
Relevance of a Novel Circuit-Level Model of Episodic Memories to Alzheimer’s Disease

Abstract
:1. Introduction
2. Results
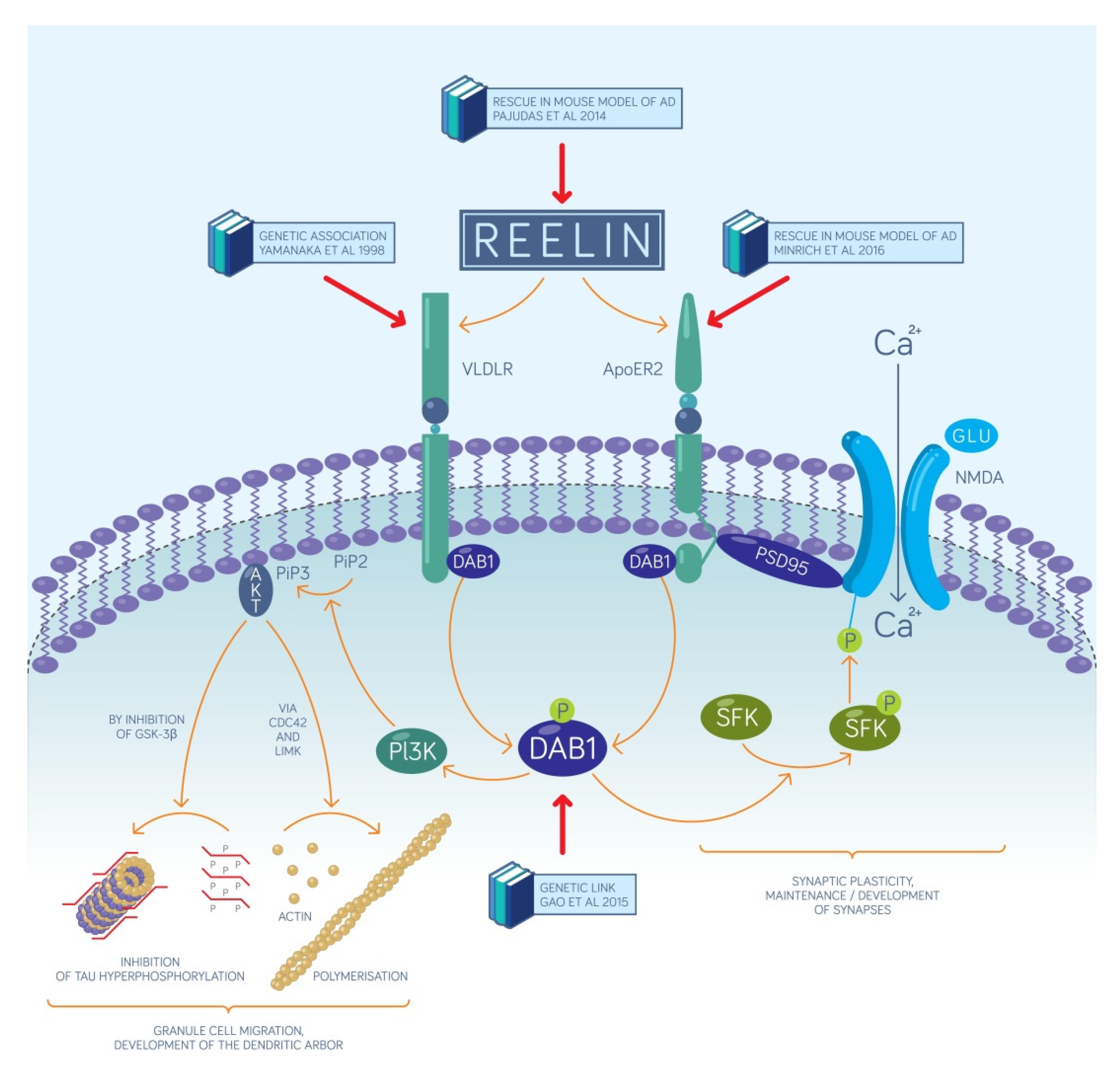
2.1. The Reelin-Positive Excitatory Entorhinal Neurons Constitute a Link between Our Recent Circuit-Level Model of Episodic Memories and the Pathomechanism of AD
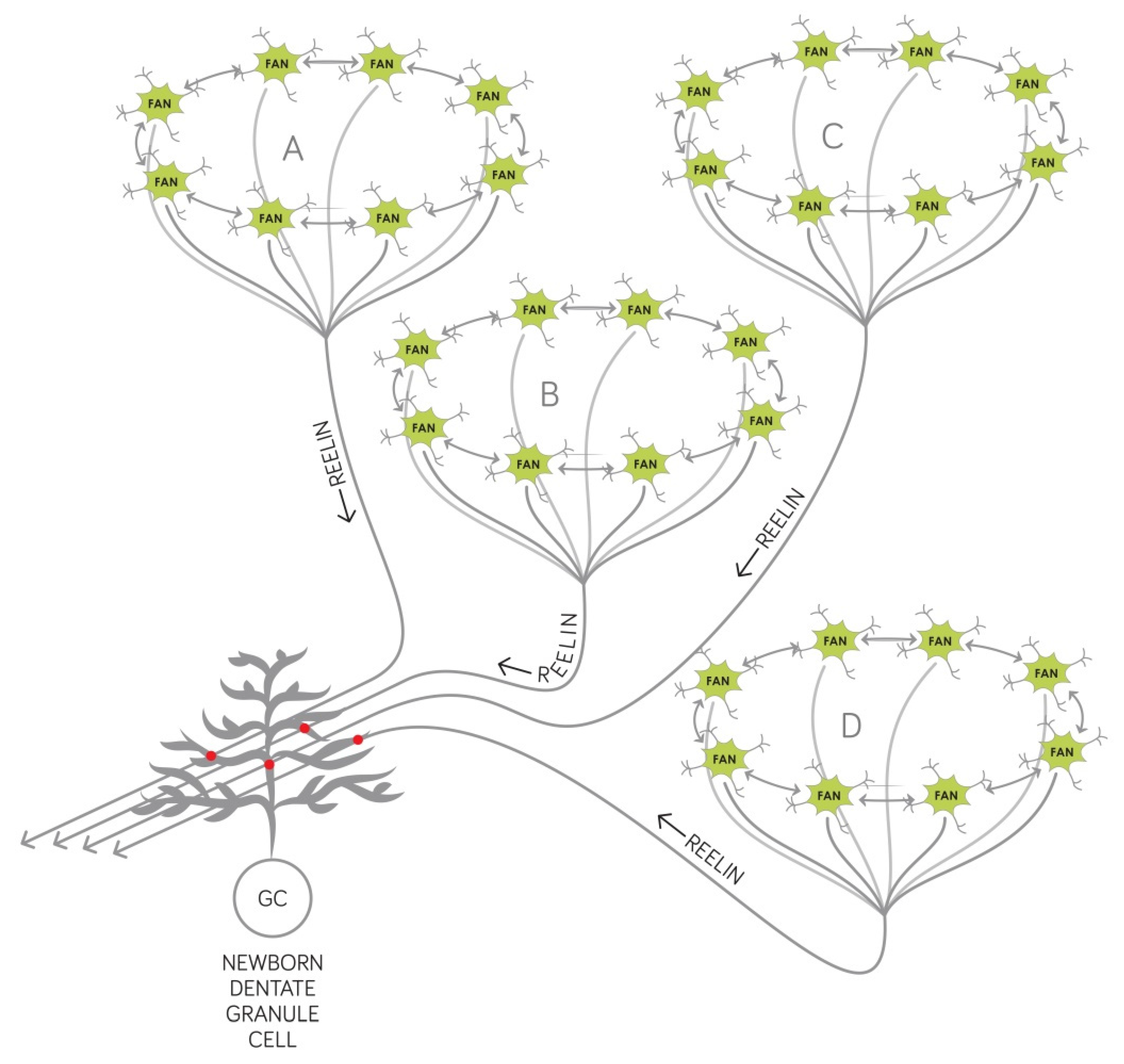
2.2. Synaptogenesis Described by Our Circuit-Level Model Underlie the Encoding of New Associations that Become Deficient Early on in AD
- (1)
- Reelin affects the proliferation in the subgranular zone where new dentate granule cells are generated. This has been demonstrated in the Reeler mouse that has no reelin expression in any of its tissues [45].
- (2)
- Reelin controls the migration of newborn dentate granule cells within the mouse dentate gyrus. It acts as an attractive signal pulling the young neurons through the already established granule cell layer and guiding them toward the marginal zone of the dentate gyrus [46].
- (3)
- Reelin signaling has an effect on the types and the morphology of the dendritic spines located on the dendritic tree of mouse dentate granule neurons [33] and cell-autonomous inactivation of the reelin receptor Dab1 specifically in developing mouse granule cells impairs their dendritic development [32].
