
Phase-Separated Subcellular Compartmentation and Related Human Diseases


Abstract
1. Introduction
2. Biomolecular Condensates
2.1. The Molecular Features of Biomolecular Aggregates
2.2. Materials Properties of Phase-Separated Condensates
2.2.1. Stress Granules
2.2.2. P Bodies
2.2.3. Nucleolus
2.2.4. Examples of Regulatory Proteins with LLPS Potential
2.3. Regulation of Condensate Assembly
2.3.1. Effects of PTMs on Protein Phase Separation
2.3.2. Effects of Chaperones on Protein Phase Separation
2.4. Functions of Phase-Separation Condensates
2.4.1. Regulation of Biological Reactions
2.4.2. Regulation of Gene Expression
2.4.3. Regulation of Viral Infection
2.4.4. Sequestration and Storage of Molecules
3. The Phase Separation of Proteins in Diseases
3.1. LLPS and Neurodegenerative Diseases
3.1.1. FUS
3.1.2. Tau
3.1.3. TDP-43
3.2. LLPS and Cancers
3.2.1. SHP2
3.2.2. YAP and TAZ
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, X.; Wang, H.; Dou, Z.; Ruan, K.; Hill, D.L.; Li, L.; Shi, Y.; Yao, X. Phase separation drives decision making in cell division. J. Biol. Chem. 2020, 295, 13419–13431. [Google Scholar] [CrossRef] [PubMed]
- Gomes, E.; Shorter, J. The molecular language of membraneless organelles. J. Biol. Chem. 2019, 294, 7115–7127. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, M.; Kato, K.; Sanbo, C.; Shigenobu, S.; Ohkawa, Y.; Fuchigami, T.; Miyanari, Y. Genomic Profiling by ALaP-Seq Reveals Transcriptional Regulation by PML Bodies through DNMT3A Exclusion. Mol. Cell 2020, 78, 493–505.e498. [Google Scholar] [CrossRef]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef]
- Dodson, A.E.; Kennedy, S. Phase Separation in Germ Cells and Development. Dev. Cell 2020, 55, 4–17. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Woodruff, J.B.; Hyman, A.A.; Boke, E. Organization and Function of Non-dynamic Biomolecular Condensates. Trends Biochem. Sci. 2018, 43, 81–94. [Google Scholar] [CrossRef]
- Case, L.B.; Zhang, X.; Ditlev, J.A.; Rosen, M.K. Stoichiometry controls activity of phase-separated clusters of actin signaling proteins. Science 2019, 363, 1093–1097. [Google Scholar] [CrossRef]
- Lu, Y.; Wu, T.; Gutman, O.; Lu, H.; Zhou, Q.; Henis, Y.I.; Luo, K. Phase separation of TAZ compartmentalizes the transcription machinery to promote gene expression. Nat. Cell Biol. 2020, 22, 453–464. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Nessling, M.; Lichter, P. Macromolecular crowding and its potential impact on nuclear function. Biochim. Biophys. Acta 2008, 1783, 2100–2107. [Google Scholar] [CrossRef] [PubMed]
- Kanekura, K.; Kuroda, M. How can we interpret the relationship between liquid-liquid phase separation and amyotrophic lateral sclerosis? Lab. Investig. 2022; online ahead of print. [Google Scholar]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 2017, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.; Dugast-Darzacq, C.; Liu, Z.; Dong, P.; Dailey, G.M.; Cattoglio, C.; Heckert, A.; Banala, S.; Lavis, L.; Darzacq, X.; et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 2018, 361, eaar2555. [Google Scholar] [CrossRef]
- Altmeyer, M.; Neelsen, K.J.; Teloni, F.; Pozdnyakova, I.; Pellegrino, S.; Grøfte, M.; Rask, M.D.; Streicher, W.; Jungmichel, S.; Nielsen, M.L.; et al. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat. Commun. 2015, 6, 8088. [Google Scholar] [CrossRef]
- Brocca, S.; Grandori, R.; Longhi, S.; Uversky, V. Liquid-Liquid Phase Separation by Intrinsically Disordered Protein Regions of Viruses: Roles in Viral Life Cycle and Control of Virus-Host Interactions. Int. J. Mol. Sci. 2020, 21, 9045. [Google Scholar] [CrossRef]
- Shen, B.; Chen, Z.; Yu, C.; Chen, T.; Shi, M.; Li, T. Computational Screening of Phase-separating Proteins. Genom. Proteom. Bioinform. 2021, 19, 13–24. [Google Scholar] [CrossRef]
- Zhang, H.; Ji, X.; Li, P.; Liu, C.; Lou, J.; Wang, Z.; Wen, W.; Xiao, Y.; Zhang, M.; Zhu, X. Liquid-liquid phase separation in biology: Mechanisms, physiological functions and human diseases. Sci. China Life Sci. 2020, 63, 953–985. [Google Scholar] [CrossRef]
- Peran, I.; Mittag, T. Molecular structure in biomolecular condensates. Curr. Opin. Struct. Biol. 2020, 60, 17–26. [Google Scholar] [CrossRef]
- Alberti, S.; Dormann, D. Liquid-Liquid Phase Separation in Disease. Annu. Rev. Genet. 2019, 53, 171–194. [Google Scholar] [CrossRef]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Mackowiak, S.D.; Niskanen, H.; Knezevic, D.; Asimi, V.; Grosswendt, S.; Geertsema, H.; Ali, S.; Jerkovic, I.; Ewers, H.; et al. Unblending of Transcriptional Condensates in Human Repeat Expansion Disease. Cell 2020, 181, 1062–1079.e30. [Google Scholar] [PubMed]
- Wang, W.; Qiao, S.; Li, G.; Cheng, J.; Yang, C.; Zhong, C.; Stovall, D.B.; Shi, J.; Teng, C.; Li, D.; et al. A histidine cluster determines YY1-compartmentalized coactivators and chromatin elements in phase-separated enhancer clusters. Nucleic Acids Res. 2022; online ahead of print. [Google Scholar]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Analyzing IDPs in Interactomes. Methods Mol. Biol. 2020, 2141, 895–945. [Google Scholar]
- Tsang, B.; Pritisanac, I.; Scherer, S.W.; Moses, A.M.; Forman-Kay, J.D. Phase Separation as a Missing Mechanism for Interpretation of Disease Mutations. Cell 2020, 183, 1742–1756. [Google Scholar] [CrossRef]
- Vacic, V.; Iakoucheva, L.M. Disease mutations in disordered regions--Exception to the rule? Mol. Biosyst. 2012, 8, 27–32. [Google Scholar] [CrossRef]
- Vacic, V.; Markwick, P.R.; Oldfield, C.J.; Zhao, X.; Haynes, C.; Uversky, V.N.; Iakoucheva, L.M. Disease-associated mutations disrupt functionally important regions of intrinsic protein disorder. PLoS Comput. Biol. 2012, 8, e1002709. [Google Scholar] [CrossRef]
