
Transgenic Mouse Models of Alzheimer’s Disease: An Integrative Analysis


 , , , ,
, , , ,  ,
,  ,
,
Abstract
1. Introduction
2. Amyloid Pathology: A Long History from an Old Friend
2.1. APP-Based Models, the Beginning

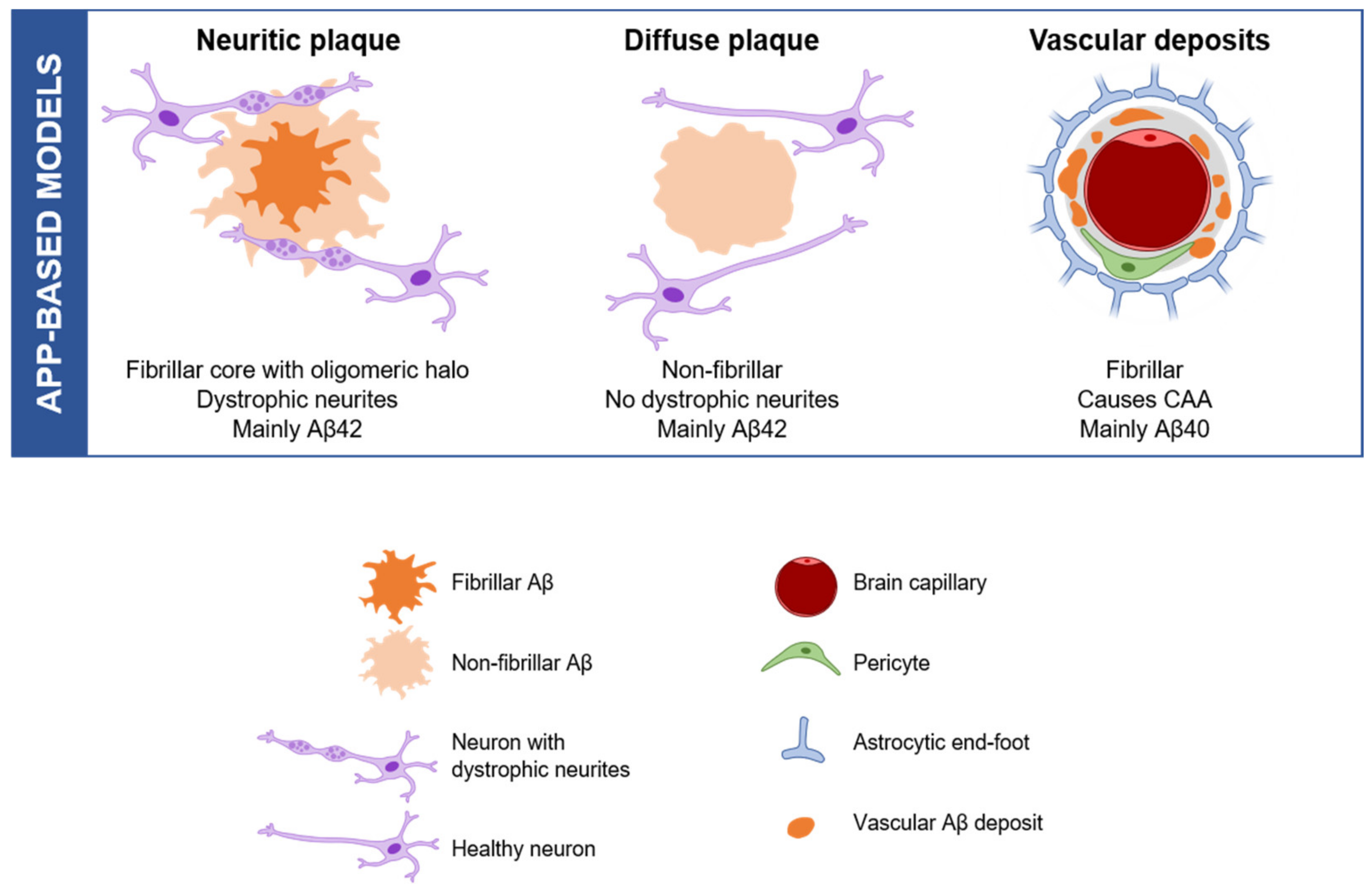{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| APP-Based Monogenic Models | Mutations | Neuropathology | Onset Cognitive Impairment | References |
|---|---|---|---|---|
| PDAPP (line 109) | APP Indiana (V717F) | Aβ deposition (neuritic and diffuse) and gliosis by 6–9 months; synaptic loss | 3 months | [3] |
| Tg2576 | APP695 Swedish (KM670/671NL) | Aβ deposition (neuritic and diffuse) and gliosis by 11–13 months; synaptic loss | 6–12 months | [21] |
| APP23 | APP751 Swedish | Aβ deposition (neuritic and diffuse) and gliosis by 6 months; neuronal and synaptic loss | 3 months | [22] |
| APP R1.40 | APP Swedish | Aβ deposition (neuritic and diffuse) and gliosis by 14 months | Unknown | [23] |
| J20 | APP Swedish, Indiana | Aβ deposition (neuritic and diffuse) and gliosis by 5–7 months; neuronal and synaptic loss | 4 months | [24] |
| APP Dutch | APP751 Dutch E693Q | CAA from 22 to 24 months | Unknown | [25] |
| TgCRND8 | APP695 Swedish, Indiana | Aβ deposition (neuritic and diffuse) and gliosis by 3 months; neuronal and synaptic loss | 3 months | [26] |
| ArcAβ | APP695 Swedish, Arctic (E693G) | Aβ deposition and CAA from 9 months | 6 months | [27] |
| Arc48 | APP Swedish, Indiana, Artic | Aβ deposition (neuritic) and gliosis by 2–3 months | 3–4 months | [28] |
| Tg-SwDI | APP770 Swedish, Dutch, Iowa (D694N) | CAA from 3 months and gliosis by 6 months | 3 months | [29] |
| App NL-F KI | Humanized App Swedish, Iberian I716F | Aβ deposition (neuritic) and gliosis by 6 months; synaptic loss | 18 months | [30] |
| App NL-G-F KI | Humanized App Swedish, Iberian, Arctic | Aβ deposition (neuritic) and gliosis by 2 months | 6 months | [30] |
| hAβ-KI | Humanized Aβ sequence | Insoluble Aβ increase (without extracellular deposition) and cytokine alterations; synaptic loss | 10–14 months | [31] |
| PS/APP | APP Swedish; PSEN1 M146L (A > C) | Aβ deposition (neuritic and diffuse) and gliosis by 6 months; neuronal loss | 3–6 months | [32] |
| APP751SL/PS1M146L | APP751 Swedish, London (V717I); PSEN1 M146L | Aβ deposition (neuritic and diffuse) and gliosis by 3 moths; neuronal and synaptic loss | 6 months | [19] |
| APP751SL/PS1-KI | APP751 Swedish, London; PSEN1 M233T, PSEN1 L235P | Aβ deposition and gliosis by 2.5 months; neuronal and synaptic loss | 6 months | [33] |
| APPswe/PS1dE9 (line 85) | APP Swedish; PSEN1 deltaE9 | Aβ deposition (neuritic) and gliosis by 6–9 months; neuronal and synaptic loss | 12 months | [34] |
| APP/PS1 | APP Swedish; PSEN1 L166P | Aβ deposition and gliosis by 1.5 months; synaptic loss | 7 months | [35] |
| 5xFAD (B6SJL) | APP Swedish, Florida (I716V), London; PSEN1 M146L (A > C), PSEN1 L286V | Aβ deposition (neuritic and diffuse) by 2 months; neuronal and synaptic loss | 4–5 months | [20] |
| PS2/APP | APP751 Swedish; PSEN2 Volga German N141I | Aβ deposition (neuritic and diffuse) and gliosis by 6 months | 8 months | [36] |
2.2. Amyloid Aggregation and Deposition: Modelling a Subset of Plaques
2.3. Aggregation and Prion-like Spreading Mechanisms
2.4. Knock-In Mice to Knock-Out the Limitations
3. Transgenic Models of Tauopathies to Unravel Alzheimer’s Disease
3.1. Tau Aggregation and Tauopathies
3.2. We Do What We Can: Generating Tau Pathology Using Tauopathy Models
| Model | Mutations | Neuropathology | Onset Cognitive Impairment | References |
|---|---|---|---|---|
| JNPL3 | P301L mutation (4R/0N) under mouse PrP promoter | NFTs and body inclusions by 4.5 months | 10 months | [60] |
| htau | Wild-type MAPT under tau promoter | NFTs by 9 months and neuronal loss | 12 months | [66] |
| rTg4510 | P301L mutation suppressed by doxycycline, under human CaMKIIα promoter | NFTs by 4 months, neuronal loss and brain atrophy | 3–5 months | [64,72] |
| Thy-Tau22 | Human (4R/1N) tau under Thy1.2-promotor | NFTs and astrogliosis by 3 months | 10 months | [65] |
| PS19 line | P301S mutation (4R/1N), under mouse PrP promoter | NFTs by 8 months, gliosis and synaptic dysfunction | 3–6 months | [62] |
