
Impact of Cerebral Amyloid Angiopathy in Two Transgenic Mouse Models of Cerebral β-Amyloidosis: A Neuropathological Study

 ,
, 
 ,
,  and
and
Abstract
1. Introduction
2. Results
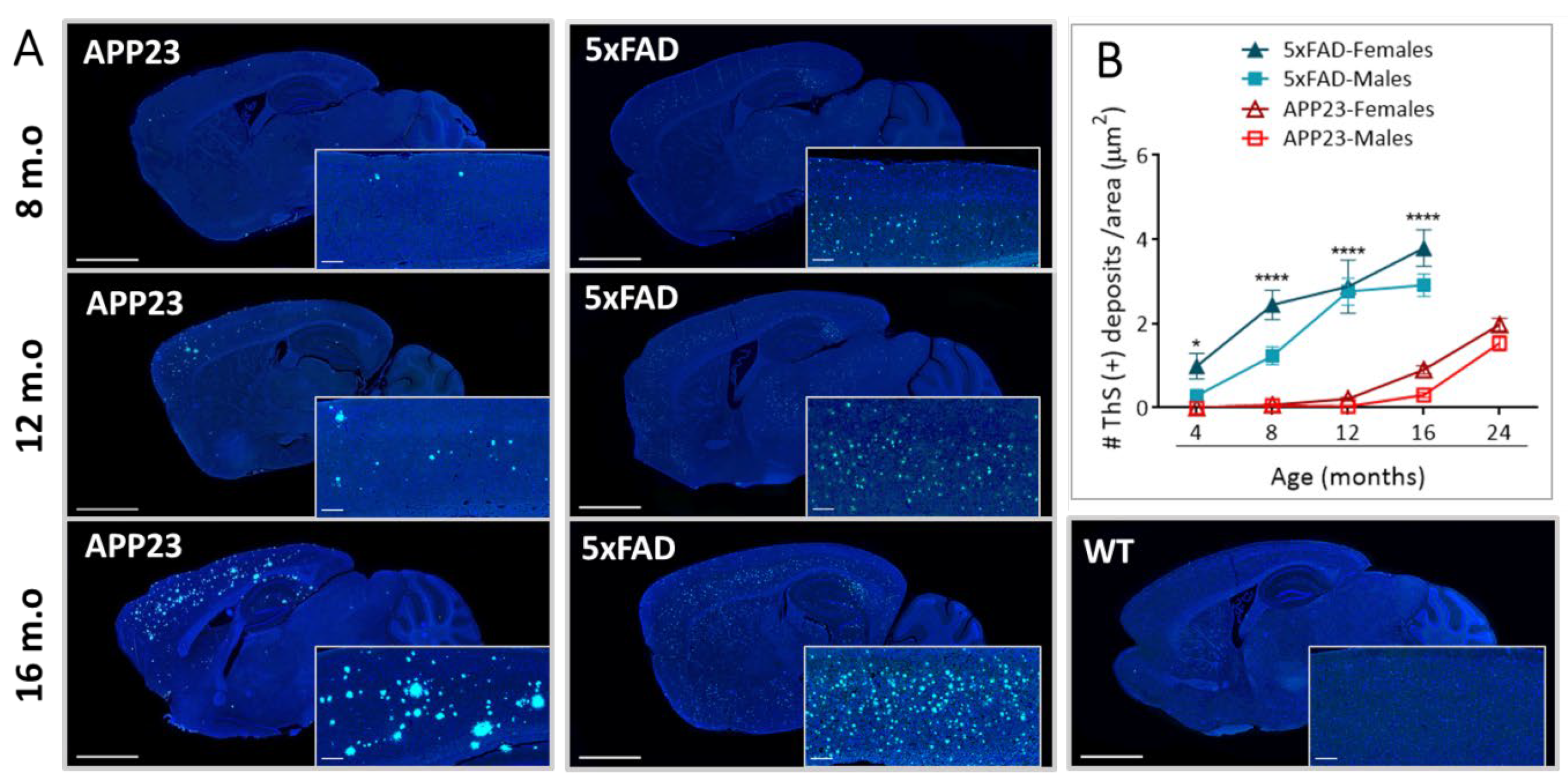
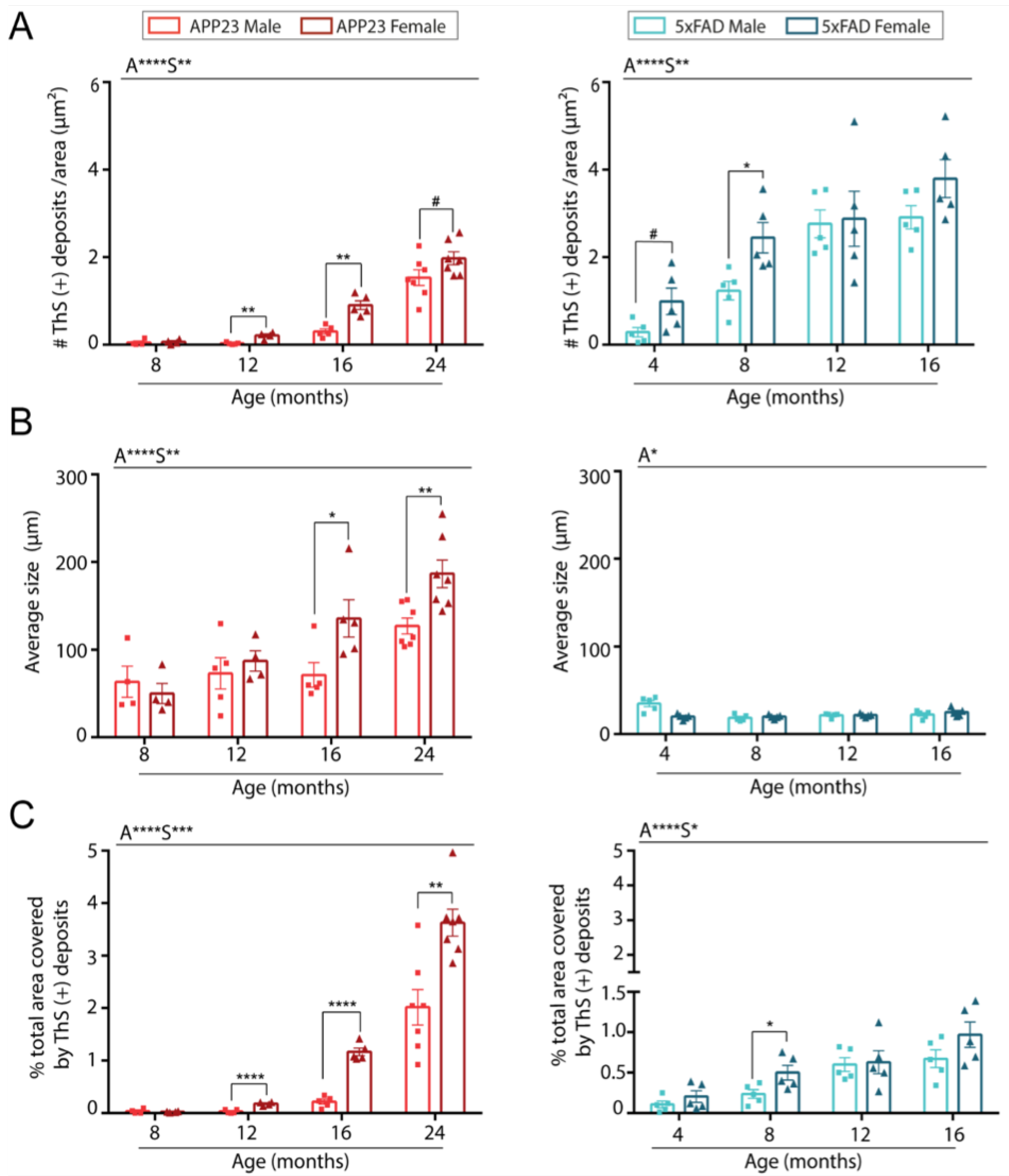
2.1. Levels of Parenchymal Aβ Deposition
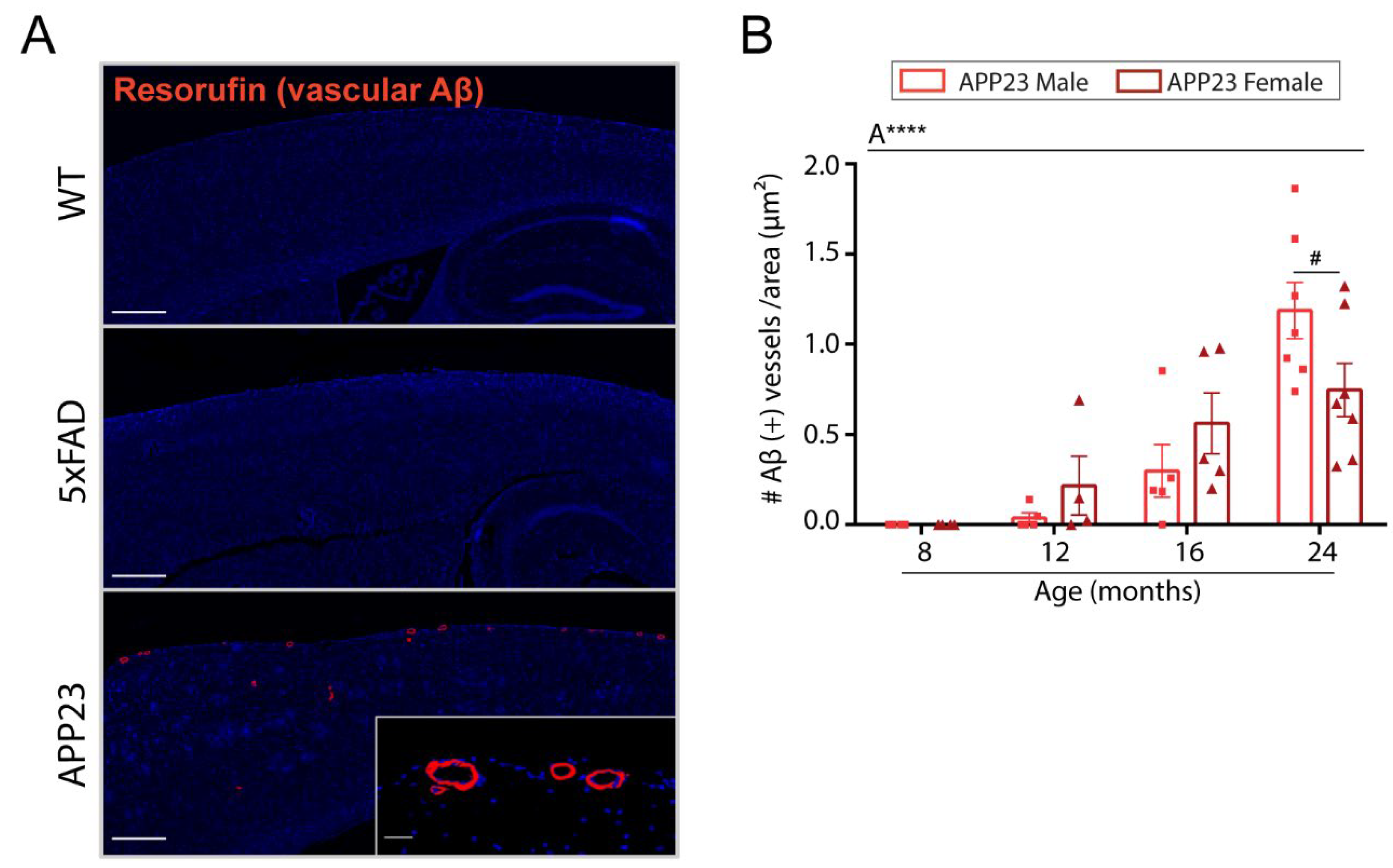
2.2. Levels of Vascular Aβ Deposition
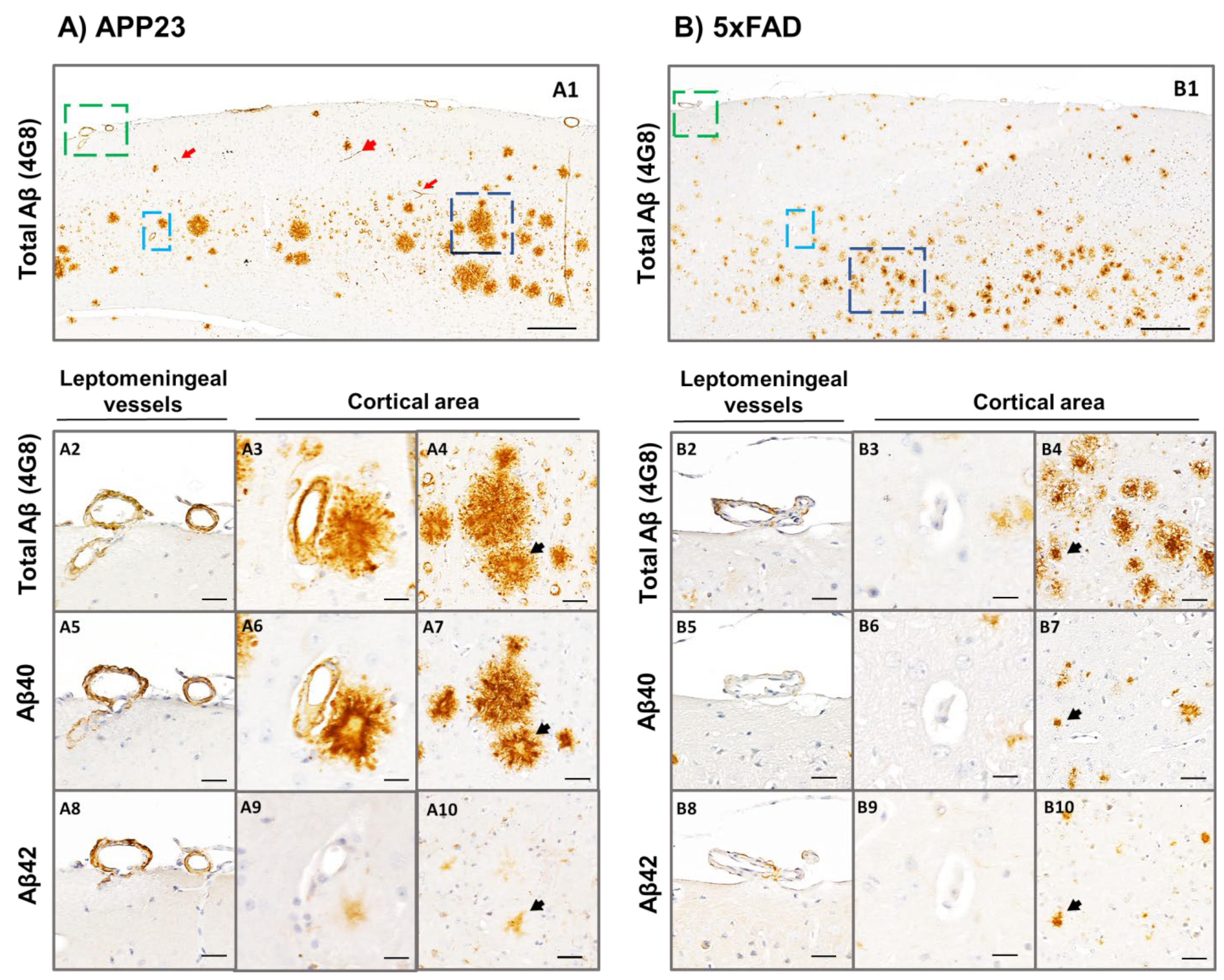
2.3. Analysis of Aβ40 and Aβ42 Deposition
2.4. Determination of CAA-Related Cerebral Microbleeds
3. Discussion
4. Material and Methods
4.1. Transgenic Animals
4.2. Tissue Processing for Histological Analysis
4.3. Tissue Processing for Magnetic Resonance Imaging
4.4. MRI Protocol and Analysis
4.5. Thioflavin S Staining
4.6. Resorufin Staining
4.7. Aβ Peptide Immunohistochemistry
4.8. Prussian Blue Staining
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weller, R.O.; Subash, M.; Preston, S.; Mazanti, I.; Carare, R.O. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 2008, 18, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.A.; Jayakody, N.; Johnston, D.A.; Bechmann, I.; Carare, R.O. Failure of Perivascular Drainage of β-amyloid in Cerebral Amyloid Angiopathy. Brain Pathol. 2014, 24, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Farid, K.; Charidimou, A.; Baron, J.-C. Amyloid positron emission tomography in sporadic cerebral amyloid angiopathy: A systematic critical update. NeuroImage: Clin. 2017, 15, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.T.; Bondolfi, L.; Herzig, M.C.; Jann, L.; Calhoun, M.E.; Wiederhold, K.-H.; Tolnay, M.; Staufenbiel, M.; Jucker, M. Spontaneous Hemorrhagic Stroke in a Mouse Model of Cerebral Amyloid Angiopathy. J. Neurosci. 2001, 21, 1619–1627. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Staufenbiel, M. Pathogenic Mechanisms of Alzheimer’s Disease Analyzed in the APP23 Transgenic Mouse Model. Ann. New York Acad. Sci. 2000, 920, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.; Mcgonigle, P. Overview of Transgenic Mouse Models for Alzheimer’s Disease. Curr. Protoc. Neurosci. 2019, 89, e81. [Google Scholar] [CrossRef]
- Eede, P.; Obst, J.; Benke, E.; Yvon-Durocher, G.; Richard, B.C.; Gimber, N.; Schmoranzer, J.; Böddrich, A.; Wanker, E.E.; Prokop, S.; et al. Interleukin-12/23 deficiency differentially affects pathology in male and female Alzheimer’s disease-like mice. EMBO Rep. 2020, 21, e48530. [Google Scholar] [CrossRef]