- Deng, H.; Gao, K.; Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef]
- McAlary, L.; Plotkin, S.S.; Yerbury, J.J.; Cashman, N.R. Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 262. [Google Scholar] [CrossRef]
- Ohsawa, T.; Sahara, T.; Muramatsu, S.; Nishimura, Y.; Yathuoka, T.; Tanaka, Y.; Yamaguchi, K.; Ishida, H.; Akagi, K. Colorectal cancer susceptibility associated with the hMLH1 V384D variant. Mol. Med. Rep. 2009, 2, 887–891. [Google Scholar] [PubMed]
- Lee, S.E.; Lee, H.S.; Kim, K.Y.; Park, J.H.; Roh, H.; Park, H.Y.; Kim, W.S. High prevalence of the MLH1 V384D germline mutation in patients with HER2-positive luminal B breast cancer. Sci. Rep. 2019, 9, 10966. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Dai, T.; Qin, Z.; Pan, T.; Chu, F.; Lou, L.; Zhang, L.; Yang, B.; Huang, H.; Lu, H.; et al. Targeting liquid-liquid phase separation of SARS-CoV-2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat. Cell Biol. 2021, 23, 718–732. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2017, 44, 18–30. [Google Scholar] [CrossRef]
- Alberti, S.; Hyman, A.A. Are aberrant phase transitions a driver of cellular aging? Bioessays 2016, 38, 959–968. [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef]
- Luo, Y.; Na, Z.; Slavoff, S.A. P-Bodies: Composition, Properties, and Functions. Biochemistry 2018, 57, 2424–2431. [Google Scholar] [CrossRef]
- Riggs, C.L.; Kedersha, N.; Ivanov, P.; Anderson, P. Mammalian stress granules and P bodies at a glance. J. Cell Sci. 2020, 133, jcs242487. [Google Scholar] [CrossRef]
- Zhang, Y.; Gu, J.; Sun, Q. Aberrant Stress Granule Dynamics and Aggrephagy in ALS Pathogenesis. Cells 2021, 10, 2247. [Google Scholar] [CrossRef]
- Voronina, E.; Seydoux, G.; Sassone-Corsi, P.; Nagamori, I. RNA granules in germ cells. Cold Spring Harb. Perspect. Biol. 2011, 3, a002774. [Google Scholar] [CrossRef]
- Zeng, M.; Chen, X.; Guan, D.; Xu, J.; Wu, H.; Tong, P.; Zhang, M. Reconstituted Postsynaptic Density as a Molecular Platform for Understanding Synapse Formation and Plasticity. Cell 2018, 174, 1172–1187.e16. [Google Scholar] [CrossRef] [PubMed]
- Formicola, N.; Vijayakumar, J.; Besse, F. Neuronal ribonucleoprotein granules: Dynamic sensors of localized signals. Traffic 2019, 20, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Jamieson-Lucy, A.; Mullins, M.C. The vertebrate Balbiani body, germ plasm, and oocyte polarity. Curr. Top. Dev. Biol. 2019, 135, 1–34. [Google Scholar] [PubMed]
- Aguilera-Gomez, A.; Rabouille, C. Membrane-bound organelles versus membrane-less compartments and their control of anabolic pathways in Drosophila. Dev. Biol. 2017, 428, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Liu, J.L. U bodies respond to nutrient stress in Drosophila. Exp. Cell Res. 2011, 317, 2835–2844. [Google Scholar] [CrossRef]
- Li, J.; Hochstrasser, M. Microautophagy regulates proteasome homeostasis. Curr. Genet. 2020, 66, 683–687. [Google Scholar] [CrossRef]
- Smalley, M.J.; Signoret, N.; Robertson, D.; Tilley, A.; Hann, A.; Ewan, K.; Ding, Y.; Paterson, H.; Dale, T.C. Dishevelled (Dvl-2) activates canonical Wnt signalling in the absence of cytoplasmic puncta. J. Cell Sci. 2005, 118, 5279–5289. [Google Scholar] [CrossRef]
- Wunder, T.; Mueller-Cajar, O. Biomolecular condensates in photosynthesis and metabolism. Curr. Opin. Plant Biol. 2020, 58, 1–7. [Google Scholar] [CrossRef]
- Sehgal, P.B. Biomolecular condensates in cancer cell biology: Interleukin-6-induced cytoplasmic and nuclear STAT3/PY-STAT3 condensates in hepatoma cells. Contemp. Oncol. 2019, 23, 16–22. [Google Scholar] [CrossRef]
- Xu, F.; Mukhopadhyay, S.; Sehgal, P.B. Live cell imaging of interleukin-6-induced targeting of “transcription factor” STAT3 to sequestering endosomes in the cytoplasm. Am. J. Physiol. Cell Physiol. 2007, 293, C1374–C1382. [Google Scholar] [CrossRef]
- Ma, W.; Mayr, C. A Membraneless Organelle Associated with the Endoplasmic Reticulum Enables 3′UTR-Mediated Protein-Protein Interactions. Cell 2018, 175, 1492–1506.e19. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Hurt, E. The nuclear pore complex: Understanding its function through structural insight. Nat. Rev. Mol. Cell Biol. 2017, 18, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Lafontaine, D.L.J.; Riback, J.A.; Bascetin, R.; Brangwynne, C.P. The nucleolus as a multiphase liquid condensate. Nat. Rev. Mol. Cell Biol. 2021, 22, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Nizami, Z.; Deryusheva, S.; Gall, J.G. The Cajal body and histone locus body. Cold Spring Harb. Perspect. Biol. 2010, 2, a000653. [Google Scholar] [CrossRef] [PubMed]
- Pessina, F.; Gioia, U.; Brandi, O.; Farina, S.; Ceccon, M.; Francia, S.; d’Adda di Fagagna, F. DNA Damage Triggers a New Phase in Neurodegeneration. Trends Genet. 2021, 37, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; de Thé, H. PML nuclear bodies: From architecture to function. Curr. Opin. Cell Biol. 2018, 52, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Biamonti, G. Nuclear stress bodies: A heterochromatin affair? Nat. Rev. Mol. Cell Biol. 2004, 5, 493–498. [Google Scholar] [CrossRef]
- Smigova, J.; Juda, P.; Cmarko, D.; Raska, I. Fine structure of the “PcG body” in human U-2 OS cells established by correlative light-electron microscopy. Nucleus 2011, 2, 219–228. [Google Scholar] [CrossRef][Green Version]
- McCluggage, F.; Fox, A.H. Paraspeckle nuclear condensates: Global sensors of cell stress? Bioessays 2021, 43, e2000245. [Google Scholar] [CrossRef]
- Pollock, C.; Huang, S. The perinucleolar compartment. Cold Spring Harb. Perspect. Biol. 2010, 2, a000679. [Google Scholar] [CrossRef]