| rTgTauEC | P301L mutation in the entorhinal cortex, under neuropsin promoter | NFTs, synaptic degeneration and neuron loss by 3 months | Unknown | [73] |
| APPswe-Tau | Tg2576 and Tau P301L mutation, under mouse PrP promoter | NFTs and neuronal loss by 6 months | Unknown | [61] |
| App/Mapt dKI | Humanized App (Swedish, Arctic and Iberian) mutations, and humanized Mapt gene under CAM kinase II promoter | ptau, Aβ pathology, dystrophic neurites and neuroinflammation by 6 months | Unknown | [71] |
| 3xTg-AD | PS1 (M146V), APP (swe) and MAPT (P301L) mutations under mouse Thy1.2 promoter (APP, MAPT) and endogenous PSEN1 promoter | Aβ plaques at 9 months, NFTs at 12 months | 3–6 months | [67] |
3.3. Seeding, Spreading and Progression of Tau Pathology: Can We Model It?
4. Modeling Synaptic Dysfunction and Neurodegeneration
4.1. Aβ Role on Synaptic Impairment
4.2. Synaptic Pathology in Tau Mice
4.3. Neurodegeneration in AD Mouse Models
4.3.1. Cholinergic Degeneration
4.3.2. GABAergic Neurodegeneration
4.3.3. Pyramidal Neurodegeneration: The Achilles’ Heel of FAD Models
4.4. Modeling Amyloid-Tau Synergy in Neurodegeneration
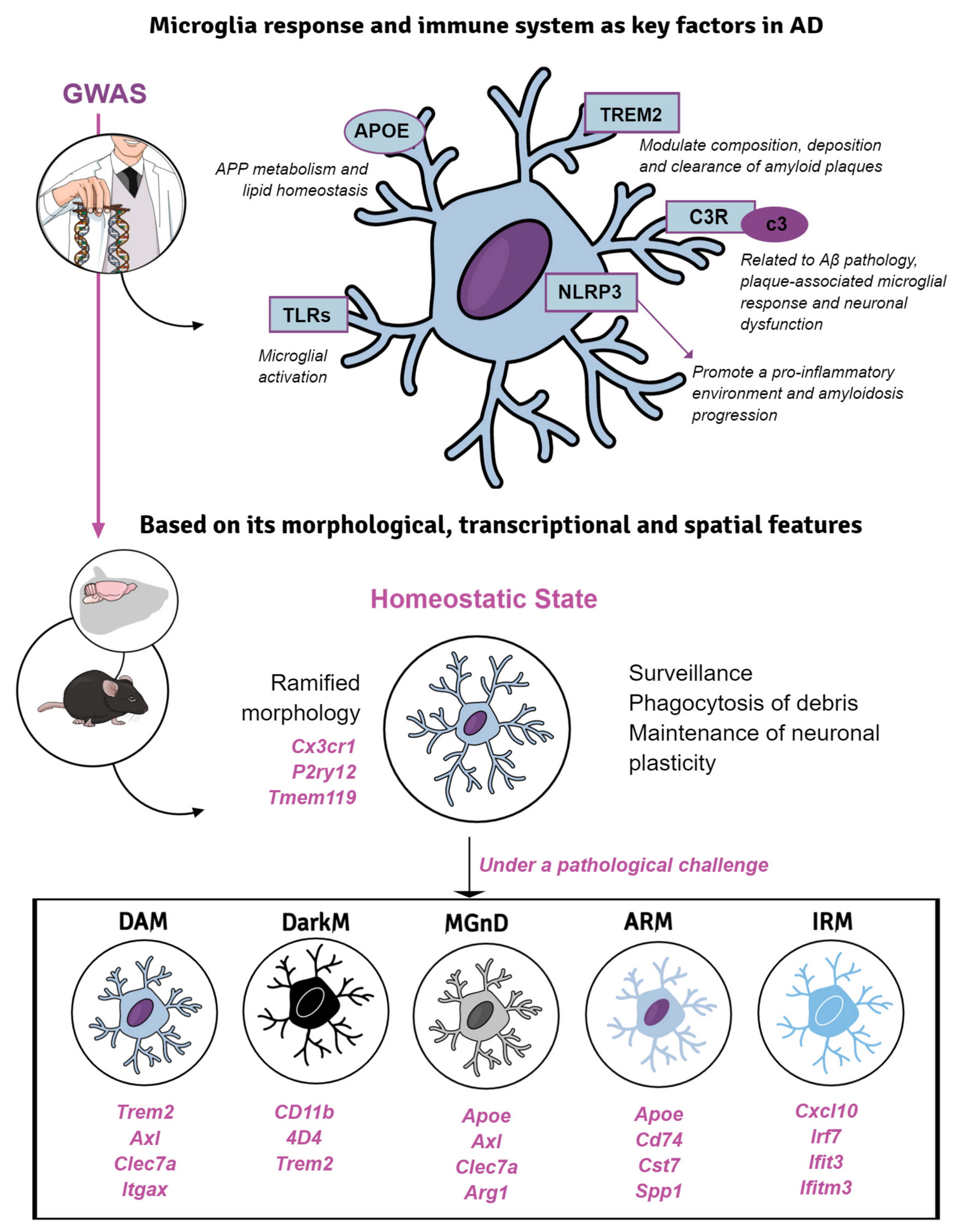
5. Microglial Response in Transgenic Mice of Alzheimer’s Disease
5.1. Microglial Phenotypes in Classic Models of Amyloidosis and Tauopathy
5.2. Introducing AD-Risk Factors for Modeling Microglial Response in sAD
5.2.1. TREM2
5.2.2. APOE, More Than a Lipoprotein
5.2.3. Toll-like Receptors, Complement Factors and Inflammasome
5.3. Targeting Microglial Cells in Animal Models
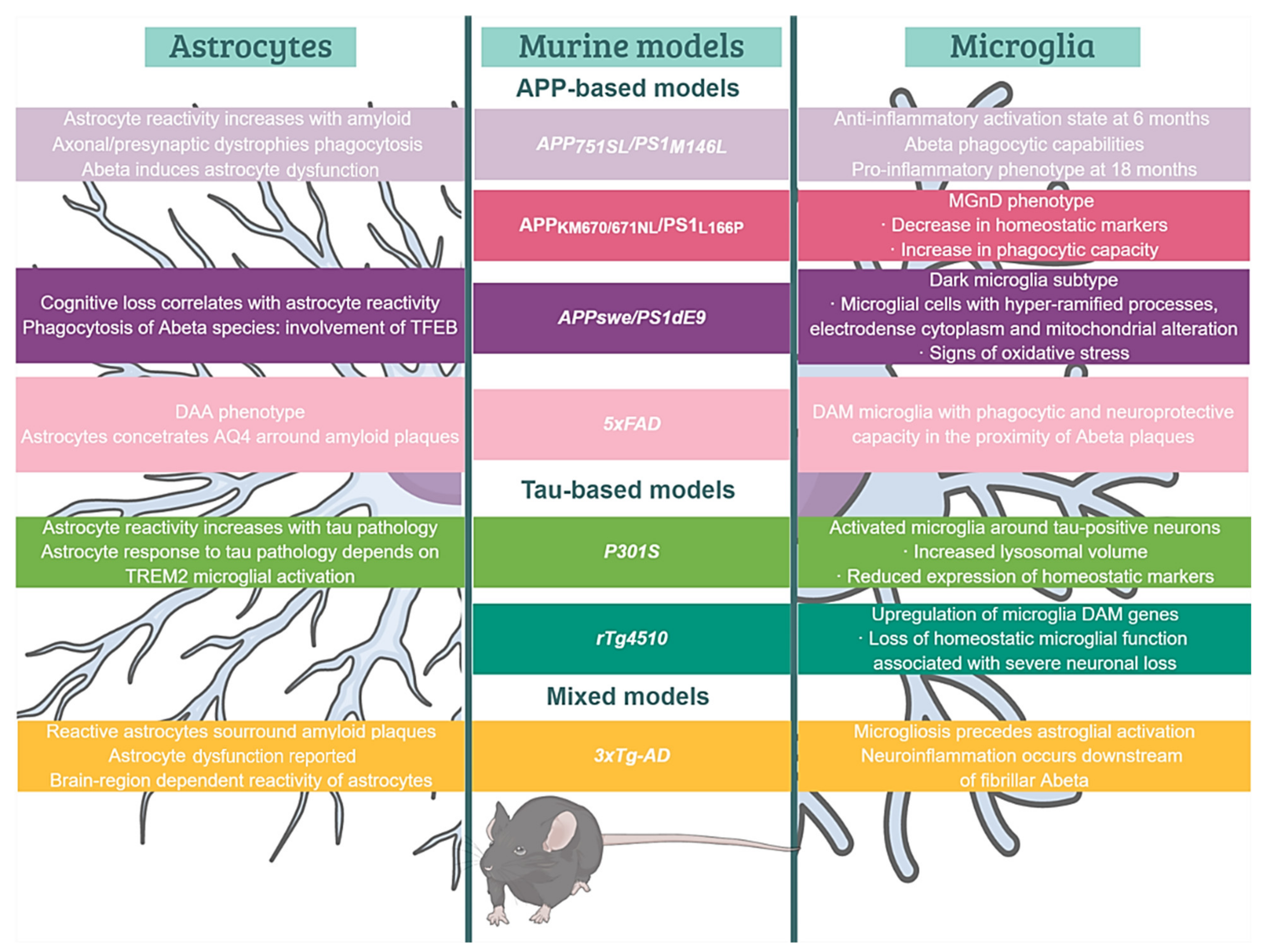
6. Astrocyte Reactivity in AD Brains and Models: Both Sides of the Story
6.1. Reactive Astrocytes in AD Brains and Models
6.2. Elimination of Amyloid-β and Ptau by Reactive Astrocytes: The Role of ApoE
6.3. Clearance through the Brain Blood Barrier
6.4. Astroglial Dysfunction and Senescence: The Dark Side of Astrocyte Reactivity
6.5. Astrocyte and Microglia Crosstalk in Mouse Models
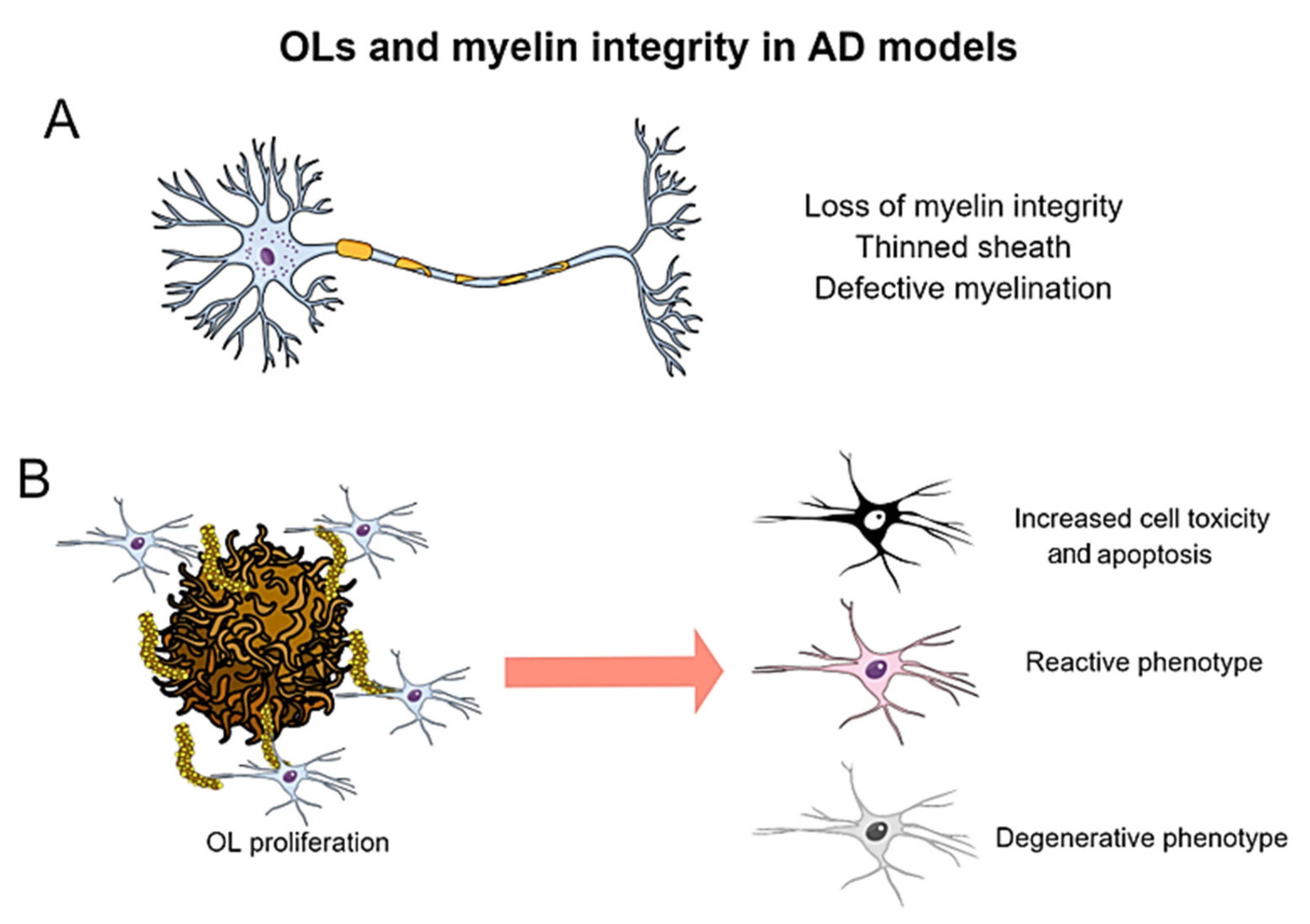
7. Oligodendrocytic Alterations in Transgenic Models of Alzheimer’s Disease
7.1. Myelin Pathology in Amyloidogenic and Tauopathy Models
7.2. Alterations in Oligodendroglial Proliferation and Behavior in Response to Aβ and Tau Pathologies
8. Transcriptomic and Proteomic Profiling to Assess Disease-Relevance of Mouse Models
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Association. Available online: https://www.alz.org/alzheimer_s_dementia (accessed on 8 April 2022).
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2021. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12179. [Google Scholar] [CrossRef] [PubMed]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Borthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; Van Eldik, L.J. Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014, 5, 88. [Google Scholar] [CrossRef]
- Martini, A.C.; Forner, S.; Trujillo-Estrada, L.; Baglietto-Vargas, D.; Laferla, F.M. Past to future: What animal models have taught us about Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 64, S365–S378. [Google Scholar] [CrossRef]
- Götz, J.; Bodea, L.-G.; Goedert, M. Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 583–598. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Andrews, S.J.; Fulton-Howard, B.; Goate, A. Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol. 2020, 19, 326–335. [Google Scholar] [CrossRef]
- De Rojas, I.; Moreno-Grau, S.; Tesi, N.; Grenier-Boley, B.; Andrade, V.; Jansen, I.E.; Pedersen, N.L.; Stringa, N.; Zettergren, A.; Hernández, I.; et al. Common variants in Alzheimer’s disease and risk stratification by polygenic risk scores. Nat. Commun. 2021, 12, 3417. [Google Scholar] [CrossRef]
- Trujillo-Estrada, L.; Sanchez-Mejias, E.; Sanchez-Varo, R.; Garcia-Leon, J.A.; Nuñez-Diaz, C.; Davila, J.C.; Vitorica, J.; LaFerla, F.M.; Moreno-Gonzalez, I.; Gutierrez, A.; et al. Animal and cellular models of Alzheimer’s disease: Progress, promise, and future approaches. Neuroscientis 2021, 10738584211001753. [Google Scholar] [CrossRef]
- Model AD. Available online: https://www.model-ad.org/ (accessed on 8 April 2022).
