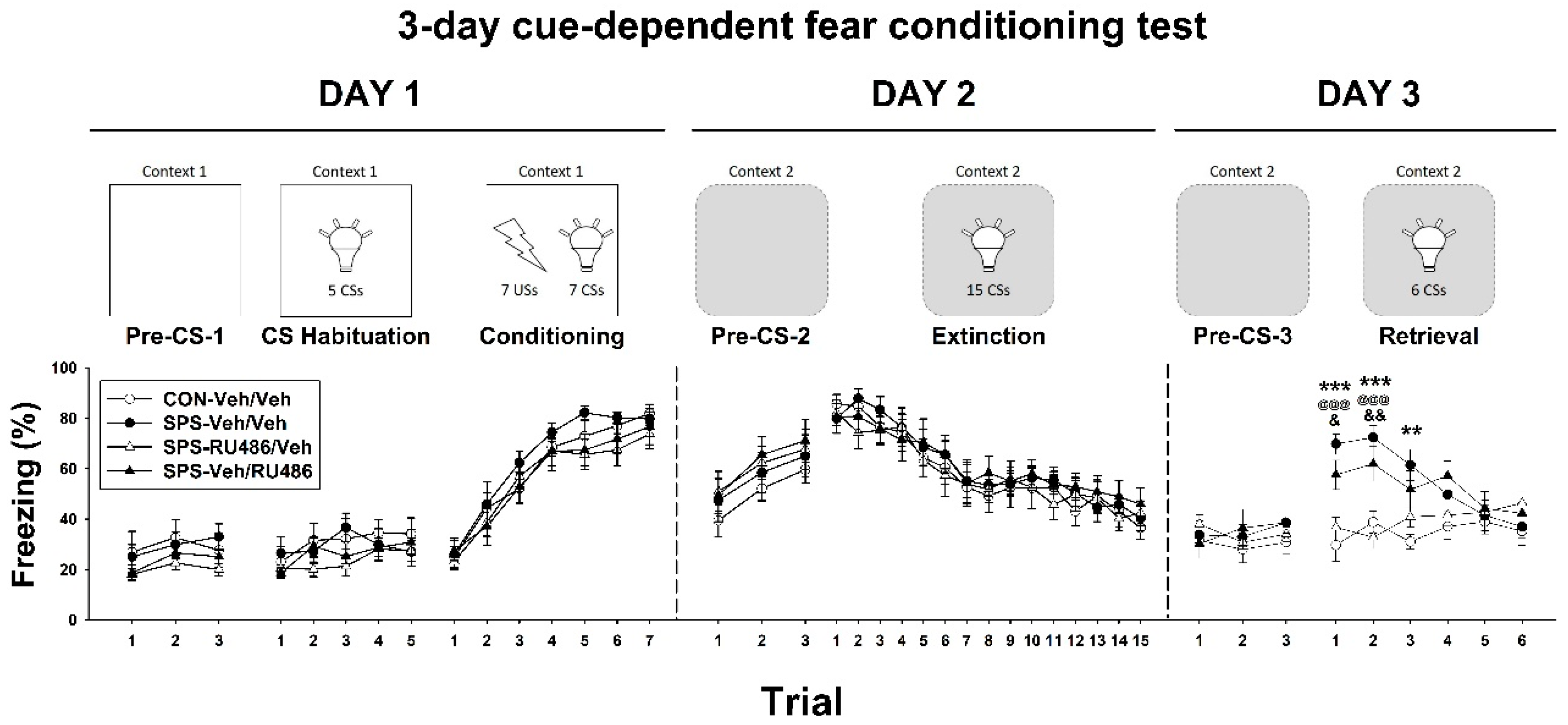
2.1. Three-Day Cue-Dependent Fear Conditioning Test
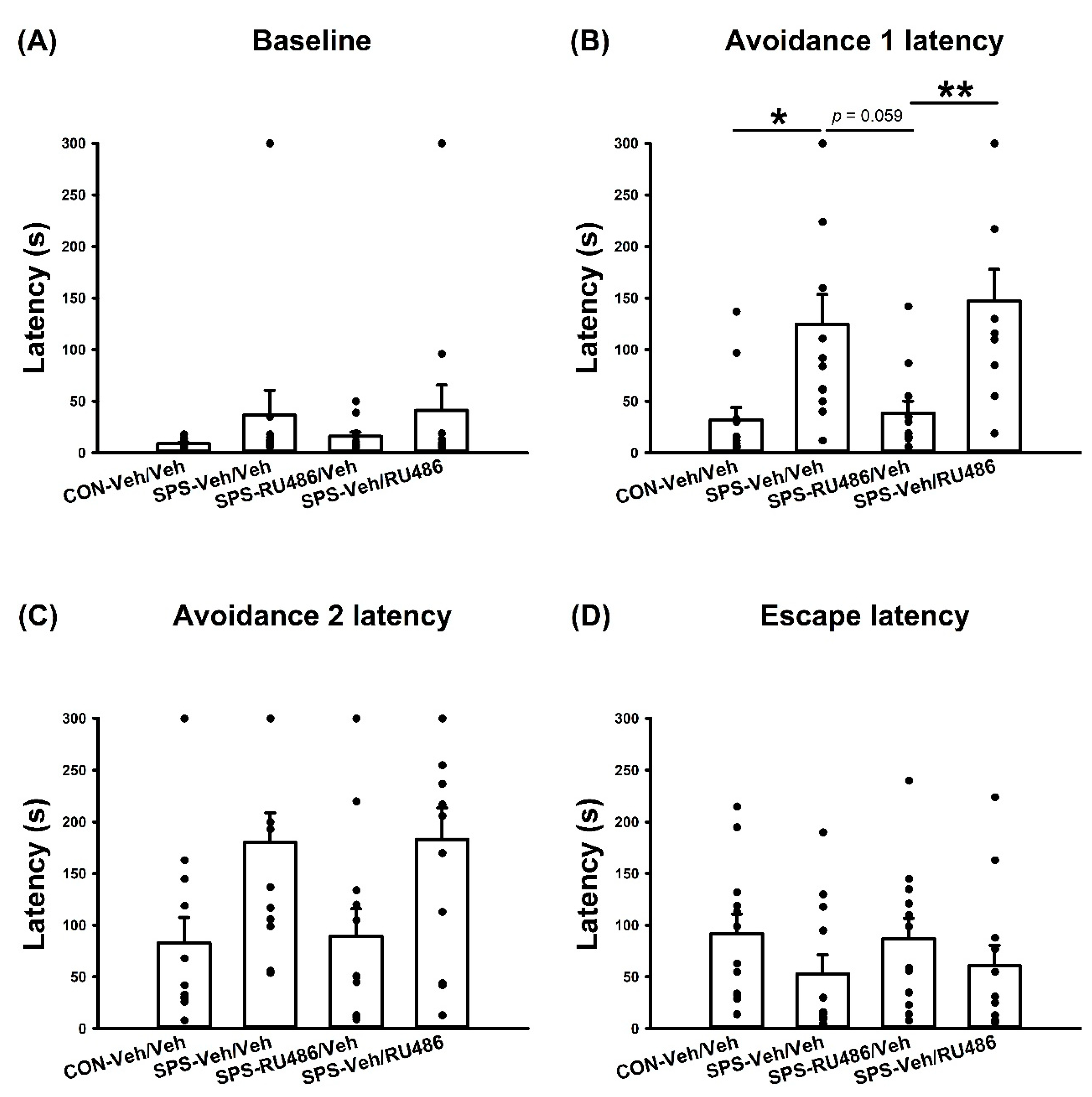
2.2. ETM Test
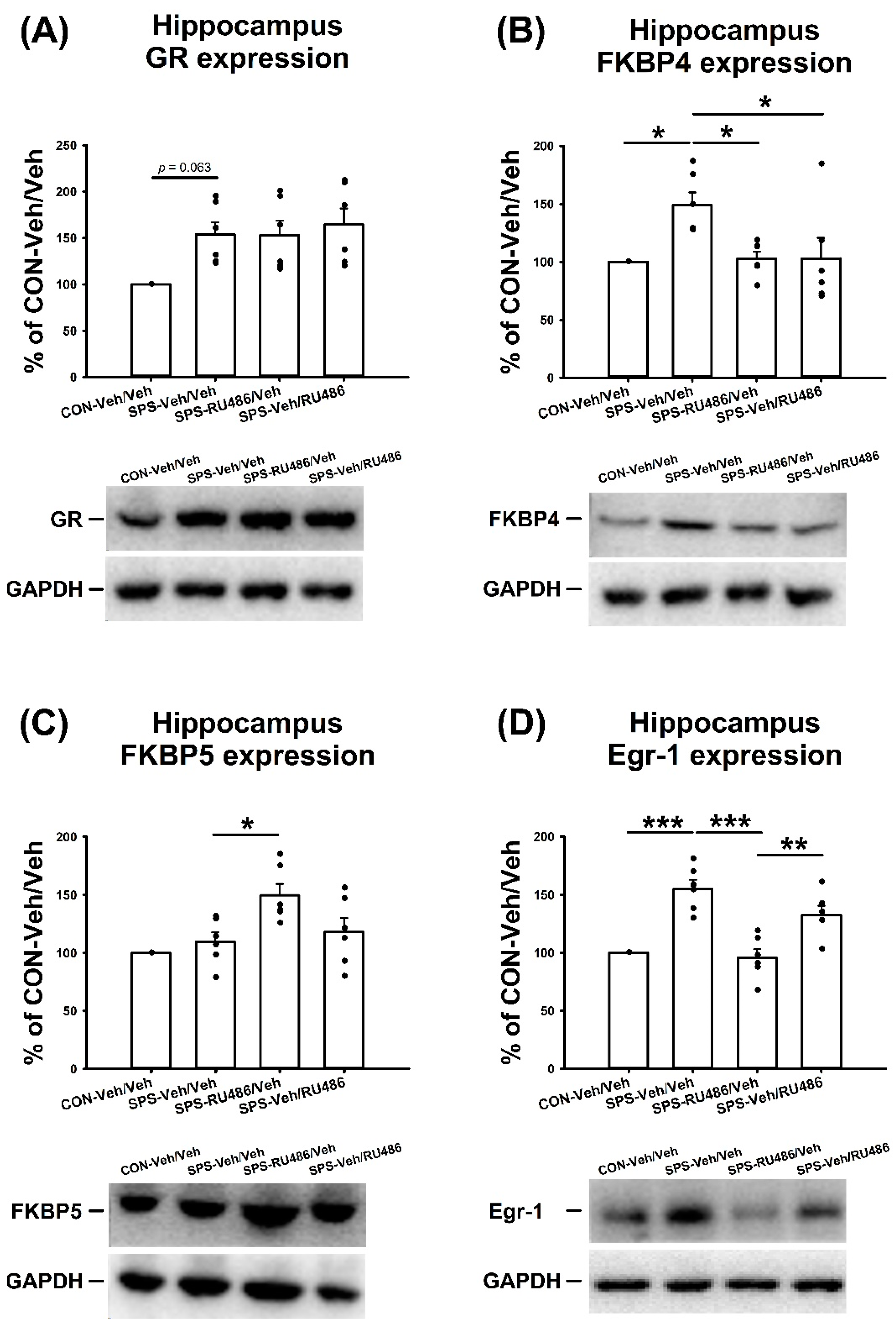
2.3. GR, FKBP4, FKBP5, and Egr-1 Expression in the Hippocampus
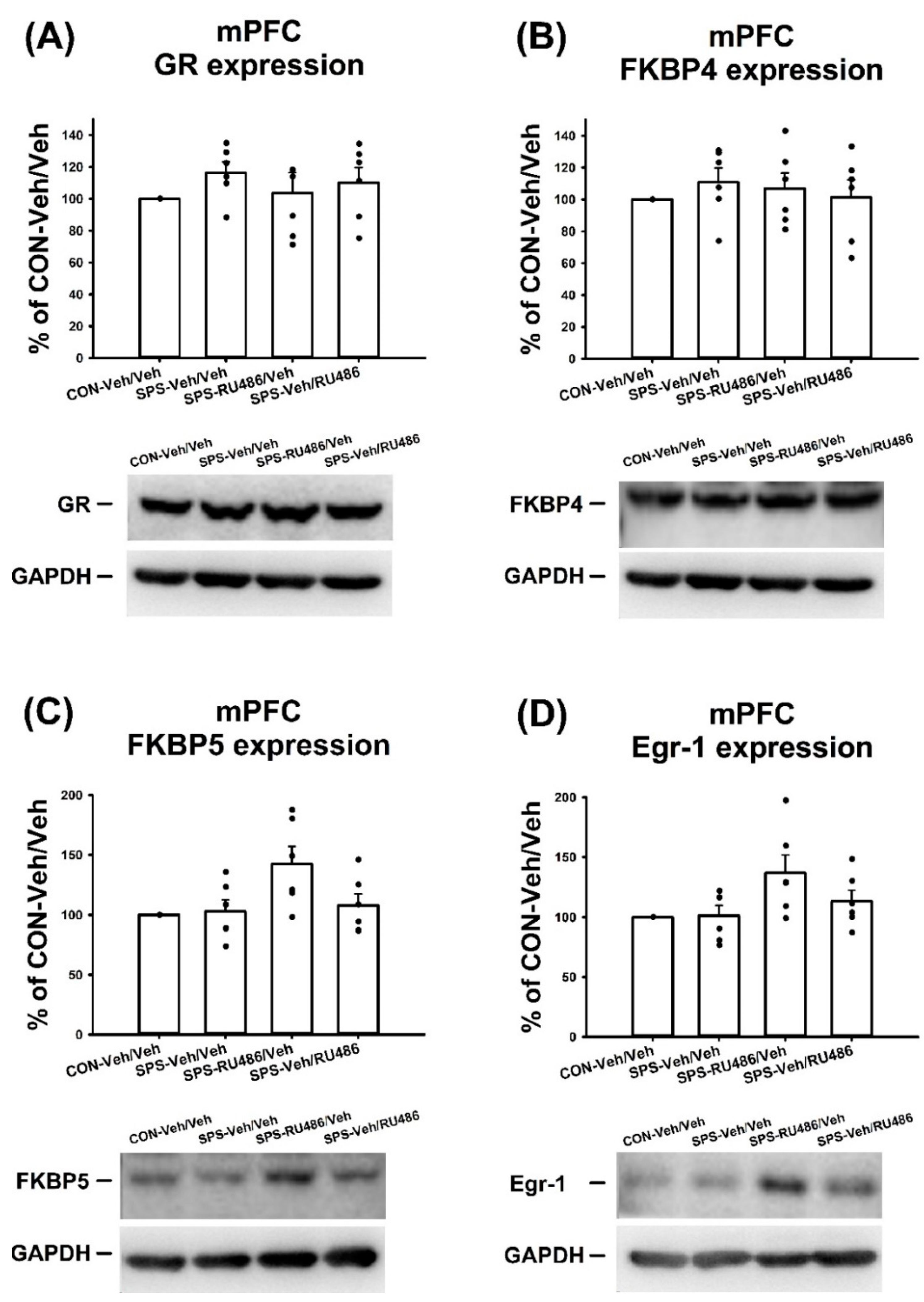
2.4. GR, FKBP4, FKBP5, and Egr-1 Expression in mPFC
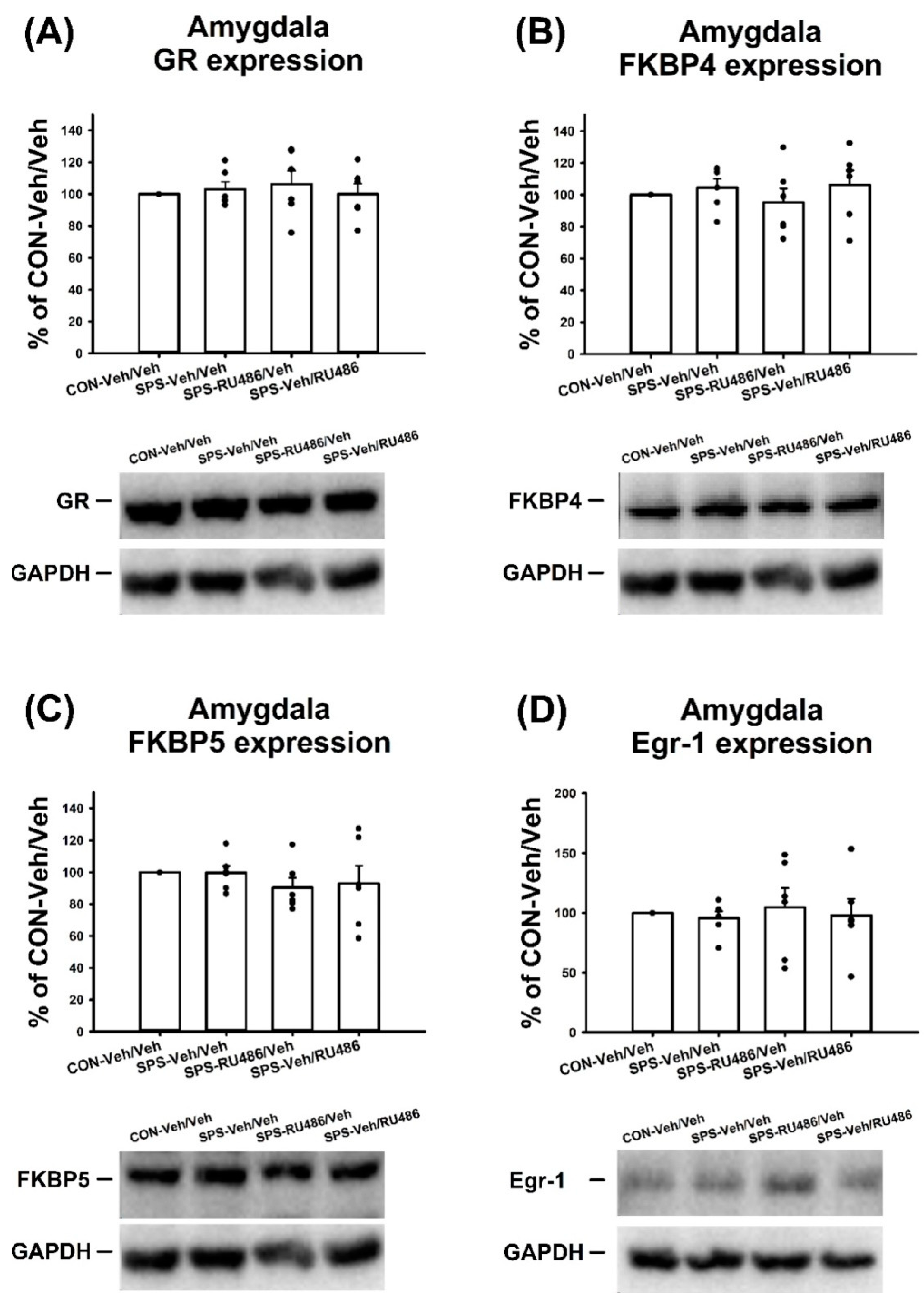
2.5. GR, FKBP4, FKBP5, and Egr-1 Expression in the Amygdala
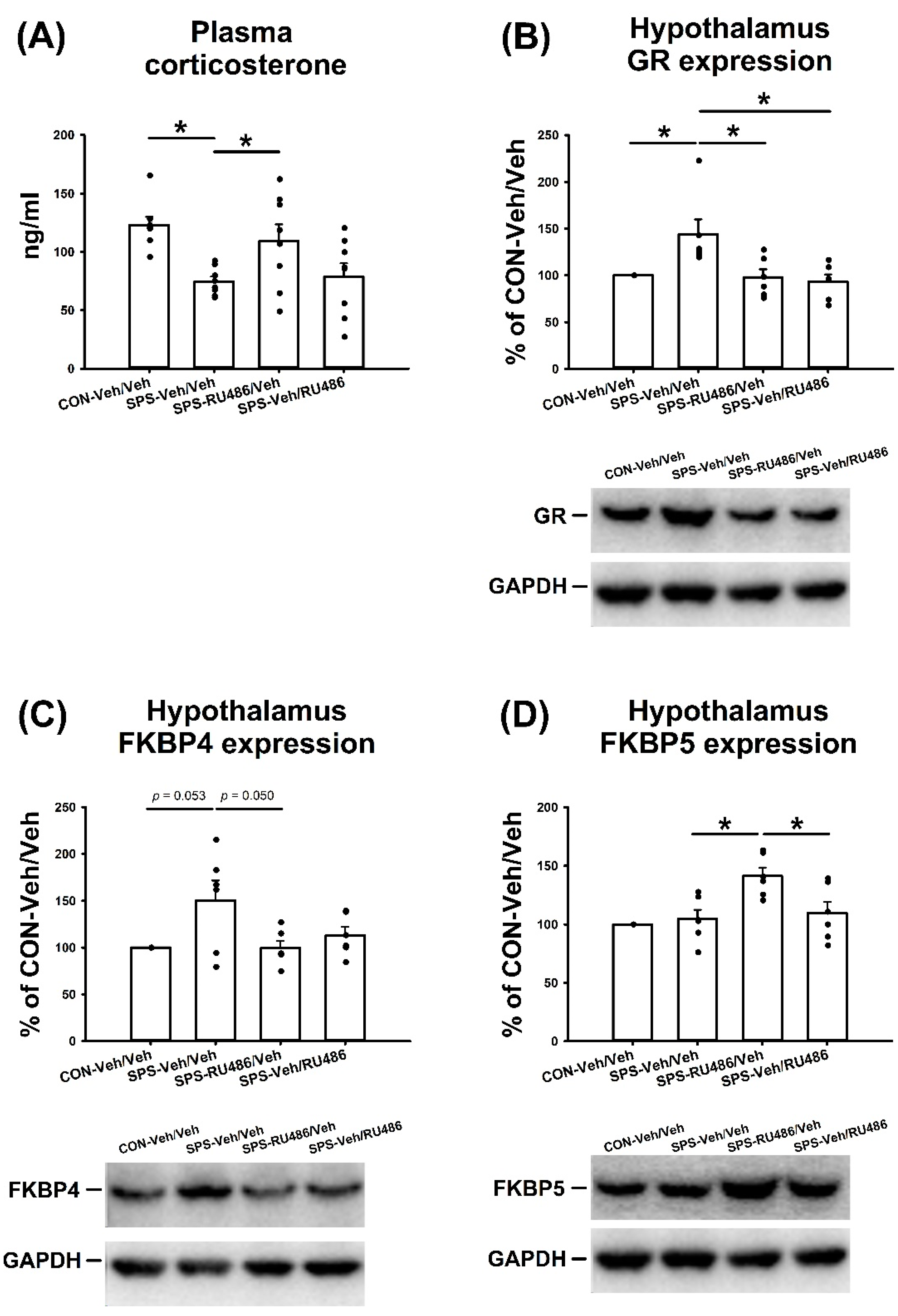
2.6. Plasma Corticosterone Level
2.7. GR, FKBP4, and FKBP5 Expression in the Hypothalamus
3. Discussion
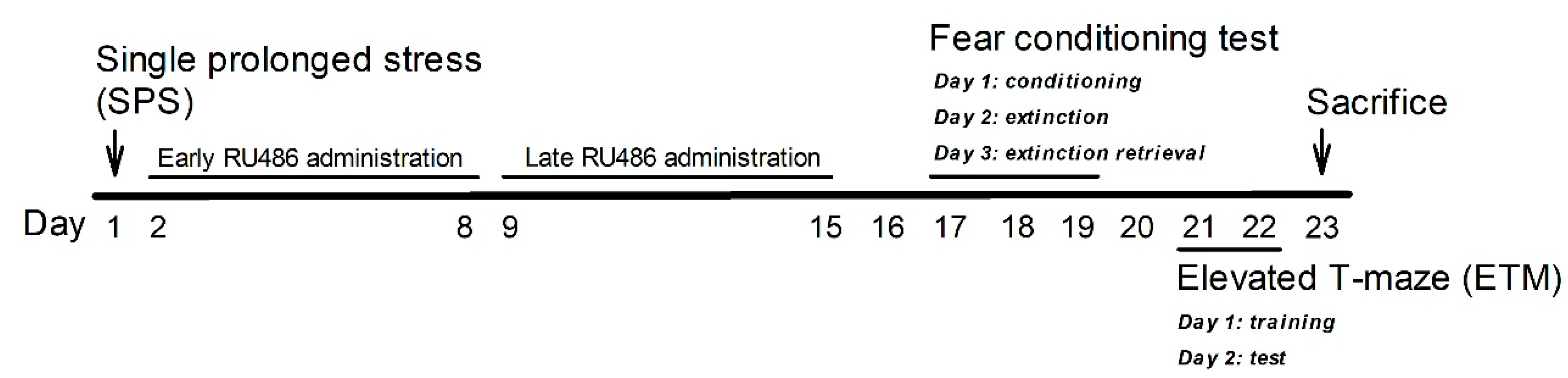
4. Materials and Methods
4.1. Animals
4.2. SPS
4.3. Drugs
4.4. Three-Day Cue-Dependent Fear Conditioning Test
4.5. ETM
4.6. Animal Euthanasia
4.7. Plasma Corticosterone Level
4.8. Western Blotting
4.9. Data Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jonas, D.E.; Cusack, K.; Forneris, C.A.; Wilkins, T.M.; Sonis, J.; Middleton, J.C.; Feltner, C.; Meredith, D.; Cavanaugh, J.; Brownley, K.A.; et al. Psychological and Pharmacological Treatments for Adults With Posttraumatic Stress Disorder (PTSD); Agency for Healthcare Research and Quality: Rockville, MD, USA, 2013. [Google Scholar]
- Yehuda, R. Post-traumatic stress disorder. N. Engl. J. Med. 2002, 346, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Diagnostic and Statistical Manual of Mental Disorders (DSM-5), 5th ed.; American Psychiatric Publishing, Inc., American Psychiatric Association: Washington, DC, USA, 2013.
- Iyadurai, L.; Visser, R.M.; Lau-Zhu, A.; Porcheret, K.; Horsch, A.; Holmes, E.A.; James, E.L. Intrusive memories of trauma: A target for research bridging cognitive science and its clinical application. Clin. Psychol. Rev. 2019, 69, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Martenyi, F.; Brown, E.B.; Caldwell, C.D. Failed efficacy of fluoxetine in the treatment of posttraumatic stress disorder: Results of a fixed-dose, placebo-controlled study. J. Clin. Psychopharmacol. 2007, 27, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Stein, D.J.; Pedersen, R.; Rothbaum, B.O.; Baldwin, D.S.; Ahmed, S.; Musgnung, J.; Davidson, J. Onset of activity and time to response on individual CAPS-SX17 items in patients treated for post-traumatic stress disorder with venlafaxine ER: A pooled analysis. Int. J. Neuropsychopharmacol. Off. Sci. J. Coll. Int. Neuropsychopharmacol. (CINP) 2009, 12, 23–31. [Google Scholar] [CrossRef]