- Bundy, J.L.; Vied, C.; Badger, C.; Nowakowski, R.S. Sex-biased hippocampal pathology in the 5XFAD mouse model of Alzheimer’s disease: A multi-omic analysis. J. Comp. Neurol. 2018, 527, 462–475. [Google Scholar] [CrossRef]
- Barnes, L.L.; Wilson, R.S.; Bienias, J.L.; Schneider, J.A.; Evans, D.A.; Bennett, D.A. Sex Differences in the Clinical Manifestations of Alzheimer Disease Pathology. Arch. Gen. Psychiatry 2005, 62, 685–691. [Google Scholar] [CrossRef]
- Corder, E.H.; Ghebremedhin, E.; Taylor, M.G.; Thal, D.R.; Ohm, T.G.; Braak, H. The Biphasic Relationship between Regional Brain Senile Plaque and Neurofibrillary Tangle Distributions: Modification by Age, Sex, and APOEPolymorphism. Ann. New York Acad. Sci. 2004, 1019, 24–28. [Google Scholar] [CrossRef]
- Nebel, R.A.; Aggarwal, N.T.; Barnes, L.L.; Gallagher, A.; Goldstein, J.M.; Kantarci, K.; Mallampalli, M.P.; Mormino, E.C.; Scott, L.; Yu, W.H.; et al. Understanding the impact of sex and gender in Alzheimer’s disease: A call to action. Alzheimer’s Dement. 2018, 14, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, A.D.M.; Silva, R.H. Sex Differences in Alzheimer’s Disease: Where Do We Stand? J. Alzheimer’s Dis. 2019, 67, 35–60. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.W.; Bennett, D.A.; Dong, H. Sexual dimorphism in predisposition to Alzheimer’s disease. Neurobiol. Aging 2018, 70, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Scheyer, O.; Rahman, A.; Hristov, H.; Berkowitz, C.; Isaacson, R.S.; Diaz Brinton, R.; Mosconi, L. Female Sex and Alzheimer’s Risk: The Menopause Connection. J. Prev. Alzheimers Dis. 2018, 5, 225–230. [Google Scholar] [CrossRef]
- Manson, J.E.; Aragaki, A.K.; Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; Lacroix, A.Z.; Chlebowski, R.T.; Howard, B.V.; Thomson, C.A.; Margolis, K.; et al. Menopausal hormone therapy and long-term all-cause and cause-specific mortality: The Women’s Health Initiative randomized trials. JAMA—J. Am. Med. Assoc. 2017, 318, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Savolainen-Peltonen, H.; Rahkola-Soisalo, P.; Hoti, F.; Vattulainen, P.; Gissler, M.; Ylikorkala, O.; Mikkola, T.S. Use of postmenopausal hormone therapy and risk of Alzheimer’s disease in Finland: Nationwide case-control study. BMJ 2019, 364, l665. [Google Scholar] [CrossRef]