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Wolfe, M.S. Structure and function of the γ-secretase complex. Biochemistry 2019, 58, 2953–2966. [Google Scholar] [CrossRef]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Younkin, L.H.; Gonzales, V.; Fadale, D.J.; Slunt, H.H.; Lester, H.A.; Younkin, S.G.; Borchelt, D.R. Rodent a beta modulates the solubility and distribution of amyloid deposits in transgenic mice. J. Biol. Chem. 2007, 282, 22707–22720. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Slunt, H.H.; Ratovitski, T.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. Co-expression of multiple transgenes in mouse CNS: A comparison of strategies. Biomol. Eng. 2001, 17, 157–165. [Google Scholar] [CrossRef]
- Blanchard, V.; Moussaoui, S.; Czech, C.; Touchet, N.; Bonici, B.; Planche, M.; Canton, T.; Jedidi, I.; Gohin, M.; Wirths, O.; et al. Time sequence of maturation of dystrophic neurites associated with Aβ Deposits in APP/PS1 transgenic mice. Exp. Neurol. 2003, 184, 247–263. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van, E.L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef]
- Lamb, B.T.; Call, L.M.; Slunt, H.H.; Bardel, K.A.; Lawler, A.M.; Eckman, C.B.; Younkin, S.G.; Holtz, G.; Wagner, S.L.; Price, D.L.; et al. Altered metabolism of familial Alzheimer’s disease linked amyloid precursor protein variants in yeast artificial chromosome transgenic mice. Hum. Mol. Genet. 1997, 6, 1535–1541. [Google Scholar] [CrossRef][Green Version]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef]
- Herzig, M.C.; Winkler, D.T.; Burgermeister, P.; Pfeifer, M.; Kohler, E.; Schmidt, S.D.; Danner, S.; Abramowski, D.; Stürchler-Pierrat, C.; Bürki, K.; et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 2004, 7, 954–960. [Google Scholar] [CrossRef]
- Chishti, M.A.; Yang, D.S.; Janus, C.; Phinney, A.L.; Horne, P.; Pearson, J.; Strome, R.; Zuker, N.; Loukides, J.; French, J.; et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 2001, 276, 21562–21570. [Google Scholar] [CrossRef]
- Knobloch, M.; Konietzko, U.; Krebs, D.C.; Nitsch, R.M. Intracellular Aβ and cognitive deficits precede β-amyloid deposition in transgenic ArcAβ Mice. Neurobiol. Aging 2007, 28, 1297–1306. [Google Scholar] [CrossRef]
- Cheng, I.H.; Scearce-Levie, K.; Legleiter, J.; Palop, J.J.; Gerstein, H.; Bien-Ly, N.; Puoliväli, J.; Lesné, S.; Ashe, K.H.; Muchowski, P.J.; et al. Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J. Biol. Chem. 2007, 282, 23818–23828. [Google Scholar] [CrossRef]
- Davis, J.; Xu, F.; Deane, R.; Romanov, G.; Previti, M.L.; Zeigler, K.; Zlokovic, B.V.; Van Nostrand, W.E. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J. Biol. Chem. 2004, 279, 20296–20306. [Google Scholar] [CrossRef]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single app knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef]
- Baglietto-Vargas, D.; Forner, S.; Cai, L.; Martini, A.C.; Trujillo-Estrada, L.; Swarup, V.; Nguyen, M.M.T.; Do Huynh, K.; Javonillo, D.I.; Tran, K.M.; et al. Generation of a humanized aβ expressing mouse demonstrating aspects of Alzheimer’s disease-like pathology. Nat. Commun. 2021, 12, 2421. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, L.; Gordon, M.N.; Mcgowan, E.; Yu, X.; Benkovic, S.; Jantzen, P.; Wright, K.; Saad, I.; Mueller, R.; Morgan, D.; et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 1998, 4, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Casas, C.; Sergeant, N.; Itier, J.M.; Blanchard, V.; Wirths, O.; Van Der Kolk, N.; Vingtdeux, V.; Van De Steeg, E.; Ret, G.; Canton, T.; et al. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Aβ42 accumulation in a novel Alzheimer transgenic model. Am. J. Pathol. 2004, 165, 1289–1300. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: Evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Ozmen, L.; Albientz, A.; Czech, C.; Jacobsen, H. Expression of transgenic APP MRNA is the key determinant for beta-amyloid deposition in PS2APP transgenic mice. Neurodegener. Dis. 2008, 6, 29–36. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The amyloid-β oligomer hypothesis: Beginning of the third decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef]
- Dear, A.J.; Michaels, T.C.T.; Meisl, G.; Klenerman, D.; Wu, S.; Perrett, S.; Linse, S.; Dobson, C.M.; Knowles, T.P.J. Kinetic diversity of amyloid oligomers. Proc. Natl. Acad. Sci. USA 2020, 117, 12087–12094. [Google Scholar] [CrossRef]
- Boon, B.D.C.; Bulk, M.; Jonker, A.J.; Morrema, T.H.J.; van den Berg, E.; Popovic, M.; Walter, J.; Kumar, S.; van der Lee, S.J.; Holstege, H.; et al. The coarse-grained plaque: A divergent Aβ Plaque-type in early-onset Alzheimer’s disease. Acta Neuropathol. 2020, 140, 811–830. [Google Scholar] [CrossRef]
- Edwards, F.A. A unifying hypothesis for Alzheimer’s disease: From plaques to neurodegeneration. Trends Neurosci. 2019, 42, 310–322. [Google Scholar] [CrossRef]
- Calderon-Garcidueñas, A.L.; Duyckaerts, C. Alzheimer disease. Handb. Clin. Neurol. 2017, 145, 325–337. [Google Scholar] [CrossRef]
- Crook, R.; Verkkoniemi, A.; Perez-Tur, J.; Mehta, N.; Baker, M.; Houlden, H.; Farrer, M.; Hutton, M.; Lincoln, S.; Hardy, J.; et al. A Variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat. Med. 1998, 4, 452–455. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1341–1349. [Google Scholar] [CrossRef]
- Gomez-Gutierrez, R.; Morales, R. The prion-like phenomenon in Alzheimer’s disease: Evidence of pathology transmission in humans. PLoS Pathog. 2020, 16, e1009004. [Google Scholar] [CrossRef]
- Kane, M.D.; Lipinski, W.J.; Callahan, M.J.; Bian, F.; Durham, R.A.; Schwarz, R.D.; Roher, A.E.; Walker, L.C. Evidence for seeding of β-amyloid by intracerebral infusion of Alzheimer brain extracts in β-amyloid precursor protein-transgenic mice. J. Neurosci. 2000, 20, 3606–3611. [Google Scholar] [CrossRef]
- Morales, R.; Duran-Aniotz, C.; Castilla, J.; Estrada, L.D.; Soto, C. De novo induction of amyloid-β deposition in vivo. Mol. Psychiatry 2012, 17, 1347–1353. [Google Scholar] [CrossRef]
- Watts, J.C.; Condello, C.; Stöhr, J.; Oehler, A.; Lee, J.; DeArmond, S.J.; Lannfelt, L.; Ingelsson, M.; Giles, K.; Prusiner, S.B. Serial propagation of distinct strains of aβ prions from Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10323–10328. [Google Scholar] [CrossRef]
- Ye, L.; Hamaguchi, T.; Fritschi, S.K.; Eisele, Y.S.; Obermüller, U.; Jucker, M.; Walker, L.C. Progression of seed-induced Aβ deposition within the limbic connectome. Brain Pathol. 2015, 25, 743–752. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks crossseed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- D’Errico, P.; Ziegler-Waldkirch, S.; Aires, V.; Hoffmann, P.; Mezö, C.; Erny, D.; Monasor, L.S.; Liebscher, S.; Ravi, V.M.; Joseph, K.; et al. Microglia contribute to the propagation of Aβ into unaffected brain tissue. Nat. Neurosci. 2022, 25, 20–25. [Google Scholar] [CrossRef]
- Huang, Y.; Happonen, K.E.; Burrola, P.G.; O’Connor, C.; Hah, N.; Huang, L.; Nimmerjahn, A.; Lemke, G. Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat. Immunol. 2021, 22, 586–594. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2016, 133, 155–175. [Google Scholar] [CrossRef]
- Oblak, A.L.; Forner, S.; Territo, P.R.; Sasner, M.; Carter, G.W.; Howell, G.R.; Sukoff-Rizzo, S.J.; Logsdon, B.A.; Mangravite, L.M.; Mortazavi, A.; et al. Model organism development and evaluation for late-onset Alzheimer’s disease: MODEL-AD. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, e12110. [Google Scholar] [CrossRef]
- Saito, T.; Saido, T.C. Neuroinflammation in mouse models of Alzheimer’s disease. Clin. Exp. Neuroimmunol. 2018, 9, 211–218. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of Tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 22–35. [Google Scholar] [CrossRef]
- Lee, V.M.Y.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Kovacs, G.G. Tauopathies. Handb. Clin. Neurol. 2018, 145, 355–368. [Google Scholar] [CrossRef]
- Johnson, K.A.; Schultz, A.; Betensky, R.A.; Becker, J.A.; Sepulcre, J.; Rentz, D.; Mormino, E.; Chhatwal, J.; Amariglio, R.; Papp, K.; et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann. Neurol. 2016, 79, 110–119. [Google Scholar] [CrossRef]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Paul Murphy, M.; Baker, M.; Yu, X.; et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) Tau protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant Tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.; McGonigle, P. Overview of Transgenic Mouse Models for Alzheimer’s Disease. Curr. Protoc. Neurosci. 2019, 89, e81. [Google Scholar] [CrossRef] [PubMed]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McCowan, E.; et al. Medicine: Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Schindowski, K.; Bretteville, A.; Leroy, K.; Bégard, S.; Brion, J.-P.; Hamdane, M.; Buée, L. Alzheimer’s disease-like Tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am. J. Pathol. 2006, 169, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Andorfer, C.; Kress, Y.; Espinoza, M.; De Silva, R.; Tucker, K.L.; Barde, Y.A.; Duff, K.; Davies, P. Hyperphosphorylation and aggregation of Tau in mice expressing normal human Tau isoforms. J. Neurochem. 2003, 86, 582–590. [Google Scholar] [CrossRef]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Aβ causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging. 2003, 24, 1063–1070. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Remolina Serrano, J.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 Deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.G.; Sauerbeck, A.D.; St-Pierre, M.-K.; Xiong, M.; Kim, N.; Remolina Serrano, J.; Tremblay, M.-È.; Kummer, T.T.; Colonna, M.; et al. Impact of TREM2R47H variant on Tau pathology-induced gliosis and neurodegeneration. J. Clin. Investig. 2020, 130, 4954–4968. [Google Scholar] [CrossRef]
- Saito, T.; Mihira, N.; Matsuba, Y.; Sasaguri, H.; Hashimoto, S.; Narasimhan, S.; Zhang, B.; Murayama, S.; Higuchi, M.; Lee, V.M.Y.; et al. Humanization of the entire murine mapt gene provides a murine model of pathological human tau propagation. J. Biol. Chem. 2019, 294, 12754–12765. [Google Scholar] [CrossRef]
- Ramsden, M.; Kotilinek, L.; Forster, C.; Paulson, J.; McGowan, E.; SantaCruz, K.; Guimaraes, A.; Yue, M.; Lewis, J.; Carlson, G.; et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J. Neurosci. 2005, 25, 10637–10647. [Google Scholar] [CrossRef]
- De Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Jouanne, M.; Rault, S.; Voisin-Chiret, A.-S. Tau protein aggregation in Alzheimer’s disease: An attractive target for the development of novel therapeutic agents. Eur. J. Med. Chem. 2017, 139, 153–167. [Google Scholar] [CrossRef]
- Vogels, T.; Leuzy, A.; Cicognola, C.; Ashton, N.J.; Smolek, T.; Novak, M.; Blennow, K.; Zetterberg, H.; Hromadka, T.; Zilka, N.; et al. Propagation of tau pathology: Integrating insights from postmortem and in vivo studies. Biol. Psychiatry 2020, 87, 808–818. [Google Scholar] [CrossRef]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef]
- McKee, A.C.; Stein, T.D.; Nowinski, C.J.; Stern, R.A.; Daneshvar, D.H.; Alvarez, V.E.; Lee, H.S.; Hall, G.; Wojtowicz, S.M.; Baugh, C.M.; et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013, 136, 43–64. [Google Scholar] [CrossRef]
- Edwards, G.; Moreno-Gonzalez, I.; Soto, C. Amyloid-beta and tau pathology following repetitive mild traumatic brain injury. Biochem. Biophys. Res. Commun. 2017, 483, 1137–1142. [Google Scholar] [CrossRef]
- Moreno-Gonzalez, I.; Soto, C. Misfolded protein aggregates: Mechanisms, structures and potential for disease transmission. Semin. Cell Dev. Biol. 2011, 22, 482–487. [Google Scholar] [CrossRef]
- Duran-Aniotz, C.; Morales, R.; Moreno-Gonzalez, I.; Hu, P.P.; Soto, C. Brains from non-Alzheimer’s individuals containing amyloid deposits accelerate Aβ deposition in vivo. Acta Neuropathol. Commun. 2014, 2, 76. [Google Scholar] [CrossRef]
- Duran-Aniotz, C.; Morales, R.; Moreno-Gonzalez, I.; Hu, P.P.; Fedynyshyn, J.; Soto, C. Aggregate-depleted brain fails to induce aβ deposition in a mouse model of Alzheimer’s disease. PLoS ONE 2014, 9, e89014. [Google Scholar] [CrossRef]
- Duran-Aniotz, C.; Moreno-Gonzalez, I.; Gamez, N.; Perez-Urrutia, N.; Vegas-Gomez, L.; Soto, C.; Morales, R. Amyloid pathology arrangements in Alzheimer’s disease brains modulate in vivo seeding capability. Acta Neuropathol. Commun. 2021, 9, 56. [Google Scholar] [CrossRef]
- Edwards, G.; Zhao, J.; Dash, P.K.; Soto, C.; Moreno-Gonzalez, I. Traumatic brain injury induces tau aggregation and spreading. J. Neurotrauma 2020, 37, 80–92. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef]