- Yehuda, R.; Hoge, C.W.; McFarlane, A.C.; Vermetten, E.; Lanius, R.A.; Nievergelt, C.M.; Hobfoll, S.E.; Koenen, K.C.; Neylan, T.C.; Hyman, S.E. Post-traumatic stress disorder. Nat. Rev. Dis. Primers 2015, 1, 15057. [Google Scholar] [CrossRef]
- Schiller, D.; Delgado, M.R. Overlapping neural systems mediating extinction, reversal and regulation of fear. Trends Cogn. Sci. 2010, 14, 268–276. [Google Scholar] [CrossRef]
- Fullana, M.A.; Harrison, B.J.; Soriano-Mas, C.; Vervliet, B.; Cardoner, N.; Avila-Parcet, A.; Radua, J. Neural signatures of human fear conditioning: An updated and extended meta-analysis of fMRI studies. Mol. Psychiatry 2016, 21, 500–508. [Google Scholar] [CrossRef]
- Knight, D.C.; Smith, C.N.; Cheng, D.T.; Stein, E.A.; Helmstetter, F.J. Amygdala and hippocampal activity during acquisition and extinction of human fear conditioning. Cogn. Affect Behav. Neurosci. 2004, 4, 317–325. [Google Scholar] [CrossRef]
- Huggins, A.A.; Weis, C.N.; Parisi, E.A.; Bennett, K.P.; Miskovic, V.; Larson, C.L. Neural substrates of human fear generalization: A 7T-fMRI investigation. NeuroImage 2021, 239, 24. [Google Scholar] [CrossRef]
- Lin, C.-S.; Wu, C.-Y.; Wu, S.-Y.; Lin, H.-H. Brain activations associated with fearful experience show common and distinct patterns between younger and older adults in the hippocampus and the amygdala. Sci. Rep. 2018, 8, 5137. [Google Scholar] [CrossRef]
- Fullana, M.A.; Albajes-Eizagirre, A.; Soriano-Mas, C.; Vervliet, B.; Cardoner, N.; Benet, O.; Radua, J.; Harrison, B.J. Fear extinction in the human brain: A meta-analysis of fMRI studies in healthy participants. Neurosci. Biobehav. Rev. 2018, 88, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Garofalo, S.; di Pellegrino, G.; Starita, F. Revaluing the Role of vmPFC in the Acquisition of Pavlovian Threat Conditioning in Humans. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 8491–8500. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S. Neurobiological advances of learned fear in humans. Adv. Clin. Exp. Med. Off. Organ Wroc. Med. Univ. 2022, 31, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.J.; Fullana, M.A.; Via, E.; Soriano-Mas, C.; Vervliet, B.; Martínez-Zalacaín, I.; Pujol, J.; Davey, C.G.; Kircher, T.; Straube, B.; et al. Human ventromedial prefrontal cortex and the positive affective processing of safety signals. NeuroImage 2017, 152, 12–18. [Google Scholar] [CrossRef]
- Battaglia, S.; Harrison, B.J.; Fullana, M.A. Does the human ventromedial prefrontal cortex support fear learning, fear extinction or both? A commentary on subregional contributions. Mol. Psychiatry 2021, 27, 784–786. [Google Scholar] [CrossRef]
