
Immune Checkpoint Blockade via PD-L1 Potentiates More CD28-Based than 4-1BB-Based Anti-Carbonic Anhydrase IX Chimeric Antigen Receptor T Cells

 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
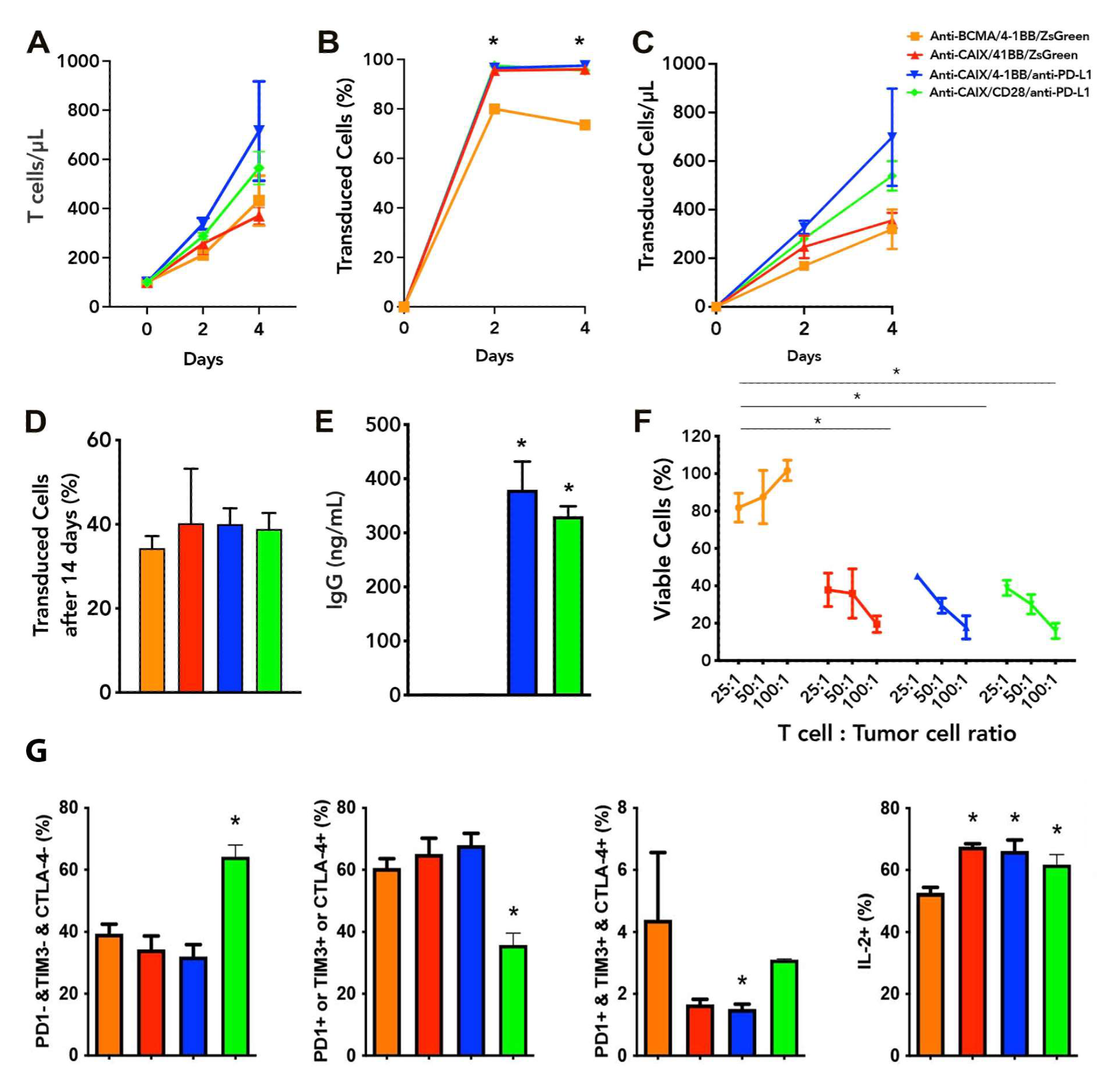
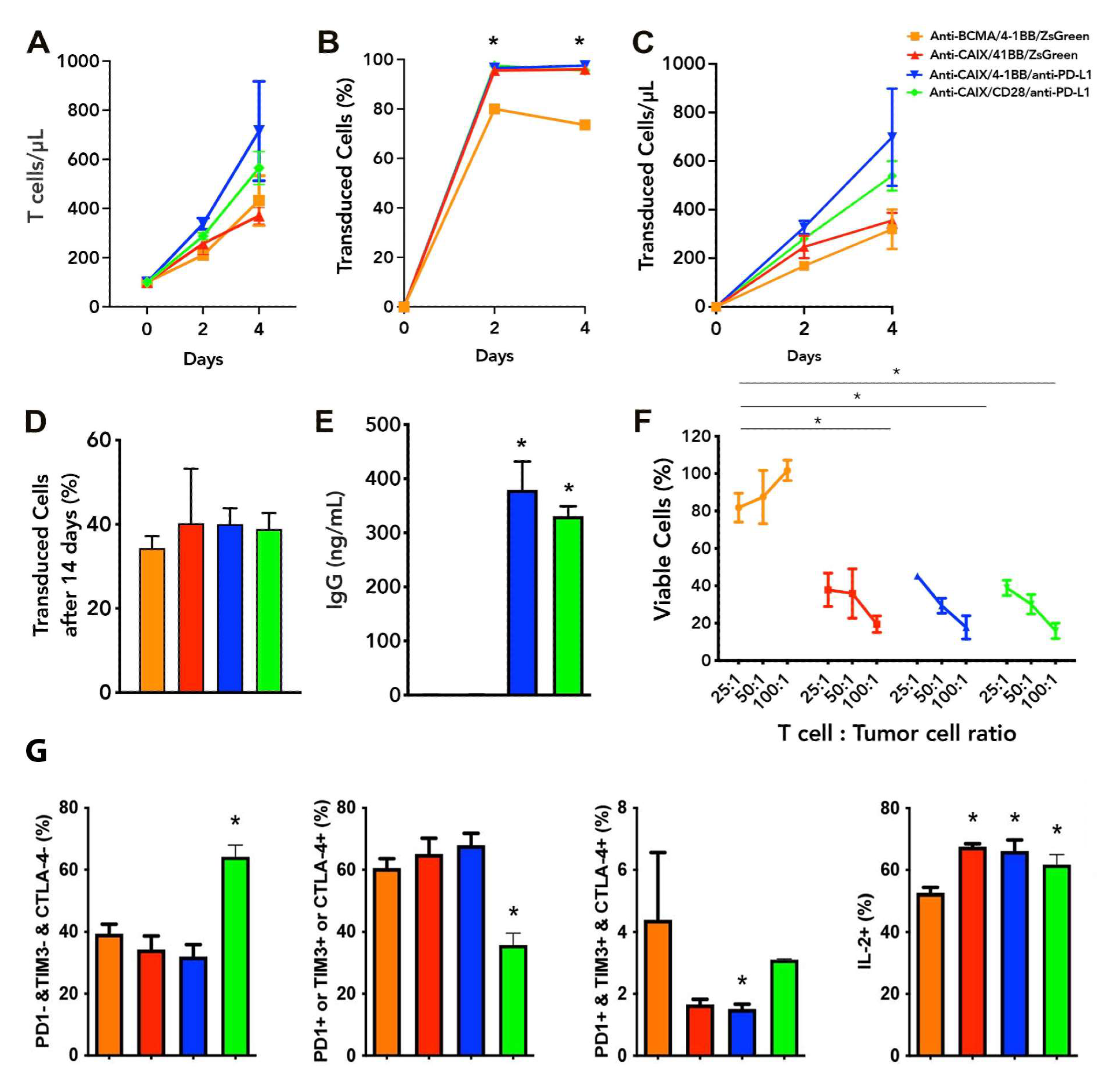
2.1. Functional Characterization and Cytotoxic Activity In Vitro of Anti-CAIX CAR T Cells CD28ζ versus CD8α 4-1BBζ Releasing Anti-PD-L1
2.2. Exhaustion Status of Anti-CAIX CAR T Cells CD28ζ versus CD8α 4-1BBζ Releasing Anti-PD-L1 IgG4 Antibodies in Co-Culture with Human ccRCC Cells In Vitro
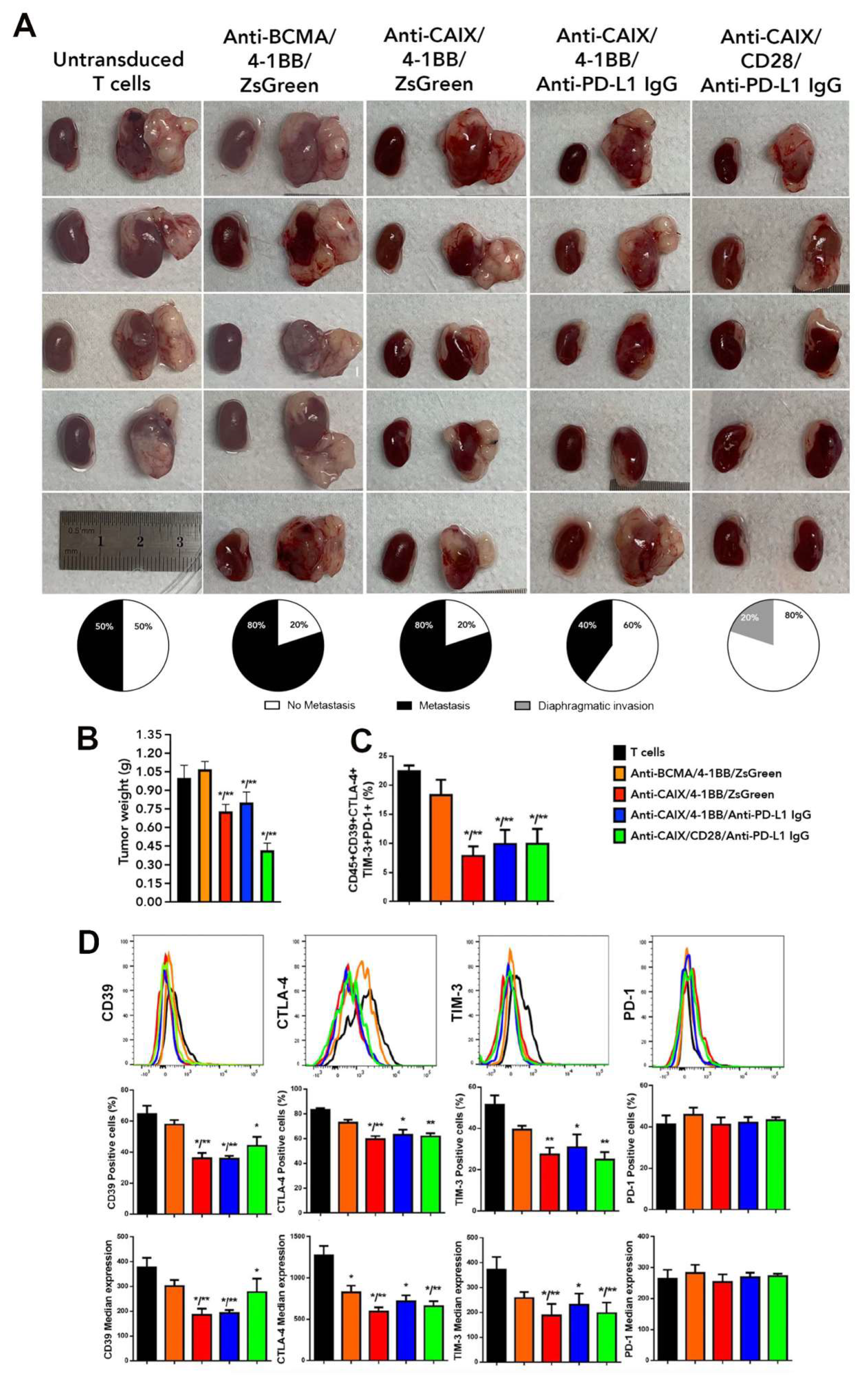
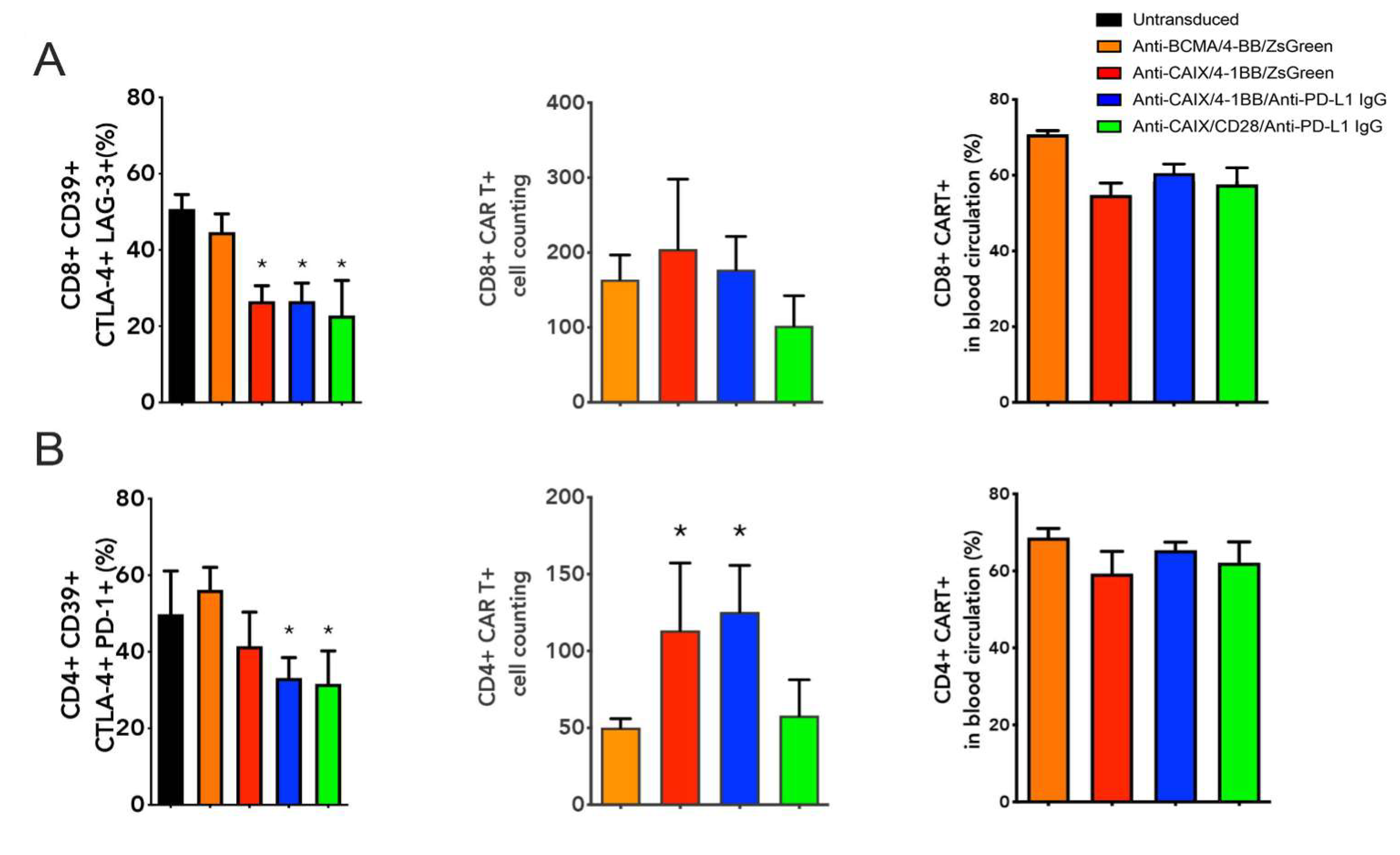
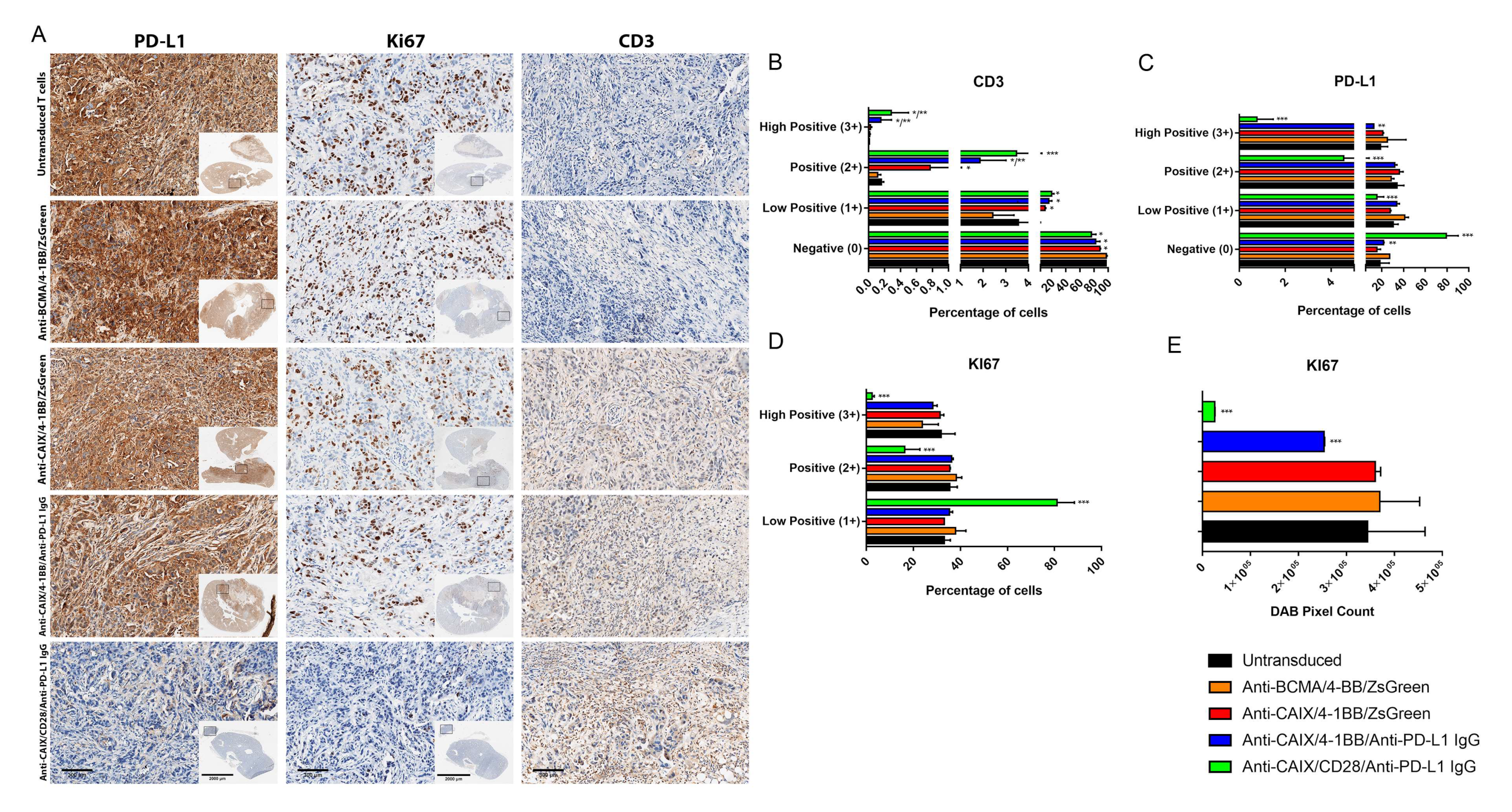
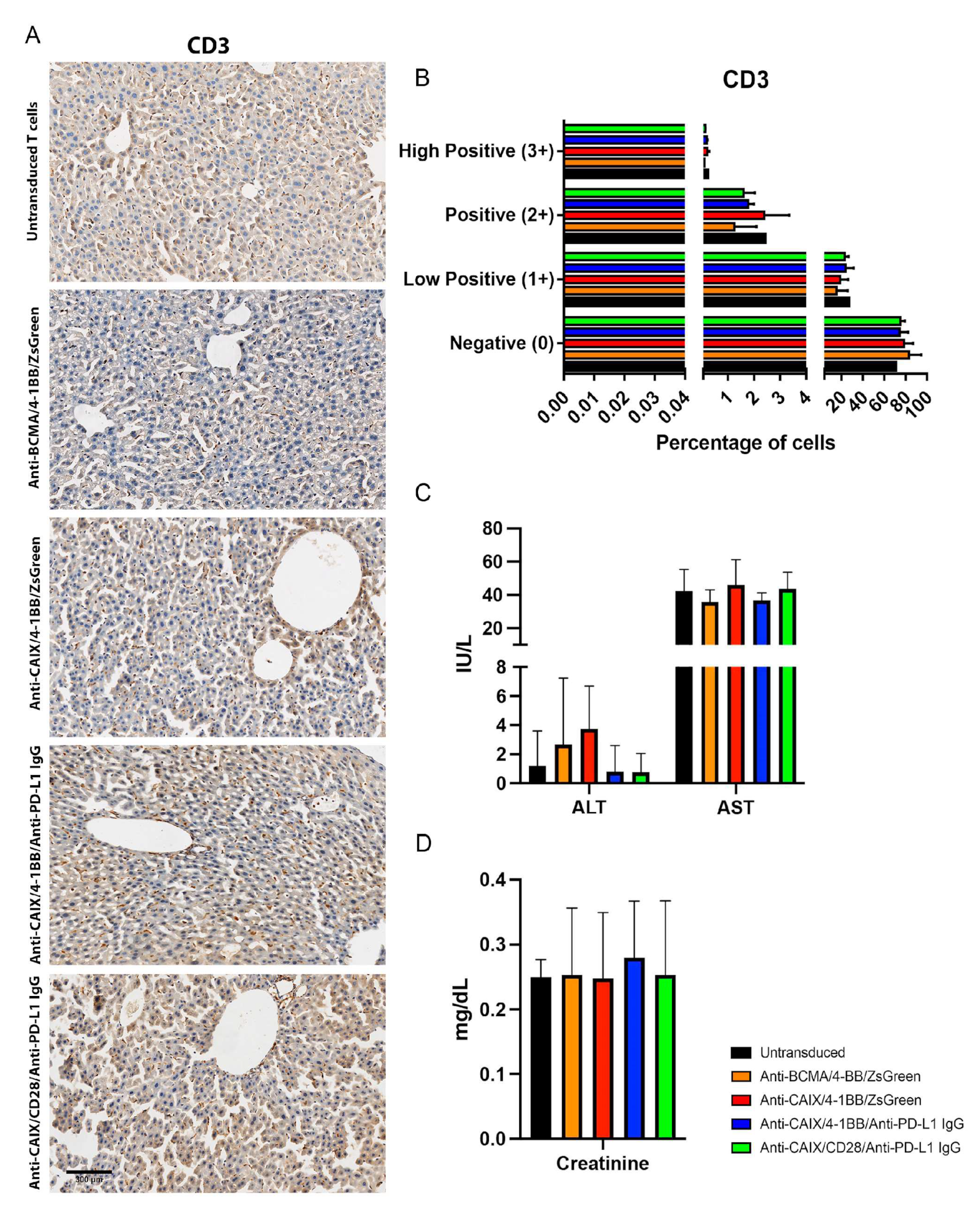
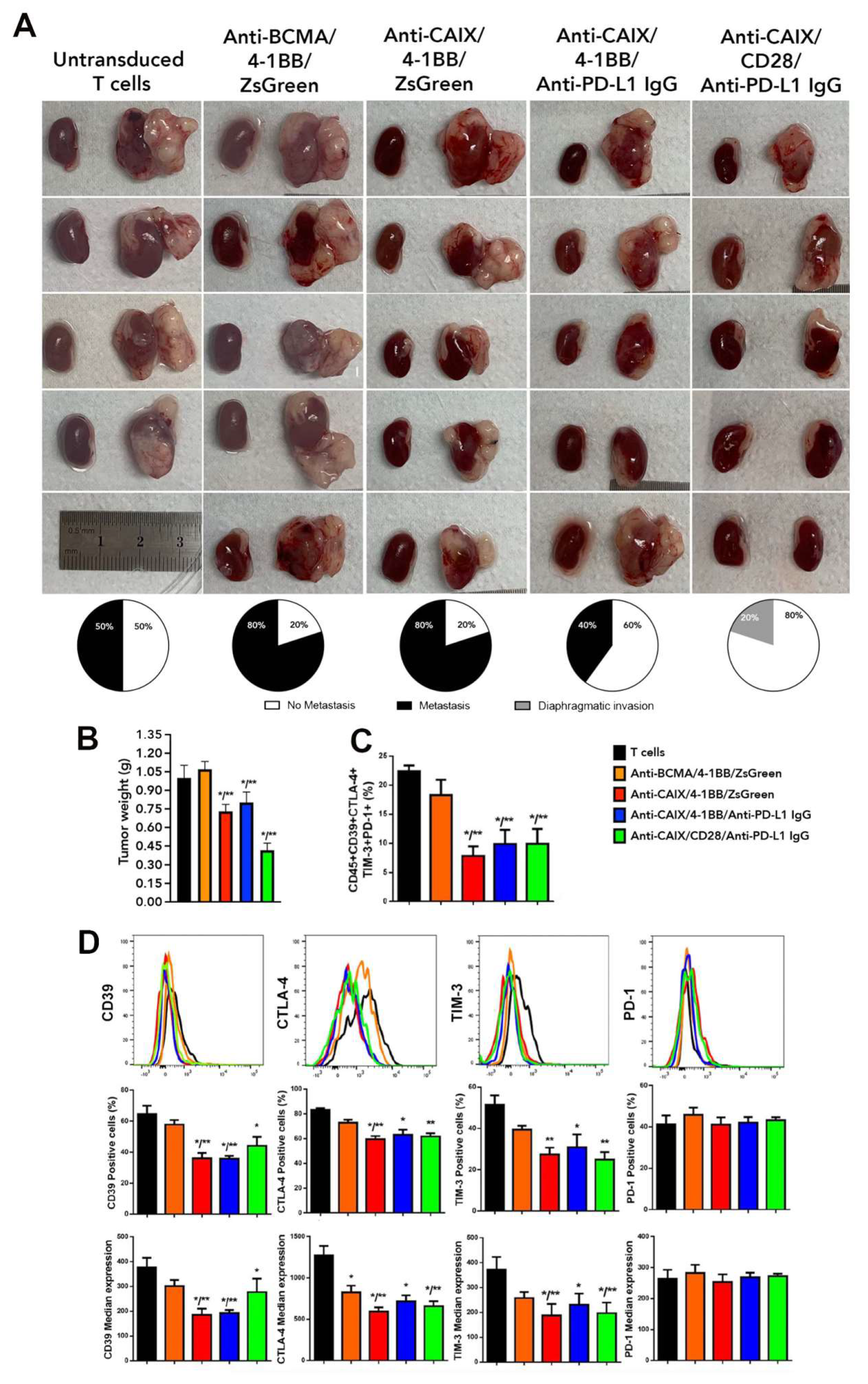
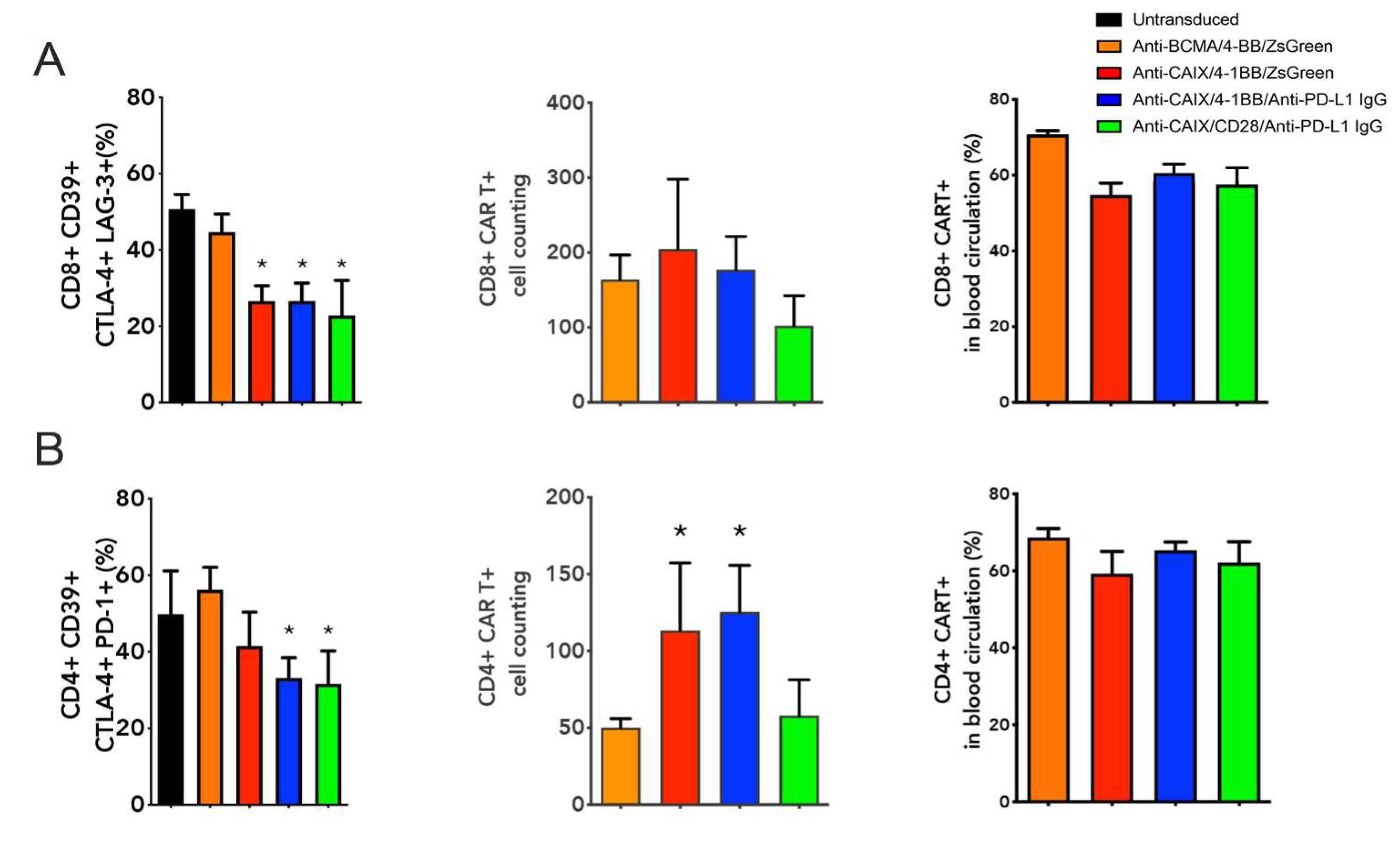
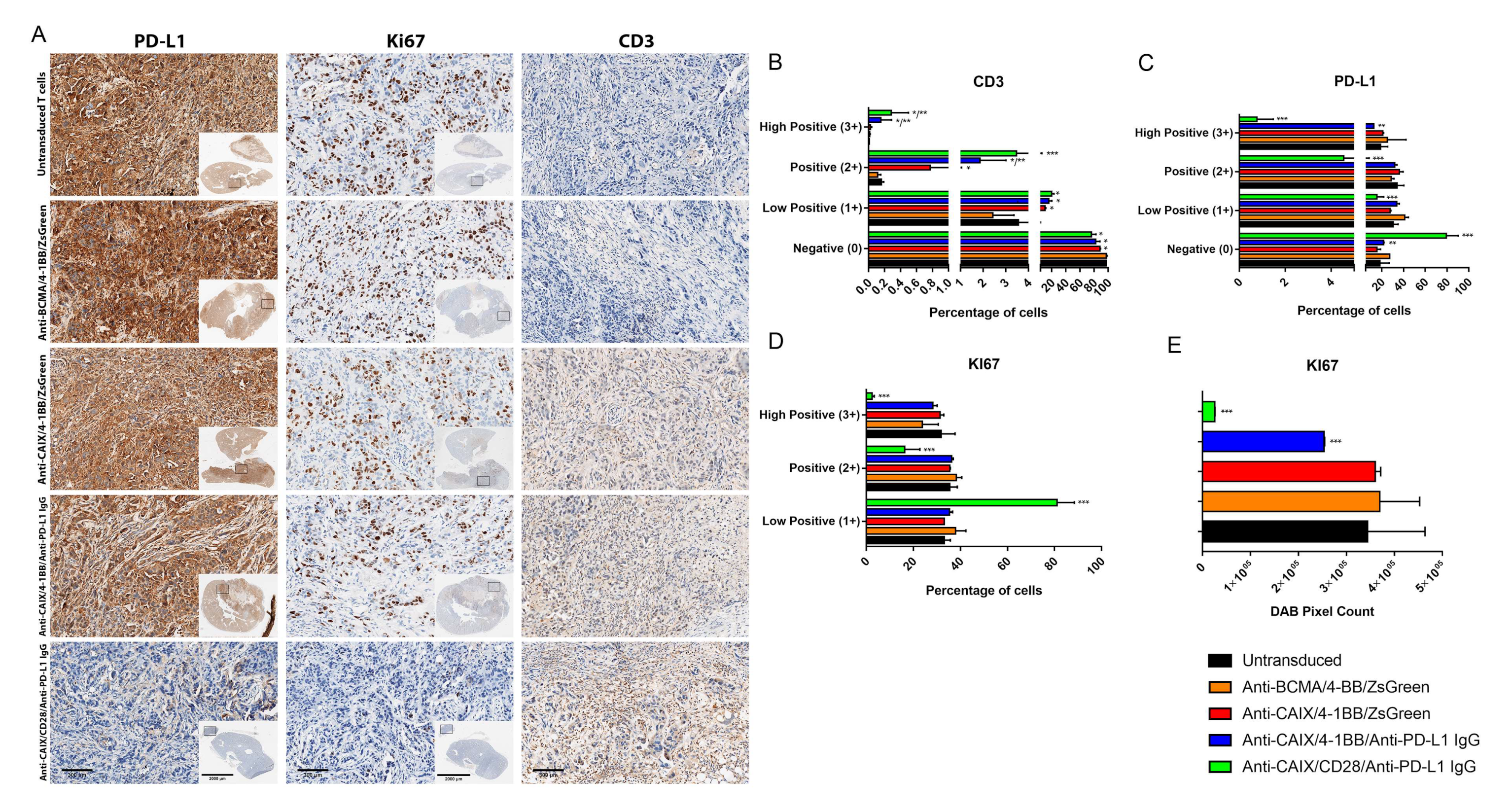
2.3. Comparative Evaluation of Anti-CAIX CAR T Cells CD28ζ versus CD8α 4-1BBζ Releasing Anti-PD-L1 IgG4 Antibodies in an Orthotopic NSG Mice Model of Human ccRCC
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Cloning of a CD8 Alpha Spacer and the 4-1BB Co-Stimulatory Domain into a Bicistronic Lentiviral Vector Encoding an Anti-CAIX CAR
4.3. Lentiviral Production
4.4. Selection, Activation, and Transduction of PBMCs
4.5. T Cell Transduction and Payload Secretion Analysis
4.6. Cytotoxic Effect of Anti-CAIX CAR T Cells Producing Anti-PD-L1 Antibodies on the Renal Carcinoma Cells
4.7. CAR T Cell Exhaustion Status in Co-Culture with ccRCC Cells