- Matera, A.G.; Frey, M.R. Coiled bodies and gems: Janus or gemini? Am. J. Hum. Genet. 1998, 63, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Belotserkovskaya, R.; Coates, J.; Dimitrova, D.S.; Polo, S.E.; Bradshaw, C.R.; Fraser, P.; Jackson, S.P. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol 2011, 193, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Veerabhadrappa, B.; Delaby, C.; Hirtz, C.; Vialaret, J.; Alcolea, D.; Lleo, A.; Fortea, J.; Santosh, M.S.; Choubey, S.; Lehmann, S. Detection of amyloid beta peptides in body fluids for the diagnosis of alzheimer’s disease: Where do we stand? Crit. Rev. Clin. Lab. Sci. 2020, 57, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gan, Y.; Cao, J.; Dong, X.; Ouyang, W. Pathophysiology of stress granules: An emerging link to diseases (Review). Int. J. Mol. Med. 2022, 49, 44. [Google Scholar] [CrossRef]
- Kim, W.J.; Kim, J.H.; Jang, S.K. Anti-inflammatory lipid mediator 15d-PGJ2 inhibits translation through inactivation of eIF4A. EMBO J. 2007, 26, 5020–5032. [Google Scholar] [CrossRef]
- Di Marco, B.; Dell’Albani, P.; D’Antoni, S.; Spatuzza, M.; Bonaccorso, C.M.; Musumeci, S.A.; Drago, F.; Bardoni, B.; Catania, M.V. Fragile X mental retardation protein (FMRP) and metabotropic glutamate receptor subtype 5 (mGlu5) control stress granule formation in astrocytes. Neurobiol. Dis. 2021, 154, 105338. [Google Scholar] [CrossRef]
- Ash, P.E.; Vanderweyde, T.E.; Youmans, K.L.; Apicco, D.J.; Wolozin, B. Pathological stress granules in Alzheimer’s disease. Brain Res. 2014, 1584, 52–58. [Google Scholar] [CrossRef]
- Yang, X.; Hu, Z.; Fan, S.; Zhang, Q.; Zhong, Y.; Guo, D.; Qin, Y.; Chen, M. Picornavirus 2A protease regulates stress granule formation to facilitate viral translation. PLoS Pathog. 2018, 14, e1006901. [Google Scholar] [CrossRef]
- Zhang, Q.; Sharma, N.R.; Zheng, Z.M.; Chen, M. Viral Regulation of RNA Granules in Infected Cells. Virol. Sin. 2019, 34, 175–191. [Google Scholar] [CrossRef]
- Balak, C.; Benard, M.; Schaefer, E.; Iqbal, S.; Ramsey, K.; Ernoult-Lange, M.; Mattioli, F.; Llaci, L.; Geoffroy, V.; Courel, M.; et al. Rare De Novo Missense Variants in RNA Helicase DDX6 Cause Intellectual Disability and Dysmorphic Features and Lead to P-Body Defects and RNA Dysregulation. Am. J. Hum. Genet. 2019, 105, 509–525. [Google Scholar] [CrossRef]
- Liu, L.; Weiss, E.; Panas, M.D.; Gotte, B.; Sellberg, S.; Thaa, B.; McInerney, G.M. RNA processing bodies are disassembled during Old World alphavirus infection. J. Gen. Virol. 2019, 100, 1375–1389. [Google Scholar] [CrossRef] [PubMed]
- Bhanji, R.A.; Eystathioy, T.; Chan, E.K.; Bloch, D.B.; Fritzler, M.J. Clinical and serological features of patients with autoantibodies to GW/P bodies. Clin. Immunol. 2007, 125, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.E.; Grassetti, A.V.; Taroni, J.N.; Lyons, S.M.; Schweppe, D.; Gordon, J.K.; Spiera, R.F.; Lafyatis, R.; Anderson, P.J.; Gerber, S.A.; et al. Stress granules and RNA processing bodies are novel autoantibody targets in systemic sclerosis. Arthritis Res. Ther. 2016, 18, 27. [Google Scholar] [CrossRef] [PubMed]
- Nunez Villacis, L.; Wong, M.S.; Ferguson, L.L.; Hein, N.; George, A.J.; Hannan, K.M. New Roles for the Nucleolus in Health and Disease. Bioessays 2018, 40, e1700233. [Google Scholar] [CrossRef]
- Scott, M.S.; Boisvert, F.M.; McDowall, M.D.; Lamond, A.I.; Barton, G.J. Characterization and prediction of protein nucleolar localization sequences. Nucleic Acids Res. 2010, 38, 7388–7399. [Google Scholar] [CrossRef]
- Frege, T.; Uversky, V.N. Intrinsically disordered proteins in the nucleus of human cells. Biochem. Biophys. Rep. 2015, 1, 33–51. [Google Scholar] [CrossRef]
- Windner, S.E.; Manhart, A.; Brown, A.; Mogilner, A.; Baylies, M.K. Nuclear Scaling Is Coordinated among Individual Nuclei in Multinucleated Muscle Fibers. Dev. Cell 2019, 49, 48–62.e43. [Google Scholar] [CrossRef]
- Walne, A.J.; Vulliamy, T.; Marrone, A.; Beswick, R.; Kirwan, M.; Masunari, Y.; Al-Qurashi, F.H.; Aljurf, M.; Dokal, I. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum. Mol. Genet. 2007, 16, 1619–1629. [Google Scholar] [CrossRef]
- Lee, K.H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788.e717. [Google Scholar] [CrossRef]
- Tiku, V.; Jain, C.; Raz, Y.; Nakamura, S.; Heestand, B.; Liu, W.; Spath, M.; Suchiman, H.E.D.; Muller, R.U.; Slagboom, P.E.; et al. Small nucleoli are a cellular hallmark of longevity. Nat. Commun. 2017, 8, 16083. [Google Scholar] [CrossRef]
- Wang, J.; Choi, J.M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174, 688–699.e16. [Google Scholar] [CrossRef]
- March, Z.M.; King, O.D.; Shorter, J. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 2016, 1647, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xu, E.S.; Leisenring, N.H.; Cardona, D.M.; Luo, L.; Ma, Y.; Ventura, A.; Kirsch, D.G. The Fusion Oncogene FUS-CHOP Drives Sarcomagenesis of High-Grade Spindle Cell Sarcomas in Mice. Sarcoma 2019, 2019, 1340261. [Google Scholar] [CrossRef] [PubMed]
- Nolan, M.; Talbot, K.; Ansorge, O. Pathogenesis of FUS-associated ALS and FTD: Insights from rodent models. Acta Neuropathol. Commun. 2016, 4, 99. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Primers 2018, 4, 5. [Google Scholar] [CrossRef]
- Ahmed, N.S.; Harrell, L.M.; Wieland, D.R.; Lay, M.A.; Thompson, V.F.; Schwartz, J.C. Fusion protein EWS-FLI1 is incorporated into a protein granule in cells. RNA 2021, 27, 920–932. [Google Scholar] [CrossRef]
- Snead, W.T.; Gladfelter, A.S. The Control Centers of Biomolecular Phase Separation: How Membrane Surfaces, PTMs, and Active Processes Regulate Condensation. Mol. Cell 2019, 76, 295–305. [Google Scholar] [CrossRef]