- Tashjian, S.M.; Zbozinek, T.D.; Mobbs, D. A Decision Architecture for Safety Computations. Trends Cogn. Sci. 2021, 25, 342–354. [Google Scholar] [CrossRef]
- LaBar, K.S.; Gatenby, J.C.; Gore, J.C.; LeDoux, J.E.; Phelps, E.A. Human amygdala activation during conditioned fear acquisition and extinction: A mixed-trial fMRI study. Neuron 1998, 20, 937–945. [Google Scholar] [CrossRef]
- Sah, P.; Westbrook, R.F. The circuit of fear. Nature 2008, 454, 589–590. [Google Scholar] [CrossRef]
- Mahan, A.L.; Ressler, K.J. Fear conditioning, synaptic plasticity and the amygdala: Implications for posttraumatic stress disorder. Trends Neurosci. 2012, 35, 24–35. [Google Scholar] [CrossRef]
- Maren, S.; Quirk, G.J. Neuronal signalling of fear memory. Nat. Rev. Neurosci. 2004, 5, 844–852. [Google Scholar] [CrossRef]
- Singewald, N.; Schmuckermair, C.; Whittle, N.; Holmes, A.; Ressler, K.J. Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol. Ther. 2015, 149, 150–190. [Google Scholar] [CrossRef] [PubMed]
- Kaouane, N.; Ducourneau, E.G.; Marighetto, A.; Segal, M.; Desmedt, A. False Opposing Fear Memories Are Produced as a Function of the Hippocampal Sector Where Glucocorticoid Receptors Are Activated. Front. Behav. Neurosci. 2020, 14, 144. [Google Scholar] [CrossRef] [PubMed]
- Revest, J.M.; Di Blasi, F.; Kitchener, P.; Rouge-Pont, F.; Desmedt, A.; Turiault, M.; Tronche, F.; Piazza, P.V. The MAPK pathway and Egr-1 mediate stress-related behavioral effects of glucocorticoids. Nat. Neurosci. 2005, 8, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Revest, J.M.; Kaouane, N.; Mondin, M.; Le Roux, A.; Rouge-Pont, F.; Vallee, M.; Barik, J.; Tronche, F.; Desmedt, A.; Piazza, P.V. The enhancement of stress-related memory by glucocorticoids depends on synapsin-Ia/Ib. Mol. Psychiatry 2010, 15, 1140–1151. [Google Scholar] [CrossRef]
- Szeszko, P.R.; Lehrner, A.; Yehuda, R. Glucocorticoids and Hippocampal Structure and Function in PTSD. Harv. Rev. Psychiatry 2018, 26, 142–157. [Google Scholar] [CrossRef]
- Liberzon, I.; Krstov, M.; Young, E.A. Stress-restress: Effects on ACTH and fast feedback. Psychoneuroendocrinology 1997, 22, 443–453. [Google Scholar] [CrossRef]
- Liberzon, I.; Lopez, J.F.; Flagel, S.B.; Vazquez, D.M.; Young, E.A. Differential regulation of hippocampal glucocorticoid receptors mRNA and fast feedback: Relevance to post-traumatic stress disorder. J. Neuroendocrinol. 1999, 11, 11–17. [Google Scholar] [CrossRef]