- Kaufman, S.K.; Sanders, D.W.; Thomas, T.L.; Ruchinskas, A.J.; Vaquer-Alicea, J.; Sharma, A.M.; Miller, T.M.; Diamond, M.I. Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron 2016, 92, 796–812. [Google Scholar] [CrossRef]
- Clavaguera, F.; Hench, J.; Lavenir, I.; Schweighauser, G.; Frank, S.; Goedert, M.; Tolnay, M. Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol. 2014, 127, 299–301. [Google Scholar] [CrossRef]
- Dugger, B.N.; Whiteside, C.M.; Maarouf, C.L.; Walker, D.G.; Beach, T.G.; Sue, L.I.; Garcia, A.; Dunckley, T.; Meechoovet, B.; Reiman, E.M.; et al. The presence of select tau species in human peripheral tissues and their relation to Alzheimer’s disease. J. Alzheimer’s Dis. 2016, 51, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.C.M.; et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Hopp, S.C.; Lin, Y.; Oakley, D.; Roe, A.D.; Devos, S.L.; Hanlon, D.; Hyman, B.T. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 269. [Google Scholar] [CrossRef]
- Pascoal, T.A.; Benedet, A.L.; Ashton, N.J.; Kang, M.S.; Therriault, J.; Chamoun, M.; Savard, M.; Lussier, F.Z.; Tissot, C.; Karikari, T.K.; et al. Microglial activation and tau propagate jointly across braak stages. Nat. Med. 2021, 27, 1592–1599. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, P-Tau and mitochondria. Ageing Res. Rev. 2021, 65, 101208. [Google Scholar] [CrossRef]
- Götz, J.; Chen, F.; Van Dorpe, J.; Nitsch, R.M. Formation of neurofibrillary tangles in P301L Tau transgenic mice induced by Aβ42 fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and Tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid β-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef]
- Huber, C.M.; Yee, C.; May, T.; Dhanala, A.; Mitchell, C.S. Cognitive decline in preclinical Alzheimer’s disease: Amyloid-beta versus tauopathy. J. Alzheimer’s Dis. 2018, 61, 265–281. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-Type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef]
- Zhong, P.; Gu, Z.; Wang, X.; Jiang, H.; Feng, J.; Yan, Z. Impaired modulation of GABAergic transmission by muscarinic receptors in a mouse transgenic model of Alzheimer’s disease. J. Biol. Chem. 2003, 278, 26888–26896. [Google Scholar] [CrossRef]
- Sanchez-Varo, R.; Sanchez-Mejias, E.; Fernandez-Valenzuela, J.J.; De Castro, V.; Mejias-Ortega, M.; Gomez-Arboledas, A.; Jimenez, S.; Sanchez-Mico, M.V.; Trujillo-Estrada, L.; Moreno-Gonzalez, I.; et al. Plaque-associated oligomeric amyloid-beta drives early synaptotoxicity in APP/PS1 mice hippocampus: Ultrastructural pathology analysis. Front. Neurosci. 2021, 15, 752594. [Google Scholar] [CrossRef]
- Bai, Y.; Li, M.; Zhou, Y.; Ma, L.; Qiao, Q.; Hu, W.; Li, W.; Wills, Z.P.; Gan, W.B. Abnormal dendritic calcium activity and synaptic depotentiation occur early in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Fiala, J.C. Mechanisms of amyloid plaque pathogenesis. Acta Neuropathol. 2007, 114, 551–571. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Varo, R.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Torres, M.; Baglietto-Vargas, D.; Moreno-Gonzalez, I.; De Castro, V.; Jimenez, S.; Ruano, D.; Vizuete, M.; et al. Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol. 2012, 123, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Sadleir, K.R.; Kandalepas, P.C.; Buggia-Prévot, V.; Nicholson, D.A.; Thinakaran, G.; Vassar, R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 235–256. [Google Scholar] [CrossRef] [PubMed]
- Sharoar, M.G.; Hu, X.; Ma, X.M.; Zhu, X.; Yan, R. Sequential Formation of Different Layers of Dystrophic Neurites in Alzheimer’s Brains. Mol. Psychiatry 2019, 24, 1369–1382. [Google Scholar] [CrossRef]
- Woodhouse, A.; Vickers, J.C.; Adlard, P.A.; Dickson, T.C. Dystrophic neurites in TgCRND8 and Tg2576 mice mimic human pathological brain aging. Neurobiol. Aging 2009, 30, 864–874. [Google Scholar] [CrossRef]
- Trujillo-Estrada, L.; Dávila, J.C.; Sánchez-Mejias, E.; Sánchez-Varo, R.; Gomez-Arboledas, A.; Vizuete, M.; Vitorica, J.; Gutiérrez, A. Early neuronal loss and axonal/presynaptic damage is associated with accelerated amyloid-β accumulation in AβPP/PS1 Alzheimer’s disease mice subiculum. J. Alzheimer’s Dis. 2014, 42, 521–541. [Google Scholar] [CrossRef]
- Fernandez-Valenzuela, J.J.; Sanchez-Varo, R.; Muñoz-Castro, C.; De Castro, V.; Sanchez-Mejias, E.; Navarro, V.; Jimenez, S.; Nuñez-Diaz, C.; Gomez-Arboledas, A.; Moreno-Gonzalez, I.; et al. Enhancing microtubule stabilization rescues cognitive deficits and ameliorates pathological phenotype in an amyloidogenic Alzheimer’s disease model. Sci. Rep. 2020, 10, 14776. [Google Scholar] [CrossRef]
- Torres, M.; Jimenez, S.; Sanchez-Varo, R.; Navarro, V.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Carmona, I.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; et al. Defective lysosomal proteolysis and axonal transport are early pathogenic events that worsen with age leading to increased APP metabolism and synaptic abeta in transgenic APP/PS1 hippocampus. Mol. Neurodegener. 2012, 7, 59. [Google Scholar] [CrossRef]
- Rosenmann, H.; Grigoriadis, N.; Eldar-Levy, H.; Avital, A.; Rozenstein, L.; Touloumi, O.; Behar, L.; Ben-Hur, T.; Avraham, Y.; Berry, E.; et al. A novel transgenic mouse expressing double mutant tau driven by its natural promoter exhibits tauopathy characteristics. Exp. Neurol. 2008, 212, 71–84. [Google Scholar] [CrossRef]
- Boekhoorn, K.; Terwel, D.; Biemans, B.; Borghgraef, P.; Wiegert, O.; Ramakers, G.J.A.; De Vos, K.; Krugers, H.; Tomiyama, T.; Mori, H.; et al. Improved long-term potentiation and memory in young Tau-P301L transgenic mice before onset of hyperphosphorylation and tauopathy. J. Neurosci. 2006, 26, 3514–3523. [Google Scholar] [CrossRef][Green Version]
- Shentu, Y.P.; Huo, Y.; Feng, X.L.; Gilbert, J.; Zhang, Q.; Liuyang, Z.Y.; Wang, X.L.; Wang, G.; Zhou, H.; Wang, X.C.; et al. CIP2A causes Tau/APP phosphorylation, synaptopathy, and memory deficits in Alzheimer’s disease. Cell Rep. 2018, 24, 713–723. [Google Scholar] [CrossRef]
- Hatch, R.J.; Wei, Y.; Xia, D.; Götz, J. Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropathol. 2017, 133, 717–730. [Google Scholar] [CrossRef]
- Levenga, J.; Krishnamurthy, P.; Rajamohamedsait, H.; Wong, H.; Franke, T.F.; Cain, P.; Sigurdsson, E.M.; Hoeffer, C.A. Tau Pathology induces loss of GABAergic interneurons leading to altered synaptic plasticity and behavioral impairments. Acta Neuropathol. Commun. 2013, 1, 34. [Google Scholar] [CrossRef]
- Van Der Jeugd, A.; Hochgräfe, K.; Ahmed, T.; Decker, J.M.; Sydow, A.; Hofmann, A.; Wu, D.; Messing, L.; Balschun, D.; D’Hooge, R.; et al. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human tau. Acta Neuropathol. 2012, 123, 787–805. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-β/fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef]
- Chabrier, M.A.; Blurton-Jones, M.; Agazaryan, A.A.; Nerhus, J.L.; Martinez-Coria, H.; LaFerla, F.M. Soluble Aβ promotes wild-type tau pathology in vivo. J. Neurosci. 2012, 32, 17345–17350. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–357. [Google Scholar] [CrossRef]
- Oddo, S.; Billings, L.; Kesslak, J.P.; Cribbs, D.H.; LaFerla, F.M. Aβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 2004, 43, 321–332. [Google Scholar] [CrossRef]
- Chesser, A.S.; Pritchard, S.M.; Johnson, G.V.W. Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front. Neurol. 2013, 4, 122. [Google Scholar] [CrossRef]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef]
- Saha, P.; Sen, N. Tauopathy: A common mechanism for neurodegeneration and brain aging. Mech. Ageing Dev. 2019, 178, 72–79. [Google Scholar] [CrossRef]
- Li, T.; Braunstein, K.E.; Zhang, J.; Lau, A.; Sibener, L.; Deeble, C.; Wong, P.C. The neuritic plaque facilitates pathological conversion of tau in an Alzheimer’s disease mouse model. Nat. Commun. 2016, 7, 12082. [Google Scholar] [CrossRef]
- Mountjoy, C.Q.; Roth, M.; Evans, N.J.R.; Evans, H.M. Cortical neuronal counts in normal elderly controls and demented patients. Neurobiol. Aging 1983, 4, 1–11. [Google Scholar] [CrossRef]
- Simic, G.; Kostovic, I.; Winblad, B.; Bogdanovic, N. Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer’s disease. J. Comp. Neurol. 1997, 379, 482–494. [Google Scholar] [CrossRef]
- Gómez-Isla, T.; Price, J.L.; McKeel, D.W.; Morris, J.C.; Growdon, J.H.; Hyman, B.T. Profound loss of layer ii entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci. 1996, 16, 4491–4500. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, P.; Herrmann, F.R.; Bussière, T.; Bouras, C.; Kövari, E.; Perl, D.P.; Morrison, J.H.; Gold, G.; Hof, P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M. Neurochemical studies of Alzheimer’s disease. Neurodegeneration 1996, 5, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Zampar, S. Neuron loss in Alzheimer’s disease: Translation in transgenic mouse models. Int. J. Mol. Sci. 2020, 21, 8144. [Google Scholar] [CrossRef] [PubMed]
- Leong, Y.Q.; Ng, K.Y.; Chye, S.M.; Ling, A.P.K.; Koh, R.Y. Mechanisms of action of amyloid-beta and its precursor protein in neuronal cell death. Metab. Brain Dis. 2020, 35, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Hemachandra Reddy, P. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 5148–5166. [Google Scholar] [CrossRef]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.M.F. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 2008, 30, 331–342. [Google Scholar] [CrossRef]
- Bancher, C.; Lassmann, H.; Breitschopf, H.; Jellinger, K.A. Mechanisms of cell death in Alzheimer’s disease. J. Neural Transm. Suppl. 1997, 50, 141–152. [Google Scholar] [CrossRef]
- Wirths, O.; Multhaup, G.; Bayer, T.A. A Modified Beta-Amyloid hypothesis: Intraneuronal accumulation of the beta-amyloid peptide—The first step of a fatal cascade. J. Neurochem. 2004, 91, 513–520. [Google Scholar] [CrossRef]
- Gouras, G.K.; Almeida, C.G.; Takahashi, R.H. Intraneuronal abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol. Aging 2005, 26, 1235–1244. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 89. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Mesulam, M.M. A Horseradish peroxidase method for the identification of the efferents of acetyl cholinesterase containing neurons. J. Histochem. Cytochem. 1976, 24, 1281–1285. [Google Scholar] [CrossRef]
- Whitehouse, P.; Price, D.; Struble, R.; Clark, A.; Coyle, J.; Delon, M. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef]
- Wong, T.P.; Debeir, T.; Duff, K.; Cuello, A.C. Reorganization of cholinergic terminals in the cerebral cortex and hippocampus in transgenic mice carrying mutated presenilin-1 and amyloid precursor protein transgenes. J. Neurosci. 1999, 19, 2706–2716. [Google Scholar] [CrossRef]
- Boncristiano, S.; Calhoun, M.E.; Kelly, P.H.; Pfeifer, M.; Bondolfi, L.; Stalder, M.; Phinney, A.L.; Abramowski, D.; Sturchler-Pierrat, C.; Enz, A.; et al. Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J. Neurosci. 2002, 22, 3234–3243. [Google Scholar] [CrossRef]
- Bales, K.R.; Tzavara, E.T.; Wu, S.; Wade, M.R.; Bymaster, F.P.; Paul, S.M.; Nomikos, G.G. Cholinergic dysfunction in a mouse model of Alzheimer disease is reversed by an Anti-Aβ Antibody. J. Clin. Investig. 2006, 116, 825–832. [Google Scholar] [CrossRef]
- German, D.C.; Yazdani, U.; Speciale, S.G.; Pasbakhsh, P.; Games, D.; Liang, C.L. Cholinergic neuropathology in a mouse model of Alzheimer’s disease. J. Comp. Neurol. 2003, 462, 371–381. [Google Scholar] [CrossRef]
- Choi, J.H.K.; Kaur, G.; Mazzella, M.J.; Morales-Corraliza, J.; Levy, E.; Mathews, P.M. Early endosomal abnormalities and cholinergic neuron degeneration in amyloid-β protein precursor transgenic mice. J. Alzheimer’s Dis. 2013, 34, 691–700. [Google Scholar] [CrossRef]
- Devi, L.; Ohno, M. Phospho-EIF2α level is important for determining abilities of BACE1 reduction to rescue cholinergic neurodegeneration and memory defects in 5XFAD Mice. PLoS ONE 2010, 5, e12974. [Google Scholar] [CrossRef]
- Christensen, D.Z.; Bayer, T.A.; Wirths, O. Intracellular Aβ triggers neuron loss in the cholinergic system of the APP/PS1KI mouse model of Alzheimer’s disease. Neurobiol. Aging 2010, 31, 1153–1163. [Google Scholar] [CrossRef]
- Foidl, B.M.; Do-Dinh, P.; Hutter-Schmid, B.; Bliem, H.R.; Humpel, C. Cholinergic neurodegeneration in an Alzheimer mouse model overexpressing amyloid-precursor protein with the Swedish-Dutch-Iowa mutations. Neurobiol. Learn. Mem. 2016, 136, 86–96. [Google Scholar] [CrossRef]