4.8. Comparative Evaluation of the Antitumor Efficacy and Exhaustion of Anti-CAIX-CAR T Cells Releasing Anti-PD-L1 Antibodies, Constructions 4-1BB versus CD28, in an Orthotopic Model of Clear Cell Renal Carcinoma
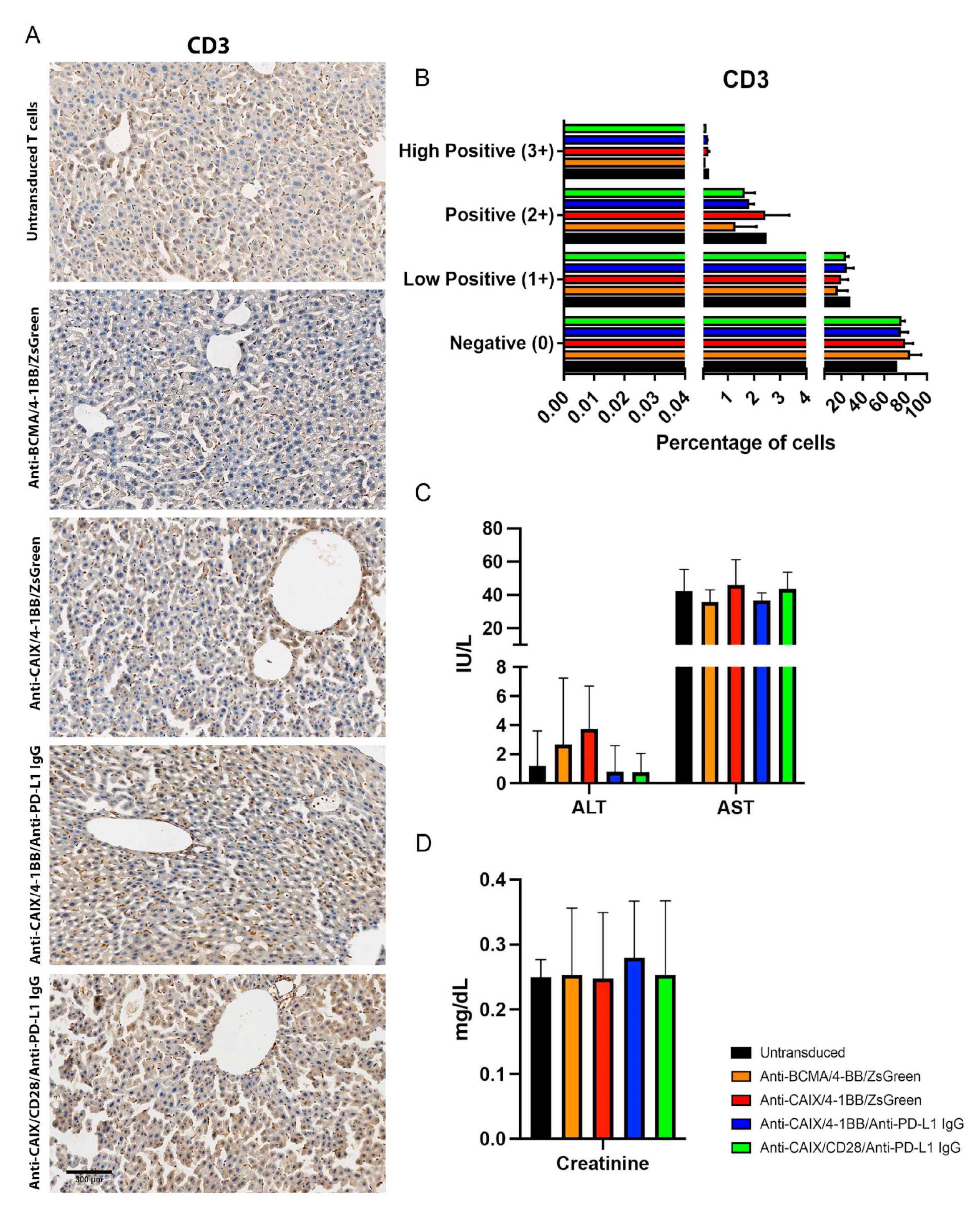
4.8.1. Hepatic and Renal Toxicity
4.8.2. Tumor Infiltrated CAR T Cells Assessment
4.8.3. Immunohistochemistry (IHC)
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gonzalez, N.M.; Zou, D.; Gu, A.; Chen, W. Schrodinger’s T Cells: Molecular Insights into Stemness and Exhaustion. Front. Immunol. 2021, 12, 725618. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Frontera, O.A.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthelemy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Tomczak, P.; Park, S.H.; Venugopal, B.; Ferguson, T.; Chang, Y.H.; Hajek, J.; Symeonides, S.N.; Lee, J.L.; Sarwar, N.; et al. Adjuvant Pembrolizumab after Nephrectomy in Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 385, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Albiges, L.; Tannir, N.M.; Burotto, M.; McDermott, D.; Plimack, E.R.; Barthelemy, P.; Porta, C.; Powles, T.; Donskov, F.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib for first-line treatment of advanced renal cell carcinoma: Extended 4-year follow-up of the phase III CheckMate 214 trial. ESMO Open 2020, 5, e001079. [Google Scholar] [CrossRef]