- Pike, C.J.; Carroll, J.C.; Rosario, E.R.; Barron, A. Protective actions of sex steroid hormones in Alzheimer’s disease. Front. Neuroendocr. 2009, 30, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Rosario, E.R.; Carroll, J.; Pike, C.J. Testosterone regulation of Alzheimer-like neuropathology in male 3xTg-AD mice involves both estrogen and androgen pathways. Brain Res. 2010, 1359, 281–290. [Google Scholar] [CrossRef]
- Lau, C.-F.; Ho, Y.-S.; Hung, C.H.-L.; Wuwongse, S.; Poon, C.-H.; Chiu, K.; Yang, X.; Chu, L.-W.; Chang, R.C.-C. Protective Effects of Testosterone on Presynaptic Terminals against Oligomericβ-Amyloid Peptide in Primary Culture of Hippocampal Neurons. BioMed Res. Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Giannoni, P.; Arango-Lievano, M.; Das Neves, I.; Rousset, M.-C.; Baranger, K.; Rivera, S.; Jeanneteau, F.; Claeysen, S.; Marchi, N. Cerebrovascular pathology during the progression of experimental Alzheimer’s disease. Neurobiol. Dis. 2016, 88, 107–117. [Google Scholar] [CrossRef]
- Xu, F.; Kotarba, A.E.; Ou-Yang, M.-H.; Fu, Z.; Davis, J.; Smith, S.O.; Van Nostrand, W.E. Early-onset Formation of Parenchymal Plaque Amyloid Abrogates Cerebral Microvascular Amyloid Accumulation in Transgenic Mice. J. Biol. Chem. 2014, 289, 17895–17908. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Imaizumi, T.; Moulin, S.; Biffi, A.; Samarasekera, N.; Yakushiji, Y.; Peeters, A.; Vandermeeren, Y.; Laloux, P.; Baron, J.-C.; et al. Brain hemorrhage recurrence, small vessel disease type, and cerebral microbleeds. Neurology 2017, 89, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Reuter, B.; Venus, A.; Heiler, P.; Schad, L.; Ebert, A.; Hennerici, M.G.; Grudzenski, S.; Fatar, M. Development of Cerebral Microbleeds in the APP23-Transgenic Mouse Model of Cerebral Amyloid Angiopathy—A 9.4 Tesla MRI Study. Front. Aging Neurosci. 2016, 8, 170. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, N.; Doelemeyer, A.; Zurbruegg, S.; Bigot, K.; Theil, D.; Frieauff, W.; Kolly, C.; Moulin, P.; Neddermann, D.; Kreutzer, R.; et al. Longitudinal noninvasive magnetic resonance imaging of brain microhemorrhages in BACE inhibitor–treated APP transgenic mice. Neurobiol. Aging 2016, 45, 50–60. [Google Scholar] [CrossRef]
- Beckmann, N.; Schuler, A.; Mueggler, T.; Meyer, E.P.; Wiederhold, K.-H.; Staufenbiel, M.; Krucker, T. Age-Dependent Cerebrovascular Abnormalities and Blood Flow Disturbances in APP23 Mice Modeling Alzheimer’s Disease. J. Neurosci. 2003, 23, 8453–8459. [Google Scholar] [CrossRef]