- Lisieski, M.J.; Eagle, A.L.; Conti, A.C.; Liberzon, I.; Perrine, S.A. Single-Prolonged Stress: A Review of Two Decades of Progress in a Rodent Model of Post-traumatic Stress Disorder. Front. Psychiatry 2018, 9, 196. [Google Scholar] [CrossRef]
- Lin, C.C.; Cheng, P.Y.; Liu, Y.P. Effects of early life social experience on fear extinction and related glucocorticoid profiles—Behavioral and neurochemical approaches in a rat model of PTSD. Behav. Brain Res. 2020, 391, 112686. [Google Scholar] [CrossRef]
- Araki, M.; Fuchikami, M.; Omura, J.; Miyagi, T.; Nagashima, N.; Okamoto, Y.; Morinobu, S. The role of glucocorticoid receptors in the induction and prevention of hippocampal abnormalities in an animal model of posttraumatic stress disorder. Psychopharmacology 2020, 237, 2125–2137. [Google Scholar] [CrossRef]
- Ding, J.; Chen, X.; da Silva, M.S.; Lingeman, J.; Han, F.; Meijer, O.C. Effects of RU486 treatment after single prolonged stress depend on the post-stress interval. Mol. Cell. Neurosci. 2020, 108, 103541. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; da Silva, M.S.; Lingeman, J.; Chen, X.; Shi, Y.; Han, F.; Meijer, O.C. Late glucocorticoid receptor antagonism changes the outcome of adult life stress. Psychoneuroendocrinology 2019, 107, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Binder, E.B. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 2009, 34 (Suppl. S1), S186–S195. [Google Scholar] [CrossRef] [PubMed]
- Maddox, S.A.; Schafe, G.E.; Ressler, K.J. Exploring epigenetic regulation of fear memory and biomarkers associated with post-traumatic stress disorder. Front. Psychiatry 2013, 4, 62. [Google Scholar] [CrossRef]
- Zannas, A.S.; Wiechmann, T.; Gassen, N.C.; Binder, E.B. Gene-Stress-Epigenetic Regulation of FKBP5: Clinical and Translational Implications. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2016, 41, 261–274. [Google Scholar] [CrossRef]
- Tatro, E.T.; Everall, I.P.; Kaul, M.; Achim, C.L. Modulation of glucocorticoid receptor nuclear translocation in neurons by immunophilins FKBP51 and FKBP52: Implications for major depressive disorder. Brain Res. 2009, 1286, 1–12. [Google Scholar] [CrossRef]
- Lim, C.P.; Jain, N.; Cao, X. Stress-induced immediate-early gene, egr-1, involves activation of p38/JNK1. Oncogene 1998, 16, 2915–2926. [Google Scholar] [CrossRef]