- Aumiller, W.M., Jr.; Keating, C.D. Experimental models for dynamic compartmentalization of biomolecules in liquid organelles: Reversible formation and partitioning in aqueous biphasic systems. Adv. Colloid Interface Sci. 2017, 239, 75–87. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H. Phase Separation, Transition, and Autophagic Degradation of Proteins in Development and Pathogenesis. Trends Cell Biol. 2019, 29, 417–427. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef]
- Tepper, K.; Biernat, J.; Kumar, S.; Wegmann, S.; Timm, T.; Hubschmann, S.; Redecke, L.; Mandelkow, E.M.; Muller, D.J.; Mandelkow, E. Oligomer formation of tau protein hyperphosphorylated in cells. J. Biol. Chem. 2014, 289, 34389–34407. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Lim, Y.J.; Liu, Z.; Long, H.; Sun, Y.; Hu, J.J.; Zhao, C.; Tao, Y.; Zhang, X.; Li, D.; et al. Parkinson’s disease-related phosphorylation at Tyr39 rearranges alpha-synuclein amyloid fibril structure revealed by cryo-EM. Proc. Natl. Acad. Sci. USA 2020, 117, 20305–20315. [Google Scholar] [CrossRef] [PubMed]
- Brahmachari, S.; Ge, P.; Lee, S.H.; Kim, D.; Karuppagounder, S.S.; Kumar, M.; Mao, X.; Shin, J.H.; Lee, Y.; Pletnikova, O.; et al. Activation of tyrosine kinase c-Abl contributes to alpha-synuclein-induced neurodegeneration. J. Clin. Investig. 2016, 126, 2970–2988. [Google Scholar] [CrossRef] [PubMed]
- Monahan, Z.; Ryan, V.H.; Janke, A.M.; Burke, K.A.; Rhoads, S.N.; Zerze, G.H.; O’Meally, R.; Dignon, G.L.; Conicella, A.E.; Zheng, W.; et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017, 36, 2951–2967. [Google Scholar] [CrossRef]
- Qamar, S.; Wang, G.; Randle, S.J.; Ruggeri, F.S.; Varela, J.A.; Lin, J.Q.; Phillips, E.C.; Miyashita, A.; Williams, D.; Strohl, F.; et al. FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-pi Interactions. Cell 2018, 173, 720–734.e15. [Google Scholar] [CrossRef]
- Piunti, A.; Shilatifard, A. The roles of Polycomb repressive complexes in mammalian development and cancer. Nat. Rev. Mol. Cell Biol. 2021, 22, 326–345. [Google Scholar] [CrossRef]
- Zhang, H.; Azevedo, R.B.; Lints, R.; Doyle, C.; Teng, Y.; Haber, D.; Emmons, S.W. Global regulation of Hox gene expression in C. elegans by a SAM domain protein. Dev. Cell 2003, 4, 903–915. [Google Scholar] [CrossRef]
- Qu, W.; Wang, Z.; Zhang, H. Phase separation of the C. elegans Polycomb protein SOP-2 is modulated by RNA and sumoylation. Protein Cell 2020, 11, 202–207. [Google Scholar] [CrossRef]
- Zhang, H.; Smolen, G.A.; Palmer, R.; Christoforou, A.; van den Heuvel, S.; Haber, D.A. SUMO modification is required for in vivo Hox gene regulation by the Caenorhabditis elegans Polycomb group protein SOP-2. Nat. Genet. 2004, 36, 507–511. [Google Scholar] [CrossRef]
- Rai, A.K.; Chen, J.X.; Selbach, M.; Pelkmans, L. Kinase-controlled phase transition of membraneless organelles in mitosis. Nature 2018, 559, 211–216. [Google Scholar] [CrossRef]
- Wippich, F.; Bodenmiller, B.; Trajkovska, M.G.; Wanka, S.; Aebersold, R.; Pelkmans, L. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell 2013, 152, 791–805. [Google Scholar] [CrossRef] [PubMed]
- Mediani, L.; Antoniani, F.; Galli, V.; Vinet, J.; Carra, A.D.; Bigi, I.; Tripathy, V.; Tiago, T.; Cimino, M.; Leo, G.; et al. Hsp90-mediated regulation of DYRK3 couples stress granule disassembly and growth via mTORC1 signaling. EMBO Rep. 2021, 22, e51740. [Google Scholar] [CrossRef] [PubMed]
- Hervas, R.; Oroz, J. Mechanistic Insights into the Role of Molecular Chaperones in Protein Misfolding Diseases: From Molecular Recognition to Amyloid Disassembly. Int. J. Mol. Sci. 2020, 21, 9186. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, S.; Gu, J.; Tong, Y.; Li, Y.; Gui, X.; Long, H.; Wang, C.; Zhao, C.; Lu, J.; et al. Hsp27 chaperones FUS phase separation under the modulation of stress-induced phosphorylation. Nat. Struct. Mol. Biol. 2020, 27, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Liu, Z.; Zhang, S.; Li, Y.; Xia, W.; Wang, C.; Xiang, H.; Liu, Z.; Tan, L.; Fang, Y.; et al. Hsp40 proteins phase separate to chaperone the assembly and maintenance of membraneless organelles. Proc. Natl. Acad. Sci. USA 2020, 117, 31123–31133. [Google Scholar] [CrossRef] [PubMed]
- Toth, M.E.; Szegedi, V.; Varga, E.; Juhasz, G.; Horvath, J.; Borbely, E.; Csibrany, B.; Alfoldi, R.; Lenart, N.; Penke, B.; et al. Overexpression of Hsp27 ameliorates symptoms of Alzheimer’s disease in APP/PS1 mice. Cell Stress Chaperones 2013, 18, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, T.; Peng, Z.; Wang, Z.; Liu, J.; Yang, T.; Wu, L.; Liu, G.; Zhou, M.; Tong, M.; et al. Histone chaperone CAF-1 promotes HIV-1 latency by leading the formation of phase-separated suppressive nuclear bodies. EMBO J. 2021, 40, e106632. [Google Scholar] [CrossRef]
- Strulson, C.A.; Molden, R.C.; Keating, C.D.; Bevilacqua, P.C. RNA catalysis through compartmentalization. Nat. Chem. 2012, 4, 941–946. [Google Scholar] [CrossRef]
- Duronio, R.J.; Marzluff, W.F. Coordinating cell cycle-regulated histone gene expression through assembly and function of the Histone Locus Body. RNA Biol. 2017, 14, 726–738. [Google Scholar] [CrossRef]
- Tatomer, D.C.; Terzo, E.; Curry, K.P.; Salzler, H.; Sabath, I.; Zapotoczny, G.; McKay, D.J.; Dominski, Z.; Marzluff, W.F.; Duronio, R.J. Concentrating pre-mRNA processing factors in the histone locus body facilitates efficient histone mRNA biogenesis. J. Cell Biol. 2016, 213, 557–570. [Google Scholar] [CrossRef]