- Ubhi, K.; Rockenstein, E.; Vazquez-Roque, R.; Mante, M.; Inglis, C.; Patrick, C.; Adame, A.; Fahnestock, M.; Doppler, E.; Novak, P.; et al. Cerebrolysin modulates pronerve growth factor/nerve growth factor ratio and ameliorates the cholinergic deficit in a transgenic model of Alzheimer’s disease. J. Neurosci. Res. 2013, 91, 167–177. [Google Scholar] [CrossRef]
- Mesulam, M.-M. Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer’s disease. J. Comp. Neurol. 2013, 521, 4124–4144. [Google Scholar] [CrossRef]
- Mesulam, M.; Shaw, P.; Mash, D.; Weintraub, S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann. Neurol. 2004, 55, 815–828. [Google Scholar] [CrossRef]
- Belarbi, K.; Burnouf, S.; Fernandez-Gomez, F.-J.; Desmercieres, J.; Troquier, L.; Brouillette, J.; Tsambou, L.; Grosjean, M.-E.; Caillierez, R.; Demeyer, D.; et al. Loss of medial septum cholinergic neurons in THY-Tau22 mouse model: What links with tau pathology? Curr. Alzheimer Res. 2011, 8, 633–638. [Google Scholar] [CrossRef]
- Cranston, A.L.; Wysocka, A.; Steczkowska, M.; Zadrożny, M.; Palasz, E.; Harrington, C.R.; Theuring, F.; Wischik, C.M.; Riedel, G.; Niewiadomska, G. Cholinergic and inflammatory phenotypes in transgenic tau mouse models of Alzheimer’s disease and frontotemporal lobar degeneration. Brain Commun. 2020, 2, fcaa033. [Google Scholar] [CrossRef]
- Pelkey, K.A.; Chittajallu, R.; Craig, M.T.; Tricoire, L.; Wester, J.C.; McBain, C.J. Hippocampal gabaergic inhibitory interneurons. Physiol. Rev. 2017, 97, 1619–1747. [Google Scholar] [CrossRef]
- Brady, D.R.; Mufson, E.J. Parvalbumin-immunoreactive neurons in the hippocampal formation of Alzheimer’s diseased brain. Neuroscience 1997, 80, 1113–1125. [Google Scholar] [CrossRef]
- Sanchez-Mejias, E.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Gomez-Arboledas, A.; Garcia-Leon, J.A.; Fernandez-Valenzuela, J.J.; Mejias-Ortega, M.; Trujillo-Estrada, L.; Baglietto-Vargas, D.; Moreno-Gonzalez, I.; et al. Distinct disease-sensitive GABAergic neurons in the perirhinal cortex of Alzheimer’s mice and patients. Brain Pathol. 2020, 30, 345–363. [Google Scholar] [CrossRef]
- Saiz-Sanchez, D.; De la Rosa-Prieto, C.; Ubeda-Banon, I.; Martinez-Marcos, A. Interneurons, tau and amyloid-β in the piriform cortex in Alzheimer’s disease. Brain Struct. Funct. 2015, 220, 2011–2025. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Katzman, R.; Terry, R.D. Reduced somatostatin-like immunoreactivity in cerebral cortex from cases of Alzheimer disease and Alzheimer senile dementa. Nature 1980, 288, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Schmid, L.C.; Mittag, M.; Poll, S.; Steffen, J.; Wagner, J.; Geis, H.-R.; Schwarz, I.; Schmidt, B.; Schwarz, M.K.; Remy, S.; et al. Dysfunction of somatostatin-positive interneurons associated with memory deficits in an Alzheimer’s disease model. Neuron 2016, 92, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Ramos, B.; Baglietto-Vargas, D.; del Rio, J.C.; Moreno-Gonzalez, I.; Santa-Maria, C.; Jimenez, S.; Caballero, C.; Lopez-Tellez, J.F.; Khan, Z.U.; Ruano, D.; et al. Early Neuropathology of somatostatin/NPY GABAergic Cells in the hippocampus of a PS1/APP transgenic model of Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1658–1672. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gonzalez, I.; Baglietto-Vargas, D.; Sanchez-Varo, R.; Jimenez, S.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Del Rio, J.C.; Torres, M.; Romero-Acebal, M.; Ruano, D.; et al. Extracellular amyloid-β and cytotoxic glial activation induce significant entorhinal neuron loss in young PS1M146L/APP751SL mice. J. Alzheimer’s Dis. 2009, 18, 755–776. [Google Scholar] [CrossRef] [PubMed]
- Baglietto-Vargas, D.; Moreno-Gonzalez, I.; Sanchez-Varo, R.; Jimenez, S.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Torres, M.; Romero-Acebal, M.; Ruano, D.; Vizuete, M.; et al. Calretinin Interneurons are early targets of extracellular amyloid-β pathology in PS1/AβPP Alzheimer mice hippocampus. J. Alzheimer’s Dis. 2010, 21, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Silva Albequerque, M.; Mahar, I.; Davoli, M.A.; Chabot, J.G.; Mechawar, N.; Quirion, R.; Krantic, S. Regional and sub-regional differences in hippocampal GABAergic neuronal vulnerability in the TgCRND8 mouse model of Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 30. [Google Scholar] [CrossRef]
- Mahar, I.; Albuquerque, M.S.; Mondragon-Rodriguez, S.; Cavanagh, C.; Davoli, M.A.; Chabot, J.-G.; Williams, S.; Mechawar, N.; Quirion, R.; Krantic, S. Phenotypic alterations in hippocampal NPY- and PV-expressing interneurons in a presymptomatic transgenic mouse model of Alzheimer’s disease. Front. Aging Neurosci. 2016, 8, 327. [Google Scholar] [CrossRef]
- Palop, J.J. Epilepsy and cognitive impairments in Alzheimer disease. Arch. Neurol. 2009, 66, 435. [Google Scholar] [CrossRef]
- Shimojo, M.; Takuwa, H.; Takado, Y.; Tokunaga, M.; Tsukamoto, S.; Minatohara, K.; Ono, M.; Seki, C.; Maeda, J.; Urushihata, T.; et al. Selective disruption of inhibitory synapses leading to neuronal hyperexcitability at an early stage of tau pathogenesis in a mouse model. J. Neurosci. 2020, 40, 3491–3501. [Google Scholar] [CrossRef]
- Rubio, S.E.; González, M.; Soriano, E.; Pascual, M.; Ávila, J.; Soler, H.; Dorca-Arévalo, J. The GABAergic septohippocampal connection is impaired in a mouse model of tauopathy. Neurobiol. Aging 2016, 49, 40–51. [Google Scholar] [CrossRef]
- Takahashi, H.; Brasnjevic, I.; Rutten, B.P.F.; Van Der Kolk, N.; Perl, D.P.; Bouras, C.; Steinbusch, H.W.M.; Schmitz, C.; Hof, P.R.; Dickstein, D.L. Hippocampal interneuron loss in an APP/PS1 double mutant mouse and in Alzheimer’s disease. Brain Struct. Funct. 2010, 214, 145–160. [Google Scholar] [CrossRef]
- Verdaguer, E.; Brox, S.; Petrov, D.; Olloquequi, J.; Romero, R.; de Lemos, M.L.; Camins, A.; Auladell, C. Vulnerability of calbindin, calretinin and parvalbumin in a transgenic/knock-in APPswe/PS1dE9 mouse model of Alzheimer disease together with disruption of hippocampal neurogenesis. Exp. Gerontol. 2015, 69, 176–188. [Google Scholar] [CrossRef]
- Hof, P.R.; Cox, K.; Young, W.G.; Celio, M.R.; Rogers, J.; Morrison, J.H. Parvalbumin-immunoreactive neurons in the neocortex are resistant to degeneration in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1991, 50, 451–462. [Google Scholar] [CrossRef]
- Mikkonen, M.; Alafuzoff, I.; Tapiola, T.; Soininen, H.; Miettinen, R. Subfield- and layer-specific changes in parvalbumin, calretinin and calbindin-D28k immunoreactivity in the entorhinal cortex in Alzheimer’s disease. Neuroscience 1999, 92, 515–532. [Google Scholar] [CrossRef]
- Satoh, J.; Tabira, T.; Sano, M.; Nakayama, H.; Tateishi, J. Parvalbumin-immunoreactive neurons in the human central nervous system are decreased in Alzheimer’s disease. Acta Neuropathol. 1991, 81, 388–395. [Google Scholar] [CrossRef]
- Dávila-Bouziguet, E.; Targa-Fabra, G.; Ávila, J.; Soriano, E.; Pascual, M. Differential accumulation of tau phosphorylated at residues Thr231, Ser262 and Thr205 in hippocampal interneurons and its modulation by tau mutations (VLW) and amyloid-β peptide. Neurobiol. Dis. 2019, 125, 232–244. [Google Scholar] [CrossRef]
- Zallo, F.; Gardenal, E.; Verkhratsky, A.; Rodríguez, J.J. Loss of calretinin and parvalbumin positive interneurones in the hippocampal CA1 of aged Alzheimer’s disease mice. Neurosci. Lett. 2018, 681, 19–25. [Google Scholar] [CrossRef]
- Davies, D.C.; Horwood, N.; Isaacs, S.L.; Mann, D.M.A. The effect of age and Alzheimer’s disease on pyramidal neuron density in the individual fields of the hippocampal formation. Acta Neuropathol. 1992, 83, 510–517. [Google Scholar] [CrossRef]
- Ter Laak, H.J.; Renkawek, K.; Van Workum, F.P.A. The olfactory bulb in Alzheimer disease: A morphologic study of neuron loss, tangles, and senile plaques in relation to olfaction. Alzheimer Dis. Assoc. Disord. 1994, 8, 38–48. [Google Scholar] [CrossRef]
- Mann, D.M.A. The topographic distribution of brain atrophy in Alzheimer’s disease. Acta Neuropathol. 1991, 83, 81–86. [Google Scholar] [CrossRef]
- Calhoun, M.E.; Wiederhold, K.-H.; Abramowski, D.; Phinney, A.L.; Probst, A.; Sturchler-Pierrat, C.; Staufenbiel, M.; Sommer, B.; Jucker, M. Neuron loss in APP transgenic mice. Nature 1998, 395, 755–756. [Google Scholar] [CrossRef]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 196.e29–196.e40. [Google Scholar] [CrossRef]
- Wegenast-Braun, B.M.; Maisch, A.F.; Eicke, D.; Radde, R.; Herzig, M.C.; Staufenbiel, M.; Jucker, M.; Calhoun, M.E. Independent effects of intra- and extracellular Aβ on learning-related gene expression. Am. J. Pathol. 2009, 175, 271–282. [Google Scholar] [CrossRef]
- Ugolini, F.; Lana, D.; Nardiello, P.; Nosi, D.; Pantano, D.; Casamenti, F.; Giovannini, M.G. Different patterns of neurodegeneration and glia activation in CA1 and CA3 hippocampal regions of TgCRND8 mice. Front. Aging Neurosci. 2018, 10, 372. [Google Scholar] [CrossRef]
- Harwell, C.S.; Coleman, M.P. Synaptophysin depletion and intraneuronal Aβ in organotypic hippocampal slice cultures from HuAPP transgenic mice. Mol. Neurodegener. 2016, 11, 44. [Google Scholar] [CrossRef]
- Schmitz, C.; Rutten, B.P.F.; Pielen, A.; Schäfer, S.; Wirths, O.; Tremp, G.; Czech, C.; Blanchard, V.; Multhaup, G.; Rezaie, P.; et al. Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer’s disease. Am. J. Pathol. 2004, 164, 1495–1502. [Google Scholar] [CrossRef]
- Breyhan, H.; Wirths, O.; Duan, K.; Marcello, A.; Rettig, J.; Bayer, T.A. APP/PS1KI bigenic mice develop early synaptic deficits and hippocampus atrophy. Acta Neuropathol. 2009, 117, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Spires, T.L.; Orne, J.D.; SantaCruz, K.; Pitstick, R.; Carlson, G.A.; Ashe, K.H.; Hyman, B.T. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am. J. Pathol. 2006, 168, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Andorfer, C.; Acker, C.M.; Kress, Y.; Hof, P.R.; Duff, K.; Davies, P. Cell-Cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 2005, 25, 5446–5454. [Google Scholar] [CrossRef] [PubMed]
- Radde, R.; Duma, C.; Goedert, M.; Jucker, M. The value of incomplete mouse models of Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, S70–S74. [Google Scholar] [CrossRef]
- Chabrier, M.A.; Cheng, D.; Castello, N.A.; Green, K.N.; LaFerla, F.M. Synergistic effects of amyloid-beta and wild-type human tau on dendritic spine loss in a floxed double transgenic model of Alzheimer’s disease. Neurobiol. Dis. 2014, 64, 107–117. [Google Scholar] [CrossRef]
- Umeda, T.; Maekawa, S.; Kimura, T.; Takashima, A.; Tomiyama, T.; Mori, H. Neurofibrillary tangle formation by introducing wild-type human tau into APP transgenic mice. Acta Neuropathol. 2014, 127, 685–698. [Google Scholar] [CrossRef]
- Ribé, E.M.; Pérez, M.; Puig, B.; Gich, I.; Lim, F.; Cuadrado, M.; Sesma, T.; Catena, S.; Sánchez, B.; Nieto, M.; et al. Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/Tau transgenic mice. Neurobiol. Dis. 2005, 20, 814–822. [Google Scholar] [CrossRef]
- Stancu, I.C.; Ris, L.; Vasconcelos, B.; Marinangeli, C.; Goeminne, L.; Laporte, V.; Haylani, L.E.; Couturier, J.; Schakman, O.; Gailly, P.; et al. Tauopathy contributes to synaptic and cognitive deficits in a murine model for Alzheimer’s disease. FASEB J. 2014, 28, 2620–2631. [Google Scholar] [CrossRef]
- Perez, S.E.; He, B.; Muhammad, N.; Oh, K.J.; Fahnestock, M.; Ikonomovic, M.D.; Mufson, E.J. Cholinotrophic basal forebrain system alterations in 3×Tg-AD transgenic mice. Neurobiol. Dis. 2011, 41, 338–352. [Google Scholar] [CrossRef]
- Orta-Salazar, E.; Aguilar-Vázquez, A.; Martínez-Coria, H.; Luquín-De Anda, S.; Rivera-Cervantes, M.; Beas-Zarate, C.; Feria-Velasco, A.; Díaz-Cintra, S. REST/NRSF-induced changes of chat protein expression in the neocortex and hippocampus of the 3×Tg-AD mouse model for Alzheimer’s disease. Life Sci. 2014, 116, 83–89. [Google Scholar] [CrossRef]
- Rapoport, M.; Dawson, H.N.; Binder, L.I.; Vitek, M.P.; Ferreira, A. Tau is essential to β-amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 6364–6369. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction of oligomeric amyloid-β with phosphorylated tau: Implications to synaptic dysfunction and neuronal damage. J. Alzheimer’s Dis. 2013, 36, 285–295. [Google Scholar] [CrossRef]
- Pickett, E.K.; Herrmann, A.G.; McQueen, J.; Abt, K.; Dando, O.; Tulloch, J.; Jain, P.; Dunnett, S.; Sohrabi, S.; Fjeldstad, M.P.; et al. Amyloid beta and tau cooperate to cause reversible behavioral and transcriptional deficits in a model of Alzheimer’s disease. Cell Rep. 2019, 29, 3592–3604.e5. [Google Scholar] [CrossRef]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef]