- Dana, H.; Chalbatani, G.M.; Jalali, S.A.; Mirzaei, H.R.; Grupp, S.A.; Suarez, E.R.; Raposo, C.; Webster, T.J. CAR-T cells: Early successes in blood cancer and challenges in solid tumors. Acta Pharm. Sin. B 2021, 11, 1129–1147. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Abken, H. Building on synthetic immunology and T cell engineering: A brief journey through the history of CARs. Hum. Gene Ther. 2021, 32, 1011–1028. [Google Scholar] [CrossRef]
- Aldera, A.P.; Govender, D. Carbonic anhydrase IX: A regulator of pH and participant in carcinogenesis. J. Clin. Pathol. 2021, 74, 350–354. [Google Scholar] [CrossRef]
- Proescholdt, M.A.; Merrill, M.J.; Stoerr, E.M.; Lohmeier, A.; Pohl, F.; Brawanski, A. Function of carbonic anhydrase IX in glioblastoma multiforme. Neuro-Oncol. 2012, 14, 1357–1366. [Google Scholar] [CrossRef]
- Hussain, S.A.; Ganesan, R.; Reynolds, G.; Gross, L.; Stevens, A.; Pastorek, J.; Murray, P.G.; Perunovic, B.; Anwar, M.S.; Billingham, L.; et al. Hypoxia-regulated carbonic anhydrase IX expression is associated with poor survival in patients with invasive breast cancer. Br. J. Cancer 2007, 96, 104–109. [Google Scholar] [CrossRef]
- McIntyre, A.; Patiar, S.; Wigfield, S.; Li, J.L.; Ledaki, I.; Turley, H.; Leek, R.; Snell, C.; Gatter, K.; Sly, W.S.; et al. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 3100–3111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, J.J.; Chen, D.; Wang, P.I.; Marker, M.; Redzematovic, A.; Chen, Y.B.; Selcuklu, S.D.; Weinhold, N.; Bouvier, N.; Huberman, K.H.; et al. Genomic Biomarkers of a Randomized Trial Comparing First-line Everolimus and Sunitinib in Patients with Metastatic Renal Cell Carcinoma. Eur. Urol. 2017, 71, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tostain, J.; Li, G.; Gentil-Perret, A.; Gigante, M. Carbonic anhydrase 9 in clear cell renal cell carcinoma: A marker for diagnosis, prognosis and treatment. Eur. J. Cancer 2010, 46, 3141–3148. [Google Scholar] [CrossRef] [PubMed]
- Genega, E.M.; Ghebremichael, M.; Najarian, R.; Fu, Y.; Wang, Y.; Argani, P.; Grisanzio, C.; Signoretti, S. Carbonic anhydrase IX expression in renal neoplasms: Correlation with tumor type and grade. Am. J. Clin. Pathol. 2010, 134, 873–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luong-Player, A.; Liu, H.; Wang, H.L.; Lin, F. Immunohistochemical reevaluation of carbonic anhydrase IX (CA IX) expression in tumors and normal tissues. Am. J. Clin. Pathol. 2014, 141, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Lamers, C.H.J.; Klaver, Y.; Gratama, J.W.; Sleijfer, S.; Debets, R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells–a completed study overview. Biochem. Soc. Trans. 2016, 44, 951–959. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef]
- Wang, Y.; Buck, A.; Grimaud, M.; Culhane, A.C.; Kodangattil, S.; Razimbaud, C.; Bonal, D.M.; Nguyen, Q.D.; Zhu, Z.; Wei, K.; et al. Anti-CAIX BBzeta CAR4/8 T cells exhibit superior efficacy in a ccRCC mouse model. Mol. Oncolytics 2022, 24, 385–399. [Google Scholar] [CrossRef]
- de Campos, N.S.P.; Souza, B.S.; da Silva, G.C.P.; Porto, V.A.; Chalbatani, G.M.; Lagreca, G.; Janji, B.; Suarez, E.R. Carbonic Anhydrase IX: A Renewed Target for Cancer Immunotherapy. Cancers 2022, 14, 1392. [Google Scholar] [CrossRef]