- Herzig, M.; Winkler, D.T.; Burgermeister, P.; Pfeifer, M.; Kohler, E.; Schmidt, S.D.; Danner, S.; Abramowski, D.; Stürchler-Pierrat, C.; Bürki, K.; et al. Aβ is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 2004, 7, 954–960. [Google Scholar] [CrossRef]
- Davis, J.; Xu, F.; Deane, R.; Romanov, G.; Previti, M.L.; Zeigler, K.; Zlokovic, B.V.; Van Nostrand, W.E. Early-onset and Robust Cerebral Microvascular Accumulation of Amyloid β-Protein in Transgenic Mice Expressing Low Levels of a Vasculotropic Dutch/Iowa Mutant Form of Amyloid β-Protein Precursor. J. Biol. Chem. 2004, 279, 20296–20306. [Google Scholar] [CrossRef]
- Cacciottolo, M.; Christensen, A.; Moser, A.; Liu, J.; Pike, C.; Smith, C.; LaDu, M.J.; Sullivan, P.M.; Morgan, T.E.; Dolzhenko, E.; et al. The APOE4 allele shows opposite sex bias in microbleeds and Alzheimer’s disease of humans and mice. Neurobiol. Aging 2015, 37, 47–57. [Google Scholar] [CrossRef]
- Cacciottolo, M.; Morgan, T.E.; Finch, C.E. Age, sex, and cerebral microbleeds in EFAD Alzheimer disease mice. Neurobiol. Aging 2021, 103, 42–51. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Briggs, M.E.; Hyman, B.T.; Kokoris, G.J.; Takis, C.; Kanter, D.S.; Kase, C.S.; Pessin, M.S. Apolipoprotein E ε4 Is Associated With the Presence and Earlier Onset of Hemorrhage in Cerebral Amyloid Angiopathy. Stroke 1996, 27, 1333–1337. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, J.T.; Wang, H.F.; Han, P.R.; Tan, C.C.; Wang, C.; Meng, X.F.; Risacher, S.L.; Saykin, A.J.; Tan, L. APOE genotype and neuroimaging markers of Alzheimer’s disease: Systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2015, 86, 127–134. [Google Scholar] [CrossRef]
- Biffi, A.; Sonni, A.; Anderson, C.D.; Kissela, B.; Jagiella, J.M.; Schmidt, H.; Jimenez-Conde, J.; Bs, B.M.H.; Fernandez-Cadenas, I.; Msc, L.C.; et al. Variants at APOE influence risk of deep and lobar intracerebral hemorrhage. Ann. Neurol. 2010, 68, 934–943. [Google Scholar] [CrossRef]
- Han, B.H.; Zhou, M.L.; Vellimana, A.K.; Milner, E.; Kim, D.H.; Greenberg, J.K.; Chu, W.; Mach, R.H.; Zipfel, G.J. Resorufin analogs preferentially bind cerebrovascular amyloid: Potential use as imaging ligands for cerebral amyloid angiopathy. Mol. Neurodegener. 