- Hammond, T.R.; Marsh, S.E.; Stevens, B. Immune signaling in neurodegeneration. Immunity 2019, 50, 955–974. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2020, 17, 157–172. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia biology: One century of evolving concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Lewcock, J.W.; Schlepckow, K.; Di Paolo, G.; Tahirovic, S.; Monroe, K.M.; Haass, C. Emerging microglia biology defines novel therapeutic approaches for Alzheimer’s disease. Neuron 2020, 108, 801–821. [Google Scholar] [CrossRef]
- Romero-Molina, C.; Navarro, V.; Jimenez, S.; Muñoz-Castro, C.; Sanchez-Mico, M.V.; Gutierrez, A.; Vitorica, J.; Vizuete, M. Should we open fire on microglia? depletion models as tools to elucidate microglial role in health and Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 9734. [Google Scholar] [CrossRef]
- Scearce-Levie, K.; Sanchez, P.E.; Lewcock, J.W. Leveraging preclinical models for the development of Alzheimer disease therapeutics. Nat. Rev. Drug Discov. 2020, 19, 447–462. [Google Scholar] [CrossRef]
- Birch, A.M.; Katsouri, L.; Sastre, M. Modulation of inflammation in transgenic models of Alzheimer’s disease. J. Neuroinflamm. 2014, 11, 25. [Google Scholar] [CrossRef]
- Miron, V.E.; Priller, J. Investigating microglia in health and disease: Challenges and opportunities. Trends Immunol. 2020, 41, 785–793. [Google Scholar] [CrossRef]
- Song, W.M.; Colonna, M. The identity and function of microglia in neurodegeneration. Nat. Immunol. 2018, 19, 1048–1058. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia heterogeneity in the single-cell era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef]
- Hashemiaghdam, A.; Mroczek, M. Microglia heterogeneity and neurodegeneration: The emerging paradigm of the role of immunity in Alzheimer’s disease. J. Neuroimmunol. 2020, 341, 577185. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-associated microglia: A universal immune sensor of neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Gabriela Sánchez, M.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gómez-Nicola, D.; Luheshi, G.; Vallières, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef]
- St-Pierre, M.K.; Šimončičová, E.; Bögi, E.; Tremblay, M.È. Shedding light on the dark side of the microglia. ASN Neuro 2020, 12, 1759091420925335. [Google Scholar] [CrossRef]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef]
- Sosna, J.; Philipp, S.; Albay, R.I.; Reyes-Ruiz, J.M.; Baglietto-Vargas, D.; LaFerla, F.M.; Glabe, C.G. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 11. [Google Scholar] [CrossRef]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.T.; Woodbury, M.E.; Srivastava, G.; et al. The major risk factors for Alzheimer’s disease: Age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. 2019, 27, 1293–1306.e6. [Google Scholar] [CrossRef]
- Liang, X.; Wu, H.; Colt, M.; Guo, X.; Pluimer, B.; Zeng, J.; Dong, S.; Zhao, Z. Microglia and its genetics in Alzheimer’s disease. Curr. Alzheimer Res. 2021, 18, 676–688. [Google Scholar] [CrossRef]
- Yang, H.S.; Onos, K.D.; Choi, K.; Keezer, K.J.; Skelly, D.A.; Carter, G.W.; Howell, G.R. Natural genetic variation determines microglia heterogeneity in wild-derived mouse models of Alzheimer’s disease. Cell Rep. 2021, 34, 1–17. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef]
- Navarro, V.; Sanchez-Mejias, E.; Jimenez, S.; Muñoz-Castro, C.; Sanchez-Varo, R.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Microglia in Alzheimer’s disease: Activated, dysfunctional or degenerative. Front. Aging Neurosci. 2018, 10, 140. [Google Scholar] [CrossRef]
- Swanson, M.; Swanson, M.; Scotter, E.L.; Murray, H.C.; Turner, C.; Faull, R.L.M.; Curtis, M.A. Identification of a dysfunctional microglial population in human Alzheimer’s disease cortex using novel single-cell histology image analysis. Acta Neuropathol. Commun. 2020, 8, 170. [Google Scholar] [CrossRef]
- Streit, W.J.; Khoshbouei, H.; Bechmann, I. Dystrophic microglia in late-onset Alzheimer’s disease. Glia 2020, 68, 845–854. [Google Scholar] [CrossRef]
- Sanchez-Mejias, E.; Navarro, V.; Jimenez, S.; Sanchez-Mico, M.; Sanchez-Varo, R.; Nu??ez-Diaz, C.; Trujillo-Estrada, L.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; et al. Soluble Phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol. 2016, 132, 897–916. [Google Scholar] [CrossRef]
- March-Diaz, R.; Lara-Ureña, N.; Romero-Molina, C.; Heras-Garvin, A.; Ortega-de San Luis, C.; Alvarez-Vergara, M.I.; Sanchez-Garcia, M.A.; Sanchez-Mejias, E.; Davila, J.C.; Rosales-Nieves, A.E.; et al. Hypoxia compromises the mitochondrial metabolism of Alzheimer’s disease microglia via HIF1. Nat. Aging 2021, 1, 385–399. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Holtzman, D.M. Glial contributions to neurodegeneration in tauopathies. Mol. Neurodegener. 2017, 12, 50. [Google Scholar] [CrossRef]
- Van Olst, L.; Verhaege, D.; Franssen, M.; Roucourt, B.; Carmans, S.; Ytebrouck, E.; Kamermans, A.; van der Pol, S.M.A.; Wever, D.; Popovic, M.; et al. Microglial activation arises after aggregation of phosphorylated-tau in a neuron specific P301S tauopathy mouse model. Neurobiol. Aging 2020, 89, 89–98. [Google Scholar] [CrossRef]
- Sobue, A.; Komine, O.; Hara, Y.; Endo, F.; Mizoguchi, H.; Watanabe, S.; Murayama, S.; Saito, T.; Saido, T.C.; Sahara, N.; et al. Microglial gene signature reveals loss of homeostatic microglia associated with neurodegeneration of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 1–17. [Google Scholar] [CrossRef]
- Belfiore, R.; Rodin, A.; Ferreira, E.; Velazquez, R.; Branca, C.; Caccamo, A.; Oddo, S. Temporal and regional progression of Alzheimer’s disease-like pathology in 3xTg-AD Mice. Aging Cell 2019, 18, e12873. [Google Scholar] [CrossRef]
- Bonham, L.W.; Sirkis, D.W.; Yokoyama, J.S. The transcriptional landscape of microglial genes in aging and neurodegenerative disease. Front. Immunol. 2019, 10, 1170. [Google Scholar] [CrossRef]
- McQuade, A.; Blurton-Jones, M. Microglia in Alzheimer’s disease: Exploring how genetics and phenotype influence risk. J. Mol. Biol. 2019, 431, 1805–1817. [Google Scholar] [CrossRef]
- Ulland, T.K.; Colonna, M. TREM2—A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Cheng-Hathaway, P.J.; Reed-geaghan, E.G.; Jay, T.R.; Casali, B.T.; Bemiller, S.M.; Puntambekar, S.S.; Saucken, V.E.V.; Williams, R.Y.; Karlo, J.C.; Moutinho, M.; et al. The TRem2 R47H variant confers loss-of- function-like phenotypes in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 29. [Google Scholar] [CrossRef]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, Microglia, and neurodegenerative diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef]
- Carmona, S.; Zahs, K.; Wu, E.; Dakin, K.; Bras, J.; Guerreiro, R. The role of TREM2 in Alzheimer’s disease and other neurodegenerative disorders. Lancet Neurol. 2018, 17, 721–730. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.-C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef]
- Song, W.; Hooli, B.; Mullin, K.; Jin, S.C.; Cella, M.; Tyler, K.; Wang, Y.; Tanzi, R.; Colonna, M. Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand dependent activation. Alzheimers Dement. 2018, 13, 381–387. [Google Scholar] [CrossRef]
- Jay, T.R.; Hirsch, A.M.; Broihier, M.L.; Miller, C.M.; Neilson, L.E.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci. 2017, 37, 637–647. [Google Scholar] [CrossRef]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Bemiller, S.M.; McCray, T.J.; Allan, K.; Formica, S.V.; Xu, G.; Wilson, G.; Kokiko-Cochran, O.N.; Crish, S.D.; Lasagna-Reeves, C.A.; Ransohoff, R.M.; et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener. 2017, 12, 74. [Google Scholar] [CrossRef]
- Sayed, F.A.; Telpoukhovskaia, M.; Kodama, L.; Li, Y.; Zhou, Y.; Le, D.; Hauduc, A.; Ludwig, C.; Gao, F.; Clelland, C.; et al. Differential effects of partial and complete loss of TREM2 on microglial injury response and tauopathy. Proc. Natl. Acad. Sci. USA 2018, 115, 10172–10177. [Google Scholar] [CrossRef]
- Karanfilian, L.; Tosto, M.G.; Malki, K. The role of TREM2 in Alzheimer’s disease; Evidence from transgenic mouse models. Neurobiol. Aging 2020, 86, 39–53. [Google Scholar] [CrossRef]
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural insights and links to Alzheimer disease pathogenesis. Neuron 2021, 109, 205–221. [Google Scholar] [CrossRef]
- Ulrich, J.D.; Ulland, T.K.; Mahan, T.E.; Nyström, S.; Peter Nilsson, K.; Song, W.M.; Zhou, Y.; Reinartz, M.; Choi, S.; Jiang, H.; et al. ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med. 2018, 215, 1047–1058. [Google Scholar] [CrossRef]
- Liu, C.-C.; Zhao, N.; Fu, Y.; Wang, N.; Linares, C.; Tsai, C.-W.; Bu, G. ApoE4 accelerates early seeding of amyloid pathology. Neuron 2017, 96, 1024–1032.e3. [Google Scholar] [CrossRef]
- Zhao, N.; Ren, Y.; Yamazaki, Y.; Qiao, W.; Li, F.; Felton, L.M.; Mahmoudiandehkordi, S.; Kueider-Paisley, A.; Sonoustoun, B.; Arnold, M.; et al. Alzheimer’s risk factors age, APOE genotype, and sex drive distinct molecular pathways. Neuron 2020, 106, 727–742.e6. [Google Scholar] [CrossRef]
- Shi, Y.; Manis, M.; Long, J.; Wang, K.; Sullivan, P.M.; Serrano, J.R.; Hoyle, R.; Holtzman, D.M. Microglia drive APOE-Dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med. 2019, 216, 2546–2561. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Momtazmanesh, S.; Perry, G.; Rezaei, N. Toll-like receptors in Alzheimer’s disease. J. Neuroimmunol. 2020, 348, 577362. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s disease (AD): From risk factors to therapeutic targeting. Cells 2020, 9, 383. [Google Scholar] [CrossRef]
- Song, M.; Jin, J.J.; Lim, J.E.; Kou, J.; Pattanayak, A.; Rehman, J.A.; Kim, H.D.; Tahara, K.; Lalonde, R.; Fukuchi, K.I. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J. Neuroinflamm. 2011, 8, 92. [Google Scholar] [CrossRef]
- Schartz, N.D.; Tenner, A.J. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J. Neuroinflamm. 2020, 17, 354. [Google Scholar] [CrossRef]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9, eaaf6295. [Google Scholar] [CrossRef]
- Litvinchuk, A.; Wan, Y.-W.; Swartzlander, D.B.; Chen, F.; Cole, A.; Propson, N.E.; Wang, Q.; Zhang, B.; Liu, Z.; Zheng, H. Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and Alzheimer’s disease. Neuron 2018, 100, 1337–1353.e5. [Google Scholar] [CrossRef]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Aβ clearance through NLRP 3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Han, X.; Li, Q.; Lan, X.; EL-Mufti, L.; Ren, H.; Wang, J. Microglial depletion with clodronate liposomes increases proinflammatory cytokine levels, induces astrocyte activation, and damages blood vessel integrity. Mol. Neurobiol. 2019, 56, 6184–6196. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Hu, W.; Tsai, J.; Li, W.; Gan, W.B. Microglia limit the expansion of β-amyloid plaques in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Bolós, M.; Llorens-Martín, M.; Perea, J.R.; Jurado-Arjona, J.; Rábano, A.; Hernández, F.; Avila, J. Absence of CX3CR1 impairs the internalization of tau by microglia. Mol. Neurodegener. 2017, 12, 59. [Google Scholar] [CrossRef]
- Martin-Estebane, M.; Gomez-Nicola, D. Targeting microglial population dynamics in Alzheimer’s disease: Are we ready for a potential impact on immune function? Front. Cell. Neurosci. 2020, 14, 149. [Google Scholar] [CrossRef]
- Spangenberg, E.E.; Green, K.N. Inflammation in Alzheimer’s disease: Lessons learned from microglia-depletion models. Brain Behav. Immun. 2017, 61, 1–11. [Google Scholar] [CrossRef]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.P.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 2016, 139, 1265–1281. [Google Scholar] [CrossRef]
- Lei, F.; Cui, N.; Zhou, C.; Chodosh, J.; Vavvas, D.G.; Paschalis, E.I. CSF1R inhibition by a small-molecule inhibitor is not microglia specific; Affecting hematopoiesis and the function of macrophages. Proc. Natl. Acad. Sci. USA 2020, 117, 23336–23338. [Google Scholar] [CrossRef]
- Navabpour, S.; Kwapis, J.L.; Jarome, T.J. A neuroscientist’s guide to transgenic mice and other genetic tools. Neurosci. Biobehav. Rev. 2020, 108, 732–748. [Google Scholar] [CrossRef]
- Van Hove, H.; Antunes, A.R.P.; De Vlaminck, K.; Scheyltjens, I.; Van Ginderachter, J.A.; Movahedi, K. Identifying the variables that drive tamoxifen-independent CreERT2 recombination: Implications for microglial fate mapping and gene deletions. Eur. J. Immunol. 2020, 50, 459–463. [Google Scholar] [CrossRef]
- Pons, V.; Lévesque, P.; Plante, M.M.; Rivest, S. Conditional genetic deletion of CSF1 receptor in microglia ameliorates the physiopathology of Alzheimer’s disease. Alzheimer’s Res. Ther. 2021, 13, 8. [Google Scholar] [CrossRef]