- Salfeld, J.G. Isotype selection in antibody engineering. Nat. Biotechnol. 2007, 25, 1369–1372. [Google Scholar] [CrossRef]
- Suarez, E.R.; Chang, D.K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, F.; Bukhari, A.B.; Malhotra, R.; De, A. IHC Profiler: An open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS ONE 2014, 9, e96801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.J.; Jeng, Y.M.; Lai, H.S.; Fong, I.U.; Sheu, F.Y.; Lai, P.L.; Yuan, R.H. Expression of hypoxic marker carbonic anhydrase IX predicts poor prognosis in resectable hepatocellular carcinoma. PLoS ONE 2015, 10, e0119181. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; van Steenbergen, S.; van Elzakker, P.; van Krimpen, B.; Groot, C.; Vulto, A.; den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, L.; Masoumi, E.; Fallah-Mehrjardi, K.; Mirzaei, H.R.; Hadjati, J. Prolonged Persistence of Chimeric Antigen Receptor (CAR) T Cell in Adoptive Cancer Immunotherapy: Challenges and Ways Forward. Front. Immunol. 2020, 11, 702. [Google Scholar] [CrossRef] [Green Version]
- Lo, A.S.; Xu, C.; Murakami, A.; Marasco, W.A. Regression of established renal cell carcinoma in nude mice using lentivirus-transduced human T-cells expressing a human anti-CAIX chimeric antigen receptor. Mol. Ther.-Oncolytics 2014, 1, 14003. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Schmetterer, K.G.; Goldhahn, K.; Ziegler, L.S.; Gerner, M.C.; Schmidt, R.L.J.; Themanns, M.; Zebedin-Brandl, E.; Trapin, D.; Leitner, J.; Pickl, W.F.; et al. Overexpression of PDE4A Acts as Checkpoint Inhibitor Against cAMP-Mediated Immunosuppression in vitro. Front. Immunol. 