- Saunderson, E.A.; Spiers, H.; Mifsud, K.R.; Gutierrez-Mecinas, M.; Trollope, A.F.; Shaikh, A.; Mill, J.; Reul, J.M. Stress-induced gene expression and behavior are controlled by DNA methylation and methyl donor availability in the dentate gyrus. Proc. Natl. Acad. Sci. USA 2016, 113, 4830–4835. [Google Scholar] [CrossRef]
- Mifsud, K.R.; Kennedy, C.L.M.; Salatino, S.; Sharma, E.; Price, E.M.; Haque, S.N.; Gialeli, A.; Goss, H.M.; Panchenko, P.E.; Broxholme, J.; et al. Distinct regulation of hippocampal neuroplasticity and ciliary genes by corticosteroid receptors. Nat. Commun. 2021, 12, 4737. [Google Scholar] [CrossRef]
- Brewin, C.R. A cognitive neuroscience account of posttraumatic stress disorder and its treatment. Behav. Res. Ther. 2001, 39, 373–393. [Google Scholar] [CrossRef]
- Carlier, I.V.; Gersons, B.P. Partial posttraumatic stress disorder (PTSD): The issue of psychological scars and the occurrence of PTSD symptoms. J. Nerv. Ment. Dis. 1995, 183, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Watkins, L.E.; Sprang, K.R.; Rothbaum, B.O. Treating PTSD: A Review of Evidence-Based Psychotherapy Interventions. Front. Behav. Neurosci. 2018, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kim, S.Y.; Artis, S.; Molfese, D.L.; Schumacher, A.; Sweatt, J.D.; Paylor, R.E.; Lubin, F.D. Histone methylation regulates memory formation. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 3589–3599. [Google Scholar] [CrossRef] [PubMed]
- Eagle, A.L.; Knox, D.; Roberts, M.M.; Mulo, K.; Liberzon, I.; Galloway, M.P.; Perrine, S.A. Single prolonged stress enhances hippocampal glucocorticoid receptor and phosphorylated protein kinase B levels. Neurosci. Res. 2013, 75, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Moulton, E.; Chamness, M.; Knox, D. Characterizing changes in glucocorticoid receptor internalization in the fear circuit in an animal model of post traumatic stress disorder. PLoS ONE 2018, 13, e0205144. [Google Scholar] [CrossRef] [PubMed]
- Bains, J.S.; Wamsteeker Cusulin, J.I.; Inoue, W. Stress-related synaptic plasticity in the hypothalamus. Nat. Rev. Neurosci. 2015, 16, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Chen, T.Y.; Cheng, P.Y.; Liu, Y.P. Early life social experience affects adulthood fear extinction deficit and associated dopamine profile abnormalities in a rat model of PTSD. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 101, 109914. [Google Scholar] [CrossRef]
- Lin, C.C.; Chang, H.A.; Tai, Y.M.; Chen, T.Y.; Wan, F.J.; Chang, C.C.; Tung, C.S.; Liu, Y.P. Subchronic administration of aripiprazole improves fear extinction retrieval of Pavlovian conditioning paradigm in rats experiencing psychological trauma. Behav. Brain Res. 2019, 362, 181–187. [Google Scholar] [CrossRef]