2011, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2017, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Attems, J.; Jellinger, K.; Thal, D.R.; Van Nostrand, W. Review: Sporadic cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 2011, 37, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, E.S.; Gurol, M.E.; Van Der Grond, J.; Haan, J.; Viswanathan, A.; Schwab, K.M.; Ayres, A.M.; Algra, A.; Rosand, J.; Van Buchem, M.A.; et al. Recurrent hemorrhage risk and mortality in hereditary and sporadic cerebral amyloid angiopathy. Neurology 2016, 87, 1482–1487. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Boulouis, G.; Gurol, M.E.; Ayata, C.; Bacskai, B.J.; Frosch, M.P.; Viswanathan, A.; Greenberg, S.M. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 2017, 140, 1829–1850. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Frosch, M.P.; Salman, R.A.-S.; Baron, J.-C.; Cordonnier, C.; Hernandez-Guillamon, M.; Linn, J.; Raposo, N.; Rodrigues, M.; Romero, J.R.; et al. Advancing diagnostic criteria for sporadic cerebral amyloid angiopathy: Study protocol for a multicenter MRI-pathology validation of Boston criteria v2.0. Int. J. Stroke 2019, 14, 956–971. [Google Scholar] [CrossRef]
- Vinters, H.V. Cerebral amyloid angiopathy. A critical review. Stroke 1987, 18, 311–324. [Google Scholar] [CrossRef]
- Boulouis, G.; Charidimou, A.; Greenberg, S.M. Sporadic Cerebral Amyloid Angiopathy: Pathophysiology, Neuroimaging Features, and Clinical Implications. Semin. Neurol. 2016, 36, 233–243. [Google Scholar] [CrossRef]
- Weller, R.O.; Preston, S.; Subash, M.; Carare, R.O.; Weller, R.O.; Preston, S.; Subash, M.; Carare, R.O. Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimer’s Res. Ther. 2009, 1, 6. [Google Scholar] [CrossRef]
- Keage, H.A.; Carare, R.O.; Friedland, R.P.; Ince, P.G.; Love, S.; A Nicoll, J.; Wharton, S.B.; Weller, R.O.; Brayne, C. Population studies of sporadic cerebral amyloid angiopathy and dementia: A systematic review. BMC Neurol. 2009, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Vemuri, P.; Rocca, W.A. Clinical Epidemiology of Alzheimer’s Disease: Assessing Sex and Gender Differences. Clin. Epidemiol. 2014, 2014, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Mazure, C.M.; Swendsen, J. Sex differences in Alzheimer’s disease and other dementias. Lancet Neurol. 2016, 15, 451–452. [Google Scholar] [CrossRef]