- Ruan, C.; Sun, L.; Kroshilina, A.; Beckers, L.; De Jager, P.; Bradshaw, E.M.; Hasson, S.A.; Yang, G.; Elyaman, W. A novel Tmem119-TdTomato reporter mouse model for studying microglia in the central nervous system. Brain. Behav. Immun. 2020, 83, 180–191. [Google Scholar] [CrossRef]
- Kaiser, T.; Feng, G. Tmem119-EGFP and Tmem119-CreERT2 transgenic mice for labeling and manipulating microglia. eNeuro 2019, 6, 1–18. [Google Scholar] [CrossRef]
- Masuda, T.; Amann, L.; Sankowski, R.; Staszewski, O.; Lenz, M.; d’Errico, P.; Snaidero, N.; Costa Jordão, M.J.; Böttcher, C.; Kierdorf, K.; et al. Novel hexb-based tools for studying microglia in the CNS. Nat. Immunol. 2020, 21, 802–815. [Google Scholar] [CrossRef]
- Trujillo-Estrada, L.; Gomez-Arboledas, A.; Forner, S.; Martini, A.C.; Gutierrez, A.; Baglietto-Vargas, D.; LaFerla, F.M. Astrocytes: From the physiology to the disease. Curr. Alzheimer Res. 2019, 16, 675–698. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V.; Vardjan, N.; Zorec, R. Physiology of astroglia. Adv. Exp. Med. Biol. 2019, 1175, 45–91. [Google Scholar]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte reactivity: Subtypes, states, and functions in CNS innate immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef]
- Giovannoni, F.; Quintana, F.J. The role of astrocytes in CNS inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef]
- Frost, G.R.; Li, Y.-M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef]
- Carter, S.F.; Herholz, K.; Rosa-Neto, P.; Pellerin, L.; Nordberg, A.; Zimmer, E.R. Astrocyte biomarkers in Alzheimer’s disease. Trends Mol. Med. 2019, 25, 77–95. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Das, S.; Li, Z.; Noori, A.; Hyman, B.T.; Serrano-Pozo, A. Meta-analysis of mouse transcriptomic studies supports a context-dependent astrocyte reaction in acute CNS injury versus neurodegeneration. J. Neuroinflamm. 2020, 17, 1–17. [Google Scholar] [CrossRef]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the astrocyte reaction in Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef]
- Gomez-Arboledas, A.; Davila, J.C.; Sanchez-Mejias, E.; Navarro, V.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Sanchez-Mico, M.V.; Trujillo-Estrada, L.; Fernandez-Valenzuela, J.J.; Vizuete, M.; et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 2018, 66, 637–653. [Google Scholar] [CrossRef]
- Spanos, F.; Liddelow, S.A. An overview of astrocyte responses in genetically induced Alzheimer’s disease mouse models. Cells 2020, 9, 2415. [Google Scholar] [CrossRef]
- Fu, H.; Hussaini, S.A.; Wegmann, S.; Profaci, C.; Daniels, J.D.; Herman, M.; Emrani, S.; Figueroa, H.Y.; Hyman, B.T.; Davies, P.; et al. 3D visualization of the temporal and spatial spread of tau pathology reveals extensive sites of tau accumulation associated with neuronal loss and recognition memory deficit in aged tau transgenic mice. PLoS ONE 2016, 11, e0159463. [Google Scholar] [CrossRef]
- Nilson, A.N.; English, K.C.; Gerson, J.E.; Whittle, T.B.; Crain, C.N.; Xue, J.; Sengupta, U.; Castillo-Carranza, D.L.; Zhang, W.; Gupta, P.; et al. Tau oligomers associate with inflammation in the brain and retina of tauopathy mice and in neurodegenerative diseases. J. Alzheimer’s Dis. 2017, 55, 1083–1099. [Google Scholar] [CrossRef]
- Wang, P.; Ye, Y. Filamentous recombinant human tau activates primary astrocytes via an integrin receptor complex. Nat. Commun. 2021, 12, 95. [Google Scholar] [CrossRef]
- Matias, I.; Morgado, J.; Gomes, F.C.A. Astrocyte heterogeneity: Impact to brain aging and disease. Front. Aging Neurosci. 2019, 11, 59. [Google Scholar] [CrossRef]
- Jiwaji, Z.; Tiwari, S.S.; Avilés-Reyes, R.X.; Hooley, M.; Hampton, D.; Torvell, M.; Johnson, D.A.; McQueen, J.; Baxter, P.; Sabari-Sankar, K.; et al. Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to tau and Aß Pathology. Nat. Commun. 2022, 13, 135. [Google Scholar] [CrossRef]
- Xiao, Q.; Yan, P.; Ma, X.; Liu, H.; Perez, R.; Zhu, A.; Gonzales, E.; Burchett, J.M.; Schuler, D.R.; Cirrito, J.R.; et al. Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 2014, 34, 9607–9620. [Google Scholar] [CrossRef]
- Sanchez-Mico, M.V.; Jimenez, S.; Gomez-Arboledas, A.; Muñoz-Castro, C.; Romero-Molina, C.; Navarro, V.; Sanchez-Mejias, E.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Galea, E.; et al. Amyloid-β impairs the phagocytosis of dystrophic synapses by astrocytes in Alzheimer’s disease. Glia 2020, 69, 997–1011. [Google Scholar] [CrossRef]
- Bales, K.R.; Verina, T.; Dodel, R.C.; Du, Y.; Altstiel, L.; Bender, M.; Hyslop, P.; Johnstone, E.M.; Little, S.P.; Cummins, D.J.; et al. Lack of apolipoprotein E dramatically reduces amyloid β-peptide deposition. Nat. Genet. 1997, 17, 263–264. [Google Scholar] [CrossRef]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef]
- Zheng, J.Y.; Sun, J.; Ji, C.M.; Shen, L.; Chen, Z.J.; Xie, P.; Sun, Y.Z.; Yu, R.T. Selective deletion of apolipoprotein E in astrocytes ameliorates the spatial learning and memory deficits in Alzheimer’s disease (APP/PS1) mice by inhibiting TGF-β/Smad2/STAT3 signaling. Neurobiol. Aging 2017, 54, 112–132. [Google Scholar] [CrossRef]
- Lewandowski, C.T.; Maldonado Weng, J.; LaDu, M.J. Alzheimer’s disease pathology in APOE transgenic mouse models: The who, what, when, where, why, and how. Neurobiol. Dis. 2020, 139, 104811. [Google Scholar] [CrossRef]
- Chung, W.S.; Verghese, P.B.; Chakraborty, C.; Joung, J.; Hyman, B.T.; Ulrich, J.D.; Holtzman, D.M.; Barres, B.A. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 10186–10191. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.-C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Prasad, H.; Rao, R. Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal PH. Proc. Natl. Acad. Sci. USA 2018, 115, E6640–E6649. [Google Scholar] [CrossRef]
- Liu, C.-C.; Hu, J.; Zhao, N.; Wang, J.; Wang, N.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Astrocytic LRP1 mediates brain Aβ clearance and impacts amyloid deposition. J. Neurosci. 2017, 37, 4023–4031. [Google Scholar] [CrossRef]
- Perea, J.R.; López, E.; Diéz-Ballesteros, J.C.; Ávila, J.; Hernández, F.; Bolós, M. Extracellular monomeric tau is internalized by astrocytes. Front. Neurosci. 2019, 13, 442. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Martini-Stoica, H.; Cole, A.L.; Swartzlander, D.B.; Chen, F.; Wan, Y.-W.; Bajaj, L.; Bader, D.A.; Lee, V.M.Y.; Trojanowski, J.Q.; Liu, Z.; et al. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J. Exp. Med. 2018, 215, 2355–2377. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, A.M.; Warren, L.; Usenovic, M.; Zhou, H.; Sugam, J.; Parmentier-Batteur, S.; Voleti, B. Astrocytic expression of the Alzheimer’s disease risk allele, ApoEε4, potentiates neuronal tau pathology in multiple preclinical models. Sci. Rep. 2021, 11, 3438. [Google Scholar] [CrossRef]
- Dorey, E.; Bamji-Mirza, M.; Najem, D.; Li, Y.; Liu, H.; Callaghan, D.; Walker, D.; Lue, L.F.; Stanimirovic, D.; Zhang, W. Apolipoprotein E isoforms differentially regulate Alzheimer’s disease and amyloid-β-induced inflammatory response in vivo and in vitro. J. Alzheimer’s Dis. 2017, 57, 1265–1279. [Google Scholar] [CrossRef]
- Profaci, C.P.; Munji, R.N.; Pulido, R.S.; Daneman, R. The blood–Brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217, e20190062. [Google Scholar] [CrossRef]
- Rainey-Smith, S.R.; Mazzucchelli, G.N.; Villemagne, V.L.; Brown, B.M.; Porter, T.; Weinborn, M.; Bucks, R.S.; Milicic, L.; Sohrabi, H.R.; Taddei, K.; et al. Genetic variation in aquaporin-4 moderates the relationship between sleep and brain Aβ-amyloid burden. Transl. Psychiatry 2018, 8, 47. [Google Scholar] [CrossRef]
- Rosu, G.C.; Catalin, B.; Balseanu, T.A.; Laurentiu, M.; Claudiu, M.; Kumar-Singh, S.; Daniel, P. Inhibition of aquaporin 4 decreases amyloid Aβ40 drainage around cerebral vessels. Mol. Neurobiol. 2020, 57, 4720. [Google Scholar] [CrossRef]
- Mader, S.; Brimberg, L. Aquaporin-4 water channel in the brain and its implication for health and disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef]
- Xu, Z.; Xiao, N.; Chen, Y.; Huang, H.; Marshall, C.; Gao, J.; Cai, Z.; Wu, T.; Hu, G.; Xiao, M. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits. Mol. Neurodegener. 2015, 10, 58. [Google Scholar] [CrossRef]
- Smith, A.J.; Duan, T.; Verkman, A.S. Aquaporin-4 reduces neuropathology in a mouse model of Alzheimer’s disease by remodeling peri-plaque astrocyte structure. Acta Neuropathol. Commun. 2019, 7, 74. [Google Scholar] [CrossRef]
- Valenza, M.; Facchinetti, R.; Steardo, L.; Scuderi, C. Altered waste disposal system in aging and Alzheimer’s disease: Focus on astrocytic aquaporin-4. Front. Pharmacol. 2020, 10, 1656. [Google Scholar] [CrossRef]
- Thal, D.R.; Larionov, S.; Abramowski, D.; Wiederhold, K.H.; Van Dooren, T.; Yamaguchi, H.; Haass, C.; Van Leuven, F.; Staufenbiel, M.; Capetillo-Zarate, E. Occurrence and Co-localization of amyloid β-protein and apolipoprotein E in perivascular drainage channels of wild-type and APP-transgenic mice. Neurobiol. Aging 2007, 28, 1221–1230. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.-C.; Qiao, W.; Bu, G. Apolipoprotein E, receptors, and modulation of Alzheimer’s disease. Biol. Psychiatry 2018, 83, 347–357. [Google Scholar] [CrossRef]
- Tachibana, M.; Holm, M.L.; Liu, C.C.; Shinohara, M.; Aikawa, T.; Oue, H.; Yamazaki, Y.; Martens, Y.A.; Murray, M.E.; Sullivan, P.M.; et al. APOE4-mediated amyloid-β pathology depends on its neuronal receptor LRP1. J. Clin. Investig. 2019, 129, 1272–1277. [Google Scholar] [CrossRef]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.W.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef]
- Arastoo, M.; Lofthouse, R.; Penny, L.K.; Harrington, C.R.; Porter, A.; Wischik, C.M.; Palliyil, S. Current progress and future directions for tau-based fluid biomarker diagnostics in Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 8673. [Google Scholar] [CrossRef]
- Harrison, I.F.; Ismail, O.; Machhada, A.; Colgan, N.; Ohene, Y.; Nahavandi, P.; Ahmed, Z.; Fisher, A.; Meftah, S.; Murray, T.K.; et al. Impaired glymphatic function and clearance of tau in an Alzheimer’s disease model. Brain 2020, 143, 2576–2593. [Google Scholar] [CrossRef]
- Levine, J.; Kwon, E.; Paez, P.; Yan, W.; Czerwieniec, G.; Loo, J.A.; Sofroniew, M.V.; Wanner, I.B. Traumatically injured astrocytes release a proteomic signature modulated by STAT3-Dependent cell survival. Glia 2016, 64, 668–694. [Google Scholar] [CrossRef]
- Reichenbach, N.; Delekate, A.; Plescher, M.; Schmitt, F.; Krauss, S.; Blank, N.; Halle, A.; Petzold, G.C. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol. Med. 2019, 11, e9665. [Google Scholar] [CrossRef]
- Pan, J.; Ma, N.; Yu, B.; Zhang, W.; Wan, J. Transcriptomic profiling of microglia and astrocytes throughout aging. J. Neuroinflamm. 2020, 17, 97. [Google Scholar] [CrossRef]
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.S.; Modrusan, Z.; Beach, T.G.; Serrano, G.E.; et al. Alzheimer’s patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Rep. 2020, 31, 107843. [Google Scholar] [CrossRef]
- Han, X.; Zhang, T.; Liu, H.; Mi, Y.; Gou, X. Astrocyte senescence and Alzheimer’s disease: A review. Front. Aging Neurosci. 2020, 12, 148. [Google Scholar] [CrossRef]
- Beauquis, J.; Vinuesa, A.; Pomilio, C.; Pavía, P.; Galván, V.; Saravia, F. Neuronal and glial alterations, increased anxiety, and cognitive impairment before hippocampal amyloid deposition in PDAPP mice, model of Alzheimer’s disease. Hippocampus 2014, 24, 257–269. [Google Scholar] [CrossRef]
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 Receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef]
- Bosson, A.; Paumier, A.; Boisseau, S.; Jacquier-Sarlin, M.; Buisson, A.; Albrieux, M. TRPA1 channels promote astrocytic Ca2+ hyperactivity and synaptic dysfunction mediated by oligomeric forms of amyloid-β peptide. Mol. Neurodegener. 2017, 12, 53. [Google Scholar] [CrossRef]
- Sidoryk-Wegrzynowicz, M.; Gerber, Y.N.; Ries, M.; Sastre, M.; Tolkovsky, A.M.; Spillantini, M.G. Astrocytes in mouse models of tauopathies acquire early deficits and lose neurosupportive functions. Acta Neuropathol. Commun. 2017, 5, 89. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and microglia: In sickness and in health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Lian, H.; Litvinchuk, A.; Chiang, A.C.A.; Aithmitti, N.; Jankowsky, J.L.; Zheng, H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J. Neurosci. 2016, 36, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Iram, T.; Ramirez-Ortiz, Z.; Byrne, M.H.; Coleman, U.A.; Kingery, N.D.; Means, T.K.; Frenkel, D.; El Khoury, J. Megf10 is a receptor for C1Q that mediates clearance of apoptotic cells by astrocytes. J. Neurosci. 2016, 36, 5185–5192. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.R.; von Saucken, V.E.; Muñoz, B.; Codocedo, J.F.; Atwood, B.K.; Lamb, B.T.; Landreth, G.E. TREM2 is required for microglial instruction of astrocytic synaptic engulfment in neurodevelopment. Glia 2019, 67, 1873–1892. [Google Scholar] [CrossRef]