- Kim, K.M.; Lee, A.; Choi, K.Y.; Lee, K.Y.; Kwak, J.J. Intestinal tuberculosis: Clinicopathologic analysis and diagnosis by endoscopic biopsy. Am. J. Gastroenterol. 1998, 93, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Trinkle-Mulcahy, L.; Sleeman, J.E. The Cajal body and the nucleolus: “In a relationship” or “It’s complicated”? RNA Biol. 2017, 14, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Berkovits, B.D.; Mayr, C. Alternative 3′ UTRs act as scaffolds to regulate membrane protein localization. Nature 2015, 522, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Lianoglou, S.; Garg, V.; Yang, J.L.; Leslie, C.S.; Mayr, C. Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev. 2013, 27, 2380–2396. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Yu, D.; Hansen, A.S.; Ganguly, S.; Liu, R.; Heckert, A.; Darzacq, X.; Zhou, Q. Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 2018, 558, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Egloff, S. CDK9 keeps RNA polymerase II on track. Cell Mol. Life Sci. 2021, 78, 5543–5567. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.K.; Spille, J.H.; Hecht, M.; Lee, C.; Li, C.; Grube, V.; Cisse, I.I. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 2018, 361, 412–415. [Google Scholar] [CrossRef]
- Burke, K.A.; Janke, A.M.; Rhine, C.L.; Fawzi, N.L. Residue-by-Residue View of In Vitro FUS Granules that Bind the C-Terminal Domain of RNA Polymerase II. Mol. Cell 2015, 60, 231–241. [Google Scholar] [CrossRef]
- Janke, A.M.; Seo, D.H.; Rahmanian, V.; Conicella, A.E.; Mathews, K.L.; Burke, K.A.; Mittal, J.; Fawzi, N.L. Lysines in the RNA Polymerase II C-Terminal Domain Contribute to TAF15 Fibril Recruitment. Biochemistry 2018, 57, 2549–2563. [Google Scholar] [CrossRef]
- Boija, A.; Klein, I.A.; Sabari, B.R.; Dall’Agnese, A.; Coffey, E.L.; Zamudio, A.V.; Li, C.H.; Shrinivas, K.; Manteiga, J.C.; Hannett, N.M.; et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 2018, 175, 1842–1855.e16. [Google Scholar] [CrossRef]
- Cai, D.; Feliciano, D.; Dong, P.; Flores, E.; Gruebele, M.; Porat-Shliom, N.; Sukenik, S.; Liu, Z.; Lippincott-Schwartz, J. Phase separation of YAP reorganizes genome topology for long-term YAP target gene expression. Nat. Cell Biol. 2019, 21, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Sommer, U.; Pranada, A.L.; Giese, B.; Kuster, A.; Haan, S.; Becker, W.; Heinrich, P.C.; Muller-Newen, G. STAT3 is enriched in nuclear bodies. J. Cell Sci. 2004, 117, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, P.B. Interleukin-6 at the Host-Tumor Interface: STAT3 in Biomolecular Condensates in Cancer Cells. Cells 2022, 11, 1164. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, P.B.; Westley, J.; Lerea, K.M.; DiSenso-Browne, S.; Etlinger, J.D. Biomolecular condensates in cell biology and virology: Phase-separated membraneless organelles (MLOs). Anal. Biochem. 2020, 597, 113691. [Google Scholar] [CrossRef]
- Nikolic, J.; Le Bars, R.; Lama, Z.; Scrima, N.; Lagaudriere-Gesbert, C.; Gaudin, Y.; Blondel, D. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 2017, 8, 58. [Google Scholar] [CrossRef]
- Geiger, F.; Acker, J.; Papa, G.; Wang, X.; Arter, W.E.; Saar, K.L.; Erkamp, N.A.; Qi, R.; Bravo, J.P.; Strauss, S.; et al. Liquid-liquid phase separation underpins the formation of replication factories in rotaviruses. EMBO J. 2021, 40, e107711. [Google Scholar] [CrossRef]
- Lahaye, X.; Vidy, A.; Pomier, C.; Obiang, L.; Harper, F.; Gaudin, Y.; Blondel, D. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: Evidence that NBs are sites of viral transcription and replication. J. Virol. 2009, 83, 7948–7958. [Google Scholar] [CrossRef]
- Hoenen, T.; Shabman, R.S.; Groseth, A.; Herwig, A.; Weber, M.; Schudt, G.; Dolnik, O.; Basler, C.F.; Becker, S.; Feldmann, H. Inclusion bodies are a site of ebolavirus replication. J. Virol. 2012, 86, 11779–11788. [Google Scholar] [CrossRef]
- Zhou, Y.; Su, J.M.; Samuel, C.E.; Ma, D. Measles Virus Forms Inclusion Bodies with Properties of Liquid Organelles. J. Virol. 2019, 93, e00948-19. [Google Scholar] [CrossRef]
- Alenquer, M.; Vale-Costa, S.; Etibor, T.A.; Ferreira, F.; Sousa, A.L.; Amorim, M.J. Influenza a virus ribonucleoproteins form liquid organelles at endoplasmic reticulum exit sites. Nat. Commun. 2019, 10, 1629. [Google Scholar] [CrossRef] [PubMed]
- Rincheval, V.; Lelek, M.; Gault, E.; Bouillier, C.; Sitterlin, D.; Blouquit-Laye, S.; Galloux, M.; Zimmer, C.; Eleouet, J.F.; Rameix-Welti, M.A. Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat. Commun. 2017, 8, 563. [Google Scholar] [CrossRef] [PubMed]
- Fouquet, B.; Nikolic, J.; Larrous, F.; Bourhy, H.; Wirblich, C.; Lagaudriere-Gesbert, C.; Blondel, D. Focal adhesion kinase is involved in rabies virus infection through its interaction with viral phosphoprotein P. J. Virol. 2015, 89, 1640–1651. [Google Scholar] [CrossRef] [PubMed]
- Raab-Traub, N. Novel mechanisms of EBV-induced oncogenesis. Curr. Opin. Virol. 2012, 2, 453–458. [Google Scholar] [CrossRef]
- Kaiser, C.; Laux, G.; Eick, D.; Jochner, N.; Bornkamm, G.W.; Kempkes, B. The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J. Virol. 1999, 73, 4481–4484. [Google Scholar] [CrossRef]
- Peng, Q.; Wang, L.; Qin, Z.; Wang, J.; Zheng, X.; Wei, L.; Zhang, X.; Zhang, X.; Liu, C.; Li, Z.; et al. Phase Separation of Epstein-Barr Virus EBNA2 and Its Coactivator EBNALP Controls Gene Expression. J. Virol. 2020, 94, e01771-19. [Google Scholar] [CrossRef]
- Pham, C.L.; Shanmugam, N.; Strange, M.; O’Carroll, A.; Brown, J.W.; Sierecki, E.; Gambin, Y.; Steain, M.; Sunde, M. Viral M45 and necroptosis-associated proteins form heteromeric amyloid assemblies. EMBO Rep. 2019, 20, e46518. [Google Scholar] [CrossRef]
- Sehgal, P.B. Metastable biomolecular condensates of interferon-inducible antiviral Mx-family GTPases: A paradigm shift in the last three years. J. Biosci. 2021, 46, 1–14. [Google Scholar] [CrossRef]