- Mayeda, E.R. Invited Commentary: Examining Sex/Gender Differences in Risk of Alzheimer Disease and Related Dementias—Challenges and Future Directions. Am. J. Epidemiol. 2019, 188, 1224–1227. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.; Robinson, A.; Snowden, J.; Davidson, Y.S.; Mann, D.M.A. Patterns of cerebral amyloid angiopathy define histopathological phenotypes in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2014, 40, 136–148. [Google Scholar] [CrossRef]
- Jäkel, L.; De Kort, A.M.; Klijn, C.J.; Schreuder, F.H.; Verbeek, M.M. Prevalence of cerebral amyloid angiopathy: A systematic review and meta-analysis. Alzheimers Dement. 2022, 18, 10–28. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef]
- Suzuki, N.; Iwatsubo, T.; Odaka, A.; Ishibashi, Y.; Kitada, C.; Ihara, Y. High tissue content of soluble β1-40 is linked to cerebral amyloid angiopathy. Am. J. Pathol. 1994, 145, 452. [Google Scholar]
- Viswanathan, A.; Greenberg, S.M. Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 2011, 70, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—one peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Howard, R.; Liu, K.Y. Questions EMERGE as Biogen claims aducanumab turnaround. Nat. Rev. Neurol. 2019, 16, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Jack, C.R., Jr.; Black, S.E.; Frosch, M.P.; Greenberg, S.M.; Hyman, B.T.; Scheltens, P.; Carrillo, M.C.; Thies, W.; Bednar, M.M.; et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimer’s Dement. 2011, 7, 367–385. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Fox, N.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.; Ferris, S.; et al. Two Phase 3 Trials of Bapineuzumab in Mild-to-Moderate Alzheimer’s Disease. New Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef]
- DiFrancesco, J.C.; Longoni, M.; Piazza, F. Anti-Aβ autoantibodies in amyloid related imaging abnormalities (ARIA): Candidate biomarker for immunotherapy in Alzheimer’s disease and cerebral amyloid angiopathy. Front. Neurol. 2015, 6, 207. [Google Scholar] [CrossRef]
- Biffi, A.; Halpin, A.; Towfighi, A.; Gilson, A.; Busl, K.; Rost, N.; Smith, E.E.; Greenberg, M.S.; Rosand, J.; Viswanathan, A. Aspirin and recurrent intracerebral hemorrhage in cerebral amyloid angiopathy. Neurology 2010, 75, 693–698. [Google Scholar] [CrossRef]
- Weber, S.A.; Patel, R.K.; Lutsep, H.L. Cerebral amyloid angiopathy: Diagnosis and potential therapies. Expert Rev. Neurother. 2018, 18, 503–513. [Google Scholar] [CrossRef]