- Kanatsou, S.; Karst, H.; Kortesidou, D.; van den Akker, R.A.; den Blaauwen, J.; Harris, A.P.; Seckl, J.R.; Krugers, H.J.; Joels, M. Overexpression of Mineralocorticoid Receptors in the Mouse Forebrain Partly Alleviates the Effects of Chronic Early Life Stress on Spatial Memory, Neurogenesis and Synaptic Function in the Dentate Gyrus. Front. Cell. Neurosci. 2017, 11, 132. [Google Scholar] [CrossRef]
- Fani, N.; Gutman, D.; Tone, E.B.; Almli, L.; Mercer, K.B.; Davis, J.; Glover, E.; Jovanovic, T.; Bradley, B.; Dinov, I.D.; et al. FKBP5 and attention bias for threat: Associations with hippocampal function and shape. JAMA Psychiatry 2013, 70, 392–400. [Google Scholar] [CrossRef]
- Sawamura, T.; Klengel, T.; Armario, A.; Jovanovic, T.; Norrholm, S.D.; Ressler, K.J.; Andero, R. Dexamethasone Treatment Leads to Enhanced Fear Extinction and Dynamic Fkbp5 Regulation in Amygdala. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2016, 41, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Golier, J.A.; Caramanica, K.; Demaria, R.; Yehuda, R. A Pilot Study of Mifepristone in Combat-Related PTSD. Depress. Res. Treat. 2012, 393251, 24. [Google Scholar] [CrossRef] [PubMed]
- Surís, A.; Holliday, R.; Adinoff, B.; Holder, N.; North, C.S. Facilitating Fear-Based Memory Extinction With Dexamethasone: A Randomized Controlled Trial in Male Veterans With Combat-Related PTSD. Psychiatry 2017, 80, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.M.; King, E.M.; Rider, C.F.; Gwozd, C.; Holden, N.S.; Eddleston, J.; Zuraw, B.; Leigh, R.; O’Byrne, P.M.; Newton, R. Corticosteroid-induced gene expression in allergen-challenged asthmatic subjects taking inhaled budesonide. Br. J. Pharmacol. 2012, 165, 1737–1747. [Google Scholar] [CrossRef]
- Vermeer, H.; Hendriks-Stegeman, B.I.; van der Burg, B.; van Buul-Offers, S.C.; Jansen, M. Glucocorticoid-induced increase in lymphocytic FKBP51 messenger ribonucleic acid expression: A potential marker for glucocorticoid sensitivity, potency, and bioavailability. J. Clin. Endocrinol. Metab. 2003, 88, 277–284. [Google Scholar] [CrossRef]
- Hansson, A.C.; Fuxe, K. Time-course of immediate early gene expression in hippocampal subregions of adrenalectomized rats after acute corticosterone challenge. Brain Res. 2008, 18, 1–10. [Google Scholar] [CrossRef]
- Fanselow, M.S.; Dong, H.W. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 2010, 65, 7–19. [Google Scholar] [CrossRef]