- Davis, D.; Yuan, H.; Liang, F.X.; Yang, Y.M.; Westley, J.; Petzold, C.; Dancel-Manning, K.; Deng, Y.; Sall, J.; Sehgal, P.B. Human Antiviral Protein MxA Forms Novel Metastable Membraneless Cytoplasmic Condensates Exhibiting Rapid Reversible Tonicity-Driven Phase Transitions. J. Virol. 2019, 93, e01014-19. [Google Scholar] [CrossRef]
- Sehgal, P.B.; Yuan, H.; Scott, M.F.; Deng, Y.; Liang, F.X.; Mackiewicz, A. Murine GFP-Mx1 forms nuclear condensates and associates with cytoplasmic intermediate filaments: Novel antiviral activity against VSV. J. Biol. Chem. 2020, 295, 18023–18035. [Google Scholar] [CrossRef]
- Gu, Z.C.; Wu, E.; Sailer, C.; Jando, J.; Styles, E.; Eisenkolb, I.; Kuschel, M.; Bitschar, K.; Wang, X.; Huang, L.; et al. Ubiquitin orchestrates proteasome dynamics between proliferation and quiescence in yeast. Mol. Biol. Cell 2017, 28, 2479–2491. [Google Scholar] [CrossRef] [PubMed]
- Enenkel, C. The paradox of proteasome granules. Curr. Genet. 2018, 64, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Yedidi, R.S.; Fatehi, A.K.; Enenkel, C. Proteasome dynamics between proliferation and quiescence stages of Saccharomyces cerevisiae. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Lavut, A.; Raveh, D. Sequestration of highly expressed mRNAs in cytoplasmic granules, P-bodies, and stress granules enhances cell viability. PLoS Genet. 2012, 8, e1002527. [Google Scholar] [CrossRef] [PubMed]
- Nostramo, R.; Xing, S.; Zhang, B.; Herman, P.K. Insights into the Role of P-Bodies and Stress Granules in Protein Quality Control. Genetics 2019, 213, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Leo, C.; Zhu, J.; Wu, X.; O’Neil, J.; Park, E.J.; Chen, J.D. Sequestration and inhibition of Daxx-mediated transcriptional repression by PML. Mol. Cell. Biol. 2000, 20, 1784–1796. [Google Scholar] [CrossRef] [PubMed]
- Shastrula, P.K.; Sierra, I.; Deng, Z.; Keeney, F.; Hayden, J.E.; Lieberman, P.M.; Janicki, S.M. PML is recruited to heterochromatin during S phase and represses DAXX-mediated histone H3.3 chromatin assembly. J. Cell Sci. 2019, 132, jcs220970. [Google Scholar] [CrossRef]
- Wang, M.; Bokros, M.; Theodoridis, P.R.; Lee, S. Nucleolar Sequestration: Remodeling Nucleoli Into Amyloid Bodies. Front. Genet. 2019, 10, 1179. [Google Scholar] [CrossRef]
- Weber, J.D.; Taylor, L.J.; Roussel, M.F.; Sherr, C.J.; Bar-Sagi, D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol. 1999, 1, 20–26. [Google Scholar] [CrossRef]
- Poyurovsky, M.V.; Jacq, X.; Ma, C.; Karni-Schmidt, O.; Parker, P.J.; Chalfie, M.; Manley, J.L.; Prives, C. Nucleotide binding by the Mdm2 RING domain facilitates Arf-independent Mdm2 nucleolar localization. Mol. Cell 2003, 12, 875–887. [Google Scholar] [CrossRef]
- Mekhail, K.; Gunaratnam, L.; Bonicalzi, M.E.; Lee, S. HIF activation by pH-dependent nucleolar sequestration of VHL. Nat. Cell Biol. 2004, 6, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Arabi, A.; Rustum, C.; Hallberg, E.; Wright, A.P. Accumulation of c-Myc and proteasomes at the nucleoli of cells containing elevated c-Myc protein levels. J. Cell Sci. 2003, 116, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.; Kusdra, L.; Collins, K. Subnuclear shuttling of human telomerase induced by transformation and DNA damage. Nat. Cell Biol. 2002, 4, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Shou, W.; Seol, J.H.; Shevchenko, A.; Baskerville, C.; Moazed, D.; Chen, Z.W.; Jang, J.; Shevchenko, A.; Charbonneau, H.; Deshaies, R.J. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell 1999, 97, 233–244. [Google Scholar] [CrossRef]
- Mehta, S.; Zhang, J. Liquid-liquid phase separation drives cellular function and dysfunction in cancer. Nat. Rev. Cancer 2022, 22, 239–252. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Takanashi, K. FUS interacts with nuclear matrix-associated protein SAFB1 as well as Matrin3 to regulate splicing and ligand-mediated transcription. Sci. Rep. 2016, 6, 35195. [Google Scholar] [CrossRef]
- Murthy, A.C.; Dignon, G.L.; Kan, Y.; Zerze, G.H.; Parekh, S.H.; Mittal, J.; Fawzi, N.L. Molecular interactions underlying liquid-liquid phase separation of the FUS low-complexity domain. Nat. Struct. Mol. Biol. 2019, 26, 637–648. [Google Scholar] [CrossRef]
- Rhoads, S.N.; Monahan, Z.T.; Yee, D.S.; Shewmaker, F.P. The Role of Post-Translational Modifications on Prion-like Aggregation and Liquid-Phase Separation of FUS. Int. J. Mol. Sci. 2018, 19, 886. [Google Scholar] [CrossRef]
- Chook, Y.M.; Suel, K.E. Nuclear import by karyopherin-betas: Recognition and inhibition. Biochim. Biophys. Acta 2011, 1813, 1593–1606. [Google Scholar] [CrossRef]
- Ishigaki, S.; Sobue, G. Importance of Functional Loss of FUS in FTLD/ALS. Front. Mol. Biosci. 2018, 5, 44. [Google Scholar] [CrossRef]
- Nomura, T.; Watanabe, S.; Kaneko, K.; Yamanaka, K.; Nukina, N.; Furukawa, Y. Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J. Biol. Chem. 2014, 289, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Dormann, D.; Madl, T.; Valori, C.F.; Bentmann, E.; Tahirovic, S.; Abou-Ajram, C.; Kremmer, E.; Ansorge, O.; Mackenzie, I.R.; Neumann, M.; et al. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012, 31, 4258–4275. [Google Scholar] [CrossRef] [PubMed]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.D.; Simons, M.; Niessing, D.; Madl, T.; et al. Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 2018, 173, 706–719.e713. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.K.; Savastano, A.; Singh, P.; Mukhopadhyay, S.; Zweckstetter, M. Liquid-liquid phase separation of tau: From molecular biophysics to physiology and disease. Protein Sci. 2021, 30, 1294–1314. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and tauopathies. Brain Res. Bull. 2016, 126, 238–292. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Savastano, A.; Flores, D.; Kadavath, H.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Disease-Associated Tau Phosphorylation Hinders Tubulin Assembly within Tau Condensates. Angew. Chem. Int. Ed. Engl. 2021, 60, 726–730. [Google Scholar] [CrossRef]