- Gatti, L.; Tinelli, F.; Scelzo, E.; Arioli, F.; Di Fede, G.; Obici, L.; Pantoni, L.; Giaccone, G.; Caroppo, P.; Parati, E.A.; et al. Understanding the Pathophysiology of Cerebral Amyloid Angiopathy. Int. J. Mol. Sci. 2020, 21, 3435. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N. Animal Models of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006320. [Google Scholar] [CrossRef]
- Jäkel, L.; Van Nostrand, W.E.; Nicoll, J.A.R.; Werring, D.J.; Verbeek, M.M. Animal models of cerebral amyloid angiopathy. Clin. Sci. 2017, 131, 2469–2488. [Google Scholar] [CrossRef]
- Van Nostrand, W.E.; Melchor, J.P.; Romanov, G.; Zeigler, K.; Davis, J. Pathogenic Effects of Cerebral Amyloid Angiopathy Mutations in the Amyloid β-Protein Precursor. Ann. N. Y. Acad. Sci. 2002, 977, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, L.; Van Duijn, C.M.; Cras, P.; Cruts, M.; Van Hul, W.; Van Harskamp, F.; Warren, A.; McInnis, M.G.; Antonarakis, S.; Martin, J.-J.; et al. Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the β–amyloid precursor protein gene. Nat. Genet. 1992, 1, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, T.J.; Cho, H.S.; Vonsattel, J.P.G.; Rebeck, G.W.; Greenberg, S.M. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 2001, 49, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.-H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.; Bondolfi, L.; Hunziker, D.; Schlecht, H.-P.; Carver, K.; Maguire, E.; Abramowski, D.; Wiederhold, K.-H.; Sturchler-Pierrat, C.; Jucker, M.; et al. Progressive age-related impairment of cognitive behavior in APP23 transgenic mice. Neurobiol. Aging 2003, 24, 365–378. [Google Scholar] [CrossRef]
- Giménez-Llort, L.; Marin-Pardo, D.; Marazuela, P.; Hernández-Guillamón, M. Survival Bias and Crosstalk between Chronological and Behavioral Age: Age- and Genotype-Sensitivity Tests Define Behavioral Signatures in Middle-Aged, Old, and Long-Lived Mice with Normal and AD-Associated Aging. Biomedicines 2021, 9, 636. [Google Scholar] [CrossRef]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 196.e29–196.e40. [Google Scholar] [CrossRef]
- Calhoun, M.E.; Burgermeister, P.; Phinney, A.L.; Stalder, M.; Tolnay, M.; Wiederhold, K.-H.; Abramowski, D.; Sturchler-Pierrat, C.; Sommer, B.; Staufenbiel, M.; et al. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc. Natl. Acad. Sci. USA 1999, 96, 14088–14093. [Google Scholar] [CrossRef]