- Lin, C.C.; Huang, K.L.; Tung, C.S.; Liu, Y.P. Hyperbaric oxygen therapy restored traumatic stress-induced dysregulation of fear memory and related neurochemical abnormalities. Behav. Brain Res. 2019, 359, 861–870. [Google Scholar] [CrossRef]
- Wang, S.C.; Lin, C.C.; Chen, C.C.; Tzeng, N.S.; Liu, Y.P. Effects of Oxytocin on Fear Memory and Neuroinflammation in a Rodent Model of Posttraumatic Stress Disorder. Int. J. Mol. Sci. 2018, 19, 3848. [Google Scholar] [CrossRef]
- Dong, L.; Wang, S.; Li, Y.; Zhao, Z.; Shen, Y.; Liu, L.; Xu, G.; Ma, C.; Li, S.; Zhang, X.; et al. RU486 Reverses Emotional Disorders by Influencing Astrocytes and Endoplasmic Reticulum Stress in Chronic Restraint Stress Challenged Rats. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 42, 1098–1108. [Google Scholar] [CrossRef]
- Frank, M.G.; Annis, J.L.; Watkins, L.R.; Maier, S.F. Glucocorticoids mediate stress induction of the alarmin HMGB1 and reduction of the microglia checkpoint receptor CD200R1 in limbic brain structures. Brain Behav. Immun. 2019, 80, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, P.R.; Raeis-Abdollahi, E.; Sameni, H.; Vafaei, A.A.; Ghanbari, A.; Rashidy-Pour, A. Protective effects of morphine in a rat model of post-traumatic stress disorder: Role of hypothalamic-pituitary-adrenal axis and beta- adrenergic system. Behav. Brain Res. 2020, 395, 19. [Google Scholar] [CrossRef] [PubMed]
- Adamec, R.; Muir, C.; Grimes, M.; Pearcey, K. Involvement of noradrenergic and corticoid receptors in the consolidation of the lasting anxiogenic effects of predator stress. Behav. Brain Res. 2007, 179, 192–207. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.Y.; Poole, R.L.; Pageler, N.M. Propylene glycol toxicity in children. J. Pediatr. Pharmacol. Ther. 2014, 19, 277–282. [Google Scholar] [CrossRef]
- Lin, C.C.; Tung, C.S.; Lin, P.H.; Huang, C.L.; Liu, Y.P. Traumatic stress causes distinctive effects on fear circuit catecholamines and the fear extinction profile in a rodent model of posttraumatic stress disorder. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2016, 26, 1484–1495. [Google Scholar] [CrossRef] [PubMed]
- Merz, C.J.; Hamacher-Dang, T.C.; Stark, R.; Wolf, O.T.; Hermann, A. Neural Underpinnings of Cortisol Effects on Fear Extinction. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2018, 43, 384–392. [Google Scholar] [CrossRef]