- Ukmar-Godec, T.; Hutten, S.; Grieshop, M.P.; Rezaei-Ghaleh, N.; Cima-Omori, M.S.; Biernat, J.; Mandelkow, E.; Soding, J.; Dormann, D.; Zweckstetter, M. Lysine/RNA-interactions drive and regulate biomolecular condensation. Nat. Commun. 2019, 10, 2909. [Google Scholar] [CrossRef]
- Ferreon, J.C.; Jain, A.; Choi, K.J.; Tsoi, P.S.; MacKenzie, K.R.; Jung, S.Y.; Ferreon, A.C. Acetylation Disfavors Tau Phase Separation. Int. J. Mol. Sci. 2018, 19, 1360. [Google Scholar] [CrossRef]
- Ou, S.H.; Wu, F.; Harrich, D.; Garcia-Martinez, L.F.; Gaynor, R.B. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 1995, 69, 3584–3596. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Dörk, T.; Zuccato, E.; Pagani, F.; Romano, M.; Baralle, F.E. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001, 20, 1774–1784. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Dhakal, S.; Wyant, C.E.; George, H.E.; Morgan, S.E.; Rangachari, V. Prion-like C-Terminal Domain of TDP-43 and alpha-Synuclein Interact Synergistically to Generate Neurotoxic Hybrid Fibrils. J. Mol. Biol. 2021, 433, 166953. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Hasegawa, M. Prion-like properties of assembled TDP-43. Curr. Opin. Neurobiol. 2020, 61, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Kwong, L.K.; Lee, E.B.; Kremmer, E.; Flatley, A.; Xu, Y.; Forman, M.S.; Troost, D.; Kretzschmar, H.A.; Trojanowski, J.Q.; et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009, 117, 137–149. [Google Scholar] [CrossRef]
- Cohen, T.J.; Hwang, A.W.; Restrepo, C.R.; Yuan, C.X.; Trojanowski, J.Q.; Lee, V.M. An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 2015, 6, 5845. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Sharpe, K.; Weerasinghe, P.P.; Algarzae, N.K.; Shekoyan, A.R.; Moussa, C.E. Parkin ubiquitinates Tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J. Biol. Chem. 2013, 288, 4103–4115. [Google Scholar] [CrossRef]
- Babinchak, W.M.; Haider, R.; Dumm, B.K.; Sarkar, P.; Surewicz, K.; Choi, J.K.; Surewicz, W.K. The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J. Biol. Chem. 2019, 294, 6306–6317. [Google Scholar] [CrossRef]
- Anselmi, M.; Calligari, P.; Hub, J.S.; Tartaglia, M.; Bocchinfuso, G.; Stella, L. Structural Determinants of Phosphopeptide Binding to the N-Terminal Src Homology 2 Domain of the SHP2 Phosphatase. J. Chem. Inf. Model. 2020, 60, 3157–3171. [Google Scholar] [CrossRef] [PubMed]
- Tajan, M.; de Rocca Serra, A.; Valet, P.; Edouard, T.; Yart, A. SHP2 sails from physiology to pathology. Eur. J. Med. Genet. 2015, 58, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Gelb, B.D. Noonan syndrome and related disorders: Genetics and pathogenesis. Annu. Rev. Genom. Hum. Genet. 2005, 6, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Digilio, M.C.; Conti, E.; Sarkozy, A.; Mingarelli, R.; Dottorini, T.; Marino, B.; Pizzuti, A.; Dallapiccola, B. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am. J. Hum. Genet. 2002, 71, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, D.; Miyamoto, M.; Takahashi, A.; Yomogita, Y.; Higashi, H.; Kondo, S.; Hatakeyama, M. Isolation of a distinct class of gain-of-function SHP-2 mutants with oncogenic RAS-like transforming activity from solid tumors. Oncogene 2008, 27, 3508–3515. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.H.; Xu, J.; Walls, C.D.; Chen, L.; Zhang, S.; Zhang, R.; Wu, L.; Wang, L.; Liu, S.; Zhang, Z.Y. Structural and mechanistic insights into LEOPARD syndrome-associated SHP2 mutations. J. Biol. Chem. 2013, 288, 10472–10482. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.H.; Zhang, R.Y.; Walls, C.D.; Chen, L.; Zhang, S.; Wu, L.; Liu, S.; Zhang, Z.Y. Molecular basis of gain-of-function LEOPARD syndrome-associated SHP2 mutations. Biochemistry 2014, 53, 4136–4151. [Google Scholar] [CrossRef]
- Zhu, G.; Xie, J.; Kong, W.; Xie, J.; Li, Y.; Du, L.; Zheng, Q.; Sun, L.; Guan, M.; Li, H.; et al. Phase Separation of Disease-Associated SHP2 Mutants Underlies MAPK Hyperactivation. Cell 2020, 183, 490–502.e18. [Google Scholar] [CrossRef]
- Boopathy, G.T.K.; Hong, W. Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis. Front. Cell Dev. Biol. 2019, 7, 49. [Google Scholar] [CrossRef]
- Masliantsev, K.; Karayan-Tapon, L.; Guichet, P.O. Hippo Signaling Pathway in Gliomas. Cells 2021, 10, 184. [Google Scholar] [CrossRef]
- Franklin, J.M.; Guan, K.L. YAP/TAZ phase separation for transcription. Nat. Cell Biol. 2020, 22, 357–358. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kang, J.G.; Kang, M.J.; Park, J.H.; Kim, Y.J.; Kweon, T.H.; Lee, H.W.; Jho, E.H.; Lee, Y.H.; Kim, S.I.; et al. O-GlcNAcylation on LATS2 disrupts the Hippo pathway by inhibiting its activity. Proc. Natl. Acad. Sci. USA 2020, 117, 14259–14269. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
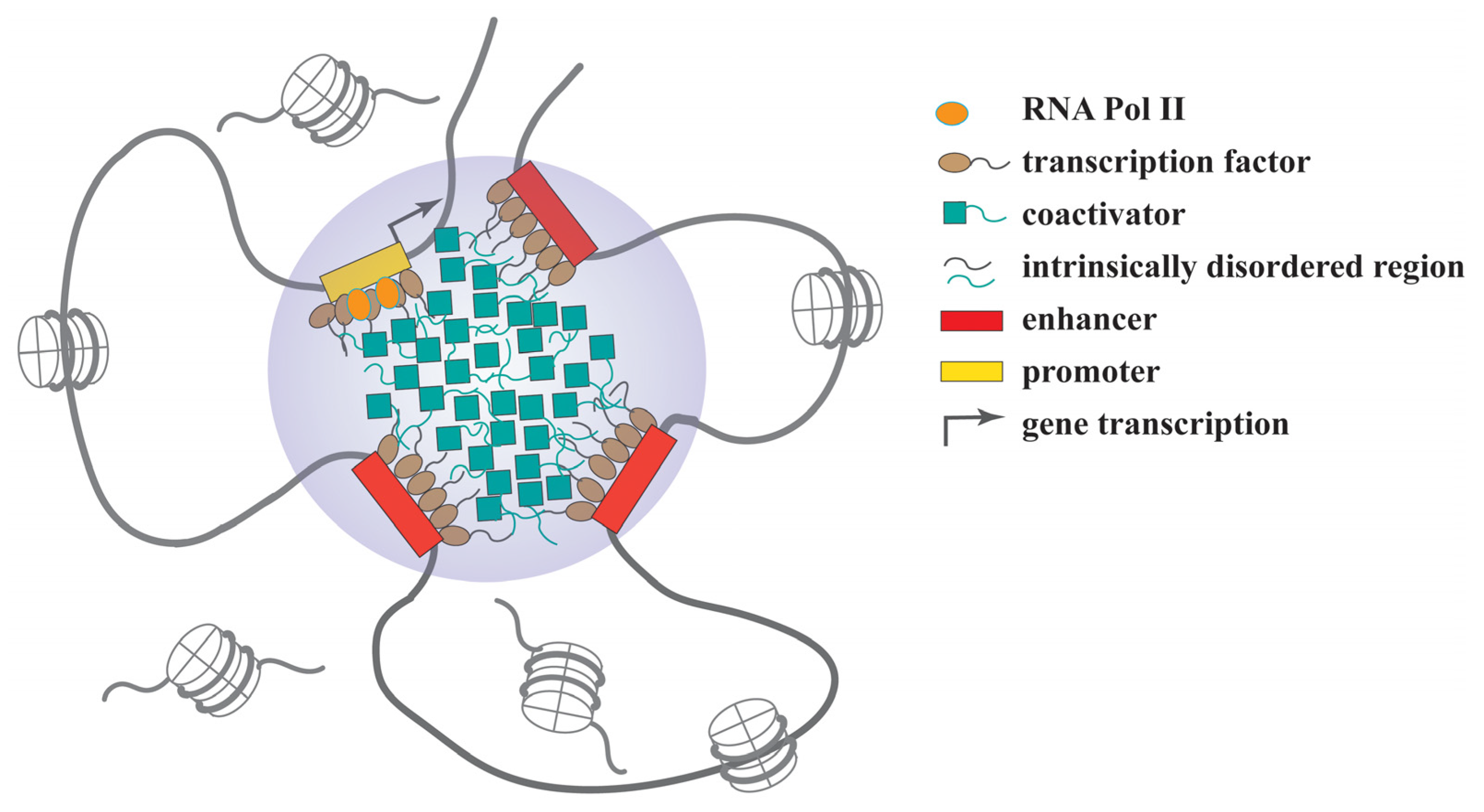{kind=link}
| Localization | Name | Alias | Size (nm) | Components | Functions | Diseases | References |
|---|---|---|---|---|---|---|---|
| Cytoplasm | P-body | GW-body, RNA processing body, decapping body | 100–300 | K63, TRAF6, Tob1, TUT4, NoBody, LSM1, GW182, DDX3, DDX6, XRN1, etc. | mRNA degradation, post-transcriptional gene silencing, response to stress, storage of translationally repressed mRNAs | viral infection, neurodegenerative diseases, autoimmune diseases. | [39,40] |