- Wattendorff, A.R.; Frangione, B.; Luyendijk, W.; Bots, G.T. Hereditary cerebral haemorrhage with amyloidosis, Dutch type (HCHWA-D): Clinicopathological studies. J. Neurol. Neurosurg. Psychiatry 1995, 58, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Nochlin, D.; Bird, T.D.; Nemens, E.J.; Ball, M.J.; Sumi, S.M. Amyloid angiopathy in a volga german family with Alzheimer’s disease and a presenilin-2 mutation (N141I). Ann. Neurol. 1998, 43, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Dermaut, B.; Kumar-Singh, S.; De Jonghe, C.; Cruts, M.; Löfgren, A.; Lübke, U.; Cras, P.; Dom, R.; De Deyn, P.P.; Martin, J.J.; et al. Cerebral amyloid angiopathy is a pathogenic lesion in Alzheimer’s disease due to a novel presenilin 1 mutation. Brain 2001, 124, 2383–2392. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT (n = 7) | 5xFAD (n = 4) | APP23 (n = 7) | p-Value | |
|---|---|---|---|---|
| Total cMBs | 0 | 0.50 (0–1.75) $ | 6 (4–8)** | <0.001 |
| Size (µm) | 0 | 195.91 (48.44–249.25) ** | 231.09 (200.13–261.58) *** | <0.001 |
| Volume (mm3) | 0 | 0.01 (0.002–0.015) | 0.01 (0.009–0.016) *** | <0.001 |
| Hemorrhagic area (mm2) | 0 | 1.47 (0–7.22) $ | 28.31 (14.68–79.82) ** | <0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marazuela, P.; Paez-Montserrat, B.; Bonaterra-Pastra, A.; Solé, M.; Hernández-Guillamon, M. Impact of Cerebral Amyloid Angiopathy in Two Transgenic Mouse Models of Cerebral β-Amyloidosis: A Neuropathological Study. Int. J. Mol. Sci. 2022, 23, 4972. https://doi.org/10.3390/ijms23094972
Marazuela P, Paez-Montserrat B, Bonaterra-Pastra A, Solé M, Hernández-Guillamon M. Impact of Cerebral Amyloid Angiopathy in Two Transgenic Mouse Models of Cerebral β-Amyloidosis: A Neuropathological Study. International Journal of Molecular Sciences. 2022; 23(9):4972. https://doi.org/10.3390/ijms23094972
Chicago/Turabian StyleMarazuela, Paula, Berta Paez-Montserrat, Anna Bonaterra-Pastra, Montse Solé, and Mar Hernández-Guillamon. 2022. "Impact of Cerebral Amyloid Angiopathy in Two Transgenic Mouse Models of Cerebral β-Amyloidosis: A Neuropathological Study" International Journal of Molecular Sciences 23, no. 9: 4972. https://doi.org/10.3390/ijms23094972
APA StyleMarazuela, P., Paez-Montserrat, B., Bonaterra-Pastra, A., Solé, M., & Hernández-Guillamon, M. (2022). Impact of Cerebral Amyloid Angiopathy in Two Transgenic Mouse Models of Cerebral β-Amyloidosis: A Neuropathological Study. International Journal of Molecular Sciences, 23(9), 4972. https://doi.org/10.3390/ijms23094972