- Goswami, S.; Cascardi, M.; Rodriguez-Sierra, O.E.; Duvarci, S.; Pare, D. Impact of predatory threat on fear extinction in Lewis rats. Learn Mem. 2010, 17, 494–501. [Google Scholar] [CrossRef]
- Keller, S.M.; Schreiber, W.B.; Stanfield, B.R.; Knox, D. Inhibiting corticosterone synthesis during fear memory formation exacerbates cued fear extinction memory deficits within the single prolonged stress model. Behav. Brain Res. 2015, 287, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Tung, C.S.; Liu, Y.P. Escitalopram reversed the traumatic stress-induced depressed and anxiety-like symptoms but not the deficits of fear memory. Psychopharmacology 2016, 233, 1135–1146. [Google Scholar] [CrossRef]
- Poltronieri, S.C.; Zangrossi, H.; de Barros Viana, M. Antipanic-like effect of serotonin reuptake inhibitors in the elevated T-maze. Behav. Brain Res. 2003, 147, 185–192. [Google Scholar] [CrossRef]
- Zangrossi, H., Jr.; Graeff, F.G. Behavioral validation of the elevated T-maze, a new animal model of anxiety. Brain Res. Bull. 1997, 44, 1–5. [Google Scholar] [CrossRef]
- Campos, A.C.; Fogaca, M.V.; Aguiar, D.C.; Guimaraes, F.S. Animal models of anxiety disorders and stress. Braz. J. Psychiatry 2013, 35 (Suppl. S2), S101–S111. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: Cambridge, MA, USA, 2008. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-C.; Cheng, P.-Y.; Hsiao, M.; Liu, Y.-P. Effects of RU486 in Treatment of Traumatic Stress-Induced Glucocorticoid Dysregulation and Fear-Related Abnormalities: Early versus Late Intervention. Int. J. Mol. Sci. 2022, 23, 5494. https://doi.org/10.3390/ijms23105494
Lin C-C, Cheng P-Y, Hsiao M, Liu Y-P. Effects of RU486 in Treatment of Traumatic Stress-Induced Glucocorticoid Dysregulation and Fear-Related Abnormalities: Early versus Late Intervention. International Journal of Molecular Sciences. 2022; 23(10):5494. https://doi.org/10.3390/ijms23105494
Chicago/Turabian StyleLin, Chen-Cheng, Pao-Yun Cheng, Michael Hsiao, and Yia-Ping Liu. 2022. "Effects of RU486 in Treatment of Traumatic Stress-Induced Glucocorticoid Dysregulation and Fear-Related Abnormalities: Early versus Late Intervention" International Journal of Molecular Sciences 23, no. 10: 5494. https://doi.org/10.3390/ijms23105494
APA StyleLin, C.-C., Cheng, P.-Y., Hsiao, M., & Liu, Y.-P. (2022). Effects of RU486 in Treatment of Traumatic Stress-Induced Glucocorticoid Dysregulation and Fear-Related Abnormalities: Early versus Late Intervention. International Journal of Molecular Sciences, 23(10), 5494. https://doi.org/10.3390/ijms23105494