- Navarro, A.; Del Valle, E.; Astudillo, A.; Del Rey, C.G.; Tolivia, J. Immunohistochemical study of distribution of apolipoproteins E and D in human cerebral β amyloid deposits. Exp. Neurol. 2003, 184, 697–704. [Google Scholar] [CrossRef]
- Rodriguez, G.A.; Tai, L.M.; LaDu, M.J.; Rebeck, G.W. Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J. Neuroinflamm. 2014, 11, 111. [Google Scholar] [CrossRef]
- Wang, C.; Xiong, M.; Gratuze, M.; Bao, X.; Shi, Y.; Andhey, P.S.; Manis, M.; Schroeder, C.; Yin, Z.; Madore, C.; et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 2021, 109, 1657–1674.e7. [Google Scholar] [CrossRef]
- Meilandt, W.J.; Ngu, H.; Gogineni, A.; Lalehzadeh, G.; Lee, S.-H.; Srinivasan, K.; Imperio, J.; Wu, T.; Weber, M.; Kruse, A.J.; et al. Trem2 deletion reduces late-stage amyloid plaque accumulation, elevates the Aβ42:Aβ40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer’s mouse model. J. Neurosci. 2020, 40, 1956–1974. [Google Scholar] [CrossRef]
- Taylor, X.; Cisternas, P.; You, Y.; You, Y.; Xiang, S.; Marambio, Y.; Zhang, J.; Vidal, R.; Lasagna-Reeves, C.A. A1 reactive astrocytes and a loss of TREM2 are associated with an early stage of pathology in a mouse model of cerebral amyloid angiopathy. J. Neuroinflamm. 2020, 17, 223. [Google Scholar] [CrossRef]
- Smit, T.; Deshayes, N.A.C.; Borchelt, D.R.; Kamphuis, W.; Middeldorp, J.; Hol, E.M. Reactive astrocytes as treatment targets in Alzheimer’s disease—Systematic review of studies using the APPswePS1dE9 mouse model. Glia 2021, 69, 1852–1881. [Google Scholar] [CrossRef]
- Nasrabady, S.E.; Rizvi, B.; Goldman, J.E.; Brickman, A.M. White matter changes in Alzheimer’s disease: A focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 2018, 6, 22. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef]
- Chen, W.; Lu, A.; Craessaerts, K.; Pavie, B.; Sala Frigerio, C.; Corthout, N.; Qian, X.; Laláková, J.; Kühnemund, M.; Voytyuk, I.; et al. Spatial transcriptomics and in situ sequencing to study Alzheimer’s disease. Cell 2020, 182, 976–991.e19. [Google Scholar] [CrossRef]
- Dean, D.C.; Hurley, S.A.; Kecskemeti, S.R.; O’Grady, J.P.; Canda, C.; Davenport-Sis, N.J.; Carlsson, C.M.; Zetterberg, H.; Blennow, K.; Asthana, S.; et al. Association of amyloid pathology with myelin alteration in preclinical Alzheimer disease. JAMA Neurol. 2017, 74, 41–49. [Google Scholar] [CrossRef]
- Papuć, E.; Rejdak, K. State of the art paper the role of myelin damage in Alzheimer’s disease pathology. Arch. Med. Sci. 2020, 16, 345–351. [Google Scholar] [CrossRef]
- Wirths, O.; Weis, J.; Szczygielski, J.; Multhaup, G.; Bayer, T.A. Axonopathy in an APP/PS1 transgenic mouse model of Alzheimer’s disease. Acta Neuropathol. 2006, 111, 312–319. [Google Scholar] [CrossRef]
- Behrendt, G.; Baer, K.; Buffo, A.; Curtis, M.A.; Faull, R.L.; Rees, M.I.; Götz, M.; Dimou, L. Dynamic changes in myelin aberrations and oligodendrocyte generation in chronic amyloidosis in mice and men. Glia 2013, 61, 273–286. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, Y.; Liu, Z.; Geng, Q.; Chen, Z.; Zhang, Y. Alterations of myelin morphology and oligodendrocyte development in early stage of Alzheimer’s disease mouse model. Neurosci. Lett. 2017, 642, 102–106. [Google Scholar] [CrossRef]
- Chu, T.H.; Cummins, K.; Sparling, J.S.; Tsutsui, S.; Brideau, C.; Nilsson, K.P.R.; Joseph, J.T.; Stys, P.K. Axonal and myelinic pathology in 5xFAD Alzheimer’s mouse spinal cord. PLoS ONE 2017, 12, e0188218. [Google Scholar] [CrossRef]
- Gu, L.; Wu, D.; Tang, X.; Qi, X.; Li, X.; Bai, F.; Chen, X. Myelin changes at the early stage of 5XFAD mice. Brain Res. Bull. 2018, 137, 285–293. [Google Scholar] [CrossRef]
- Wu, D.; Tang, X.; Gu, L.H.; Li, X.L.; Qi, X.Y.; Bai, F.; Chen, X.C.; Wang, J.Z.; Ren, Q.G.; Zhang, Z.J. LINGO-1 antibody ameliorates myelin impairment and spatial memory deficits in the early stage of 5XFAD mice. CNS Neurosci. Ther. 2018, 24, 381–393. [Google Scholar] [CrossRef]
- Ferreira, S.; Pitman, K.A.; Wang, S.; Summers, B.S.; Bye, N.; Young, K.M.; Cullen, C.L. Amyloidosis is associated with thicker myelin and increased oligodendrogenesis in the adult mouse brain. J. Neurosci. Res. 2020, 98, 1905–1932. [Google Scholar] [CrossRef]
- Sahara, N.; Perez, P.D.; Lin, W.-L.; Dickson, D.W.; Ren, Y.; Zeng, H.; Lewis, J.; Febo, M. Age-related decline in white matter integrity in a mouse model of tauopathy: An in vivo diffusion tensor magnetic resonance imaging study. Neurobiol. Aging 2014, 35, 1364–1374. [Google Scholar] [CrossRef]
- Jackson, J.; Bianco, G.; Rosa, A.O.; Cowan, K.; Bond, P.; Anichtchik, O.; Fern, R. White matter tauopathy: Transient functional loss and novel myelin remodeling. Glia 2018, 66, 813–827. [Google Scholar] [CrossRef]
- Higuchi, M.; Zhang, B.; Forman, M.S.; Yoshiyama, Y.; Trojanowski, J.Q.; Lee, V.M. Axonal degeneration induced by targeted expression of mutant human tau in oligodendrocytes of transgenic mice that model glial tauopathies. J. Neurosci. 2005, 25, 9434–9443. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Lin, W.L.; Sanchez, L.; Ceballos, C.; Polydoro, M.; Spires-Jones, T.L.; Hyman, B.T.; Dickson, D.W.; Sahara, N. Endogenous tau aggregates in oligodendrocytes of RTg4510 mice induced by human P301L tau. J. Alzheimer’s Dis. 2014, 38, 589–600. [Google Scholar] [CrossRef]
- Desai, M.K.; Sudol, K.L.; Janelsins, M.C.; Mastrangelo, M.A.; Frazer, M.E.; Bowers, W.J. Triple-transgenic Alzheimer’s disease mice exhibit region-specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia 2009, 57, 54–65. [Google Scholar] [CrossRef]
- Desai, M.K.; Mastrangelo, M.A.; Ryan, D.A.; Sudol, K.L.; Narrow, W.C.; Bowers, W.J. Early oligodendrocyte/myelin pathology in Alzheimer’s disease mice constitutes a novel therapeutic target. Am. J. Pathol. 2010, 177, 1422–1435. [Google Scholar] [CrossRef]
- Vanzulli, I.; Papanikolaou, M.; De-La-Rocha, I.C.; Pieropan, F.; Rivera, A.D.; Gomez-Nicola, D.; Verkhratsky, A.; Rodríguez, J.J.; Butt, A.M. Disruption of oligodendrocyte progenitor cells is an early sign of pathology in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2020, 94, 130–139. [Google Scholar] [CrossRef]
- Han, X.; Holtzman, D.M.; McKeel, D.W.; Kelley, J.; Morris, J.C. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: Potential role in disease pathogenesis. J. Neurochem. 2002, 82, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.X.; Zhang, H.Y.; Li, H.Y.; Liu, P.H.; Sui, Y.; Sun, X.H. Association between Alzheimer’s disease pathogenesis and early demyelination and oligodendrocyte dysfunction. Neural Regen. Res. 2018, 13, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Teo, J.D.; Song, H.; McEwen, H.P.; Yup Lee, J.; Couttas, T.A.; Duncan, T.; Chesworth, R.; Bertz, J.; Przybyla, M.; et al. Sphingosine kinase 2 potentiates amyloid deposition but protects against hippocampal volume loss and demyelination in a mouse model of Alzheimer’s disease. J. Neurosci. 2019, 39, 9645–9659. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Li, W.; Tang, Y.; Fan, Z.; Meng, Y.; Yang, G.; Luo, J.; Ke, Z.J. Autophagy is involved in oligodendroglial precursor-mediated clearance of amyloid peptide. Mol. Neurodegener. 2013, 8, 27. [Google Scholar] [CrossRef]
- Cha, M.-Y.; Kwon, Y.-W.; Ahn, H.-S.; Jeong, H.; Lee, Y.Y.; Moon, M.; Baik, S.H.; Kim, D.K.; Song, H.; Yi, E.C.; et al. Protein-induced pluripotent stem cells ameliorate cognitive dysfunction and reduce Aβ deposition in a mouse model of Alzheimer’s disease. Stem Cells Transl. Med. 2017, 6, 293–305. [Google Scholar] [CrossRef]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Kamphuis, W.; Orre, M.; Kooijman, L.; Dahmen, M.; Hol, E.M. Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer’s disease mouse model. Glia 2012, 60, 615–629. [Google Scholar] [CrossRef]
- Honjo, Y.; Ayaki, T.; Tomiyama, T.; Horibe, T.; Ito, H.; Mori, H.; Takahashi, R.; Kawakami, K. Increased GADD34 in oligodendrocytes in Alzheimer’s disease. Neurosci. Lett. 2015, 602, 50–55. [Google Scholar] [CrossRef]
- Ossola, B.; Zhao, C.; Compston, A.; Pluchino, S.; Franklin, R.J.M.; Spillantini, M.G. Neuronal expression of pathological tau accelerates oligodendrocyte progenitor cell differentiation. Glia 2016, 64, 457–471. [Google Scholar] [CrossRef]
- Ferreira, S.; Pitman, K.A.; Summers, B.S.; Wang, S.; Young, K.M.; Cullen, C.L. Oligodendrogenesis increases in hippocampal grey and white matter prior to locomotor or memory impairment in an adult mouse model of tauopathy. Eur. J. Neurosci. 2020, 54, 5762–5784. [Google Scholar] [CrossRef]
- Desai, M.K.; Guercio, B.J.; Narrow, W.C.; Bowers, W.J. An Alzheimer’s disease-relevant presenilin-1 mutation augments amyloid-beta-induced oligodendrocyte dysfunction. Glia 2011, 59, 627–640. [Google Scholar] [CrossRef]
- Kastyak-Ibrahim, M.Z.; Di Curzio, D.L.; Buist, R.; Herrera, S.L.; Albensi, B.C.; Del Bigio, M.R.; Martin, M. Neurofibrillary tangles and plaques are not accompanied by white matter pathology in aged triple transgenic-Alzheimer disease mice. Magn. Reson. Imaging 2013, 31, 1515–1521. [Google Scholar] [CrossRef]
- Bagyinszky, E.; Giau, V.V.; An, S.A. Transcriptomics in Alzheimer’s disease: Aspects and challenges. Int. J. Mol. Sci. 2020, 21, 3517. [Google Scholar] [CrossRef]
- Hampel, H.; Nisticò, R.; Seyfried, N.T.; Levey, A.I.; Modeste, E.; Lemercier, P.; Baldacci, F.; Toschi, N.; Garaci, F.; Perry, G.; et al. Omics sciences for systems biology in Alzheimer’s disease: State-of-the-art of the evidence. Ageing Res. Rev. 2021, 69, 101346. [Google Scholar] [CrossRef]
- Wan, Y.W.; Al-Ouran, R.; Mangleburg, C.G.; Perumal, T.M.; Lee, T.V.; Allison, K.; Swarup, V.; Funk, C.C.; Gaiteri, C.; Allen, M.; et al. Meta-analysis of the Alzheimer’s disease human brain transcriptome and functional dissection in mouse models. Cell Rep. 2020, 32, 107908. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef]
- Rexach, J.E.; Polioudakis, D.; Yin, A.; Swarup, V.; Chang, T.S.; Nguyen, T.; Sarkar, A.; Chen, L.; Huang, J.; Lin, L.C.; et al. Tau pathology drives dementia risk-associated gene networks toward chronic inflammatory states and immunosuppression. Cell Rep. 2020, 33, 108398. [Google Scholar] [CrossRef]
- Kim, D.K.; Park, J.; Han, D.; Yang, J.; Kim, A.; Woo, J.; Kim, Y.; Mook-Jung, I. Molecular and functional signatures in a novel Alzheimer’s disease mouse model assessed by quantitative proteomics. Mol. Neurodegener. 2018, 13, 2. [Google Scholar] [CrossRef]
- Hedl, T.J.; Gil, R.S.; Cheng, F.; Rayner, S.L.; Davidson, J.M.; De Luca, A.; Villalva, M.D.; Ecroyd, H.; Walker, A.K.; Lee, A. Proteomics approaches for biomarker and drug target discovery in ALS and FTD. Front. Neurosci. 2019, 13, 548. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez-Varo, R.; Mejias-Ortega, M.; Fernandez-Valenzuela, J.J.; Nuñez-Diaz, C.; Caceres-Palomo, L.; Vegas-Gomez, L.; Sanchez-Mejias, E.; Trujillo-Estrada, L.; Garcia-Leon, J.A.; Moreno-Gonzalez, I.; et al. Transgenic Mouse Models of Alzheimer’s Disease: An Integrative Analysis. Int. J. Mol. Sci. 2022, 23, 5404. https://doi.org/10.3390/ijms23105404
Sanchez-Varo R, Mejias-Ortega M, Fernandez-Valenzuela JJ, Nuñez-Diaz C, Caceres-Palomo L, Vegas-Gomez L, Sanchez-Mejias E, Trujillo-Estrada L, Garcia-Leon JA, Moreno-Gonzalez I, et al. Transgenic Mouse Models of Alzheimer’s Disease: An Integrative Analysis. International Journal of Molecular Sciences. 2022; 23(10):5404. https://doi.org/10.3390/ijms23105404
Chicago/Turabian StyleSanchez-Varo, Raquel, Marina Mejias-Ortega, Juan Jose Fernandez-Valenzuela, Cristina Nuñez-Diaz, Laura Caceres-Palomo, Laura Vegas-Gomez, Elisabeth Sanchez-Mejias, Laura Trujillo-Estrada, Juan Antonio Garcia-Leon, Ines Moreno-Gonzalez, and et al. 2022. "Transgenic Mouse Models of Alzheimer’s Disease: An Integrative Analysis" International Journal of Molecular Sciences 23, no. 10: 5404. https://doi.org/10.3390/ijms23105404
APA StyleSanchez-Varo, R., Mejias-Ortega, M., Fernandez-Valenzuela, J. J., Nuñez-Diaz, C., Caceres-Palomo, L., Vegas-Gomez, L., Sanchez-Mejias, E., Trujillo-Estrada, L., Garcia-Leon, J. A., Moreno-Gonzalez, I., Vizuete, M., Vitorica, J., Baglietto-Vargas, D., & Gutierrez, A. (2022). Transgenic Mouse Models of Alzheimer’s Disease: An Integrative Analysis. International Journal of Molecular Sciences, 23(10), 5404. https://doi.org/10.3390/ijms23105404