| Stress granule | — | 1000–2000 | RBPs, non-RBPs, TDRD3, TDP43, G3BP1, eIF3, eIF4G, PABPC1, etc. | translational regulation, response to stresses, antiviral defense, response to stresses, store mRNA and proteins | amyotrophic lateral sclerosis, frontotemporal lobar degeneration, cancer, viral infection, inflammatory diseases | [5,41] | |
| Germ granule | P-granule, chromatoid body, polar granule | 250–4000 | MEG-3, PGL, RNA, etc. | post-transcriptional regulation, regulation of Germ cell development and function, cell division | Germ cell development | [42] | |
| Synaptic density | Postsynaptic density | 500 | PSD-95, GKAP, Shank, Homer, etc. | responsible for signal processing | neuropsychiatric diseases | [43] | |
| RNA transport granule | Neuronal RNA granule | 500–1000 | Sam68, RNG105, SMN, etc. | mRNA storage and transport | neurodegenerative diseases | [44] | |
| Balbiani Body | Balbiani’s vesicle, the yolk body of Balbiani, yolk nucleus | 50–250,000 | RNA, mitochondria, Golgi, endoplasmic reticulum, etc. | store RNA, proteins and mitochondria | — | [45] | |
| Sec body | — | 1000 | COPII components, Sec16, etc. | response to the nutrient stress of amino acid starvation, protect ERES components from degradation | — | [46] | |
| U-body | Uridine-rich snRNP body | 500 | SnRNP, SMN, etc. | storage and assembly of snRNPs | spinal muscular atrophy | [47] | |
| PSG | — | 500 | proteasomes, free ubiquitin, etc. | protein-specific degradation, store proteasome | aging and age-related disease | [48] | |
| Signaling puncta | Dvl puncta | 500–1000 | Dvl-2, etc. | signal transduction | — | [49] | |
| Metabolic granule | G-body | 1000–5000 | glycolytic enzymes, etc. | glycolysis and storage | — | [50] | |
| STAT3 cytoplasmic body | STAT3 sequestering endosomes | — | STAT3 | prolongation of signaling and/or cross talk | hepatoma | [51,52] | |
| TIS granule | — | 1000–5000 | TIS11B, membrane protein-encoding mRNAs | 3′UTR-dependent nurturing of nascent proteins | — | [53] | |
| Nuclear membrane | Nuclear pore complex | — | 40–100 | nucleoporins, NDC1, GP210, POM121 etc. | facilitate nucleocytoplasmic transport, chromatin organization | neurological disorders and the aging brain, viral infections and immunity, the development and progression of cancers | [54] |
| Nucleus | Nucleolus | — | 1000–10,000 | Nucleolin, rRNA, rDNA, etc. | ribosome biogenesis | Werner syndrome, Bloom syndrome, Treacher Collins syndrome, dyskeratosis congenita syndrome, Rothmund–Thomson syndrome | [55] |
| HLB | — | 1000 | NPAT, FLASH, SLBP, p220NPAT, NELF, symplekin, etc. | processing of the histone pre-mRNAs, histone gene transcription | breast cancer | [56] | |
| DNA damage foci | — | 500 | γH2AX, ATM, 53BP1, RAD51, etc. | response to DNA damage | neurodegenerative diseases | [57] | |
| PML body | PML oncogenic domain, nuclear dot, Kremer body, | 250–500 | UBC9, RNF4, SP100, P53, DAXX, SUMO, PML, RNF168, etc. | transcription regulation, apoptosis signaling, epigenetic gene silencing, sequester partner proteins, SUMOylation sites | Acute Promyelocytic Leukemia, liver fibrosis | [58] | |
| Nuclear stress body | Peroxisome granule (PG) | 300–3000 | HSF1, HAP, SAM68, etc. | response to stress, control of gene expression and RNA splicing activities | metabolic syndrome | [59] | |
| Cajal body | accessory body | 100–2000 | RNA, snRNPs, scaRNAs, Coilin, SMN, etc. | pre-mRNA and pre-rRNA processing | amyotrophic lateral sclerosis, spinal muscular atrophy | [56] | |
| PcG body | — | 200–1500 | PRC1, PRC2, EZH2, etc. | transcriptional repression | malignant lymphomas, epithelial tumors | [60] | |
| CNB | — | 1000–3000 | CBP, SUMO-1, etc. | response to DNA damage, protein SUMOylation | — | [36] | |
| Paraspeckle | — | 500–1000 | CTN-RNA, PSP1, p54nrb, NEAT1, NONO, etc. | regulate gene expression, RNA processing | breast cancer, hepatocellular carcinoma, viral infection, neurodegenerative diseases | [61] | |
| PNC | — | 250–4000 | CUGBP, KSRP, polymerase III, Nucleolin, PTB, SRP RNA, etc. | transcriptional regulation, RNA metabolism | breast cancer, ovarian cancer | [62] | |
| Nuclear gem | Gemini of Cajal body, Gemini of coiled body | 100–2000 | SMN, etc. | mRNA processing | spinal muscular atrophy | [63] | |
| OPT domain body | 53P1-OPT domain | 1000–1500 | Nascent mRNA, transcription factors, etc. | transcriptional regulation, response to the replication stress | — | [64] | |
| STAT3 nuclear body | — | — | STAT3, CREB binding protein (CBP), acetylated histone H4 | activation of target genes | hepatoma | [51] | |
| Nucleolus | Amyloid body | A-body | 500–2000 | Amyloid beta peptides, etc. | store proteins | neurodegenerative diseases | [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Wang, S.; Wang, W.; Shi, J.; Stovall, D.B.; Li, D.; Sui, G. Phase-Separated Subcellular Compartmentation and Related Human Diseases. Int. J. Mol. Sci. 2022, 23, 5491. https://doi.org/10.3390/ijms23105491
Zhang L, Wang S, Wang W, Shi J, Stovall DB, Li D, Sui G. Phase-Separated Subcellular Compartmentation and Related Human Diseases. International Journal of Molecular Sciences. 2022; 23(10):5491. https://doi.org/10.3390/ijms23105491
Chicago/Turabian StyleZhang, Lin, Shubo Wang, Wenmeng Wang, Jinming Shi, Daniel B. Stovall, Dangdang Li, and Guangchao Sui. 2022. "Phase-Separated Subcellular Compartmentation and Related Human Diseases" International Journal of Molecular Sciences 23, no. 10: 5491. https://doi.org/10.3390/ijms23105491
APA StyleZhang, L., Wang, S., Wang, W., Shi, J., Stovall, D. B., Li, D., & Sui, G. (2022). Phase-Separated Subcellular Compartmentation and Related Human Diseases. International Journal of Molecular Sciences, 23(10), 5491. https://doi.org/10.3390/ijms23105491