2019, 10, 1790. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 765. [Google Scholar] [CrossRef]
- Miggelbrink, A.M.; Jackson, J.D.; Lorrey, S.J.; Srinivasan, E.S.; Waibl-Polania, J.; Wilkinson, D.S.; Fecci, P.E. CD4 T-Cell Exhaustion: Does It Exist and What Are Its Roles in Cancer? Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 5742–5752. [Google Scholar] [CrossRef]
- Banta, K.L.; Xu, X.; Chitre, A.S.; Au-Yeung, A.; Takahashi, C.; O’Gorman, W.E.; Wu, T.D.; Mittman, S.; Cubas, R.; Comps-Agrar, L.; et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8(+) T cell responses. Immunity 2022, 55, 512–526.e9. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.H.; Epstein, D.J.; Tamaresis, J.S.; Ehlinger, Z.; Spiegel, J.Y.; Craig, J.; Claire, G.K.; Frank, M.J.; Muffly, L.; Shiraz, P.; et al. Immune reconstitution and infectious complications following axicabtagene ciloleucel therapy for large B-cell lymphoma. Blood Adv. 2021, 5, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Iacoboni, G.; Villacampa, G.; Martinez-Cibrian, N.; Bailén, R.; Lopez Corral, L.; Sanchez, J.M.; Guerreiro, M.; Caballero, A.C.; Mussetti, A.; Sancho, J.M.; et al. Real-world evidence of tisagenlecleucel for the treatment of relapsed or refractory large B-cell lymphoma. Cancer Med. 2021, 10, 3214–3223. [Google Scholar] [CrossRef] [PubMed]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.; Gileadi, T.; et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Sci. Transl. Med. 2020, 12, eabd6169. [Google Scholar] [CrossRef]
- Clinical Trials.gov. CAR T Cell Receptor Immunotherapy Targeting EGFRvIII for Patients with Malignant Gliomas Expressing EGFRvIII. Available online: https://clinicaltrials.gov/show/NCT01454596. (accessed on 8 March 2022).
- Hsu, C.Y.; Uludag, H. A simple and rapid nonviral approach to efficiently transfect primary tissue-derived cells using polyethylenimine. Nat. Protoc. 2012, 7, 935–945. [Google Scholar] [CrossRef]
- Kutner, R.H.; Zhang, X.Y.; Reiser, J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc. 2009, 4, 495–505. [Google Scholar] [CrossRef]
- Mardiana, S.; John, L.B.; Henderson, M.A.; Slaney, C.Y.; von Scheidt, B.; Giuffrida, L.; Davenport, A.J.; Trapani, J.A.; Neeson, P.J.; Loi, S.; et al. A Multifunctional Role for Adjuvant Anti-4-1BB Therapy in Augmenting Antitumor Response by Chimeric Antigen Receptor T Cells. Cancer Res. 2017, 77, 1296–1309. [Google Scholar] [CrossRef] [Green Version]
- The Jackson Laboratory. NOD.CG-PRKDCSCID IL2RGTM1WJL/SZJ Body Weight Chart. Available online: https://www.jax.org/jax-mice-and-services/strain-data-sheet-pages/body-weight-chart-005557 (accessed on 8 March 2022).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Campos, N.S.P.; de Oliveira Beserra, A.; Pereira, P.H.B.; Chaves, A.S.; Fonseca, F.L.A.; da Silva Medina, T.; dos Santos, T.G.; Wang, Y.; Marasco, W.A.; Suarez, E.R. Immune Checkpoint Blockade via PD-L1 Potentiates More CD28-Based than 4-1BB-Based Anti-Carbonic Anhydrase IX Chimeric Antigen Receptor T Cells. Int. J. Mol. Sci. 2022, 23, 5448. https://doi.org/10.3390/ijms23105448
de Campos NSP, de Oliveira Beserra A, Pereira PHB, Chaves AS, Fonseca FLA, da Silva Medina T, dos Santos TG, Wang Y, Marasco WA, Suarez ER. Immune Checkpoint Blockade via PD-L1 Potentiates More CD28-Based than 4-1BB-Based Anti-Carbonic Anhydrase IX Chimeric Antigen Receptor T Cells. International Journal of Molecular Sciences. 2022; 23(10):5448. https://doi.org/10.3390/ijms23105448
Chicago/Turabian Stylede Campos, Najla Santos Pacheco, Adriano de Oliveira Beserra, Pedro Henrique Barbosa Pereira, Alexandre Silva Chaves, Fernando Luiz Affonso Fonseca, Tiago da Silva Medina, Tiago Goss dos Santos, Yufei Wang, Wayne Anthony Marasco, and Eloah Rabello Suarez. 2022. "Immune Checkpoint Blockade via PD-L1 Potentiates More CD28-Based than 4-1BB-Based Anti-Carbonic Anhydrase IX Chimeric Antigen Receptor T Cells" International Journal of Molecular Sciences 23, no. 10: 5448. https://doi.org/10.3390/ijms23105448
APA Stylede Campos, N. S. P., de Oliveira Beserra, A., Pereira, P. H. B., Chaves, A. S., Fonseca, F. L. A., da Silva Medina, T., dos Santos, T. G., Wang, Y., Marasco, W. A., & Suarez, E. R. (2022). Immune Checkpoint Blockade via PD-L1 Potentiates More CD28-Based than 4-1BB-Based Anti-Carbonic Anhydrase IX Chimeric Antigen Receptor T Cells. International Journal of Molecular Sciences, 23(10), 5448. https://doi.org/10.3390/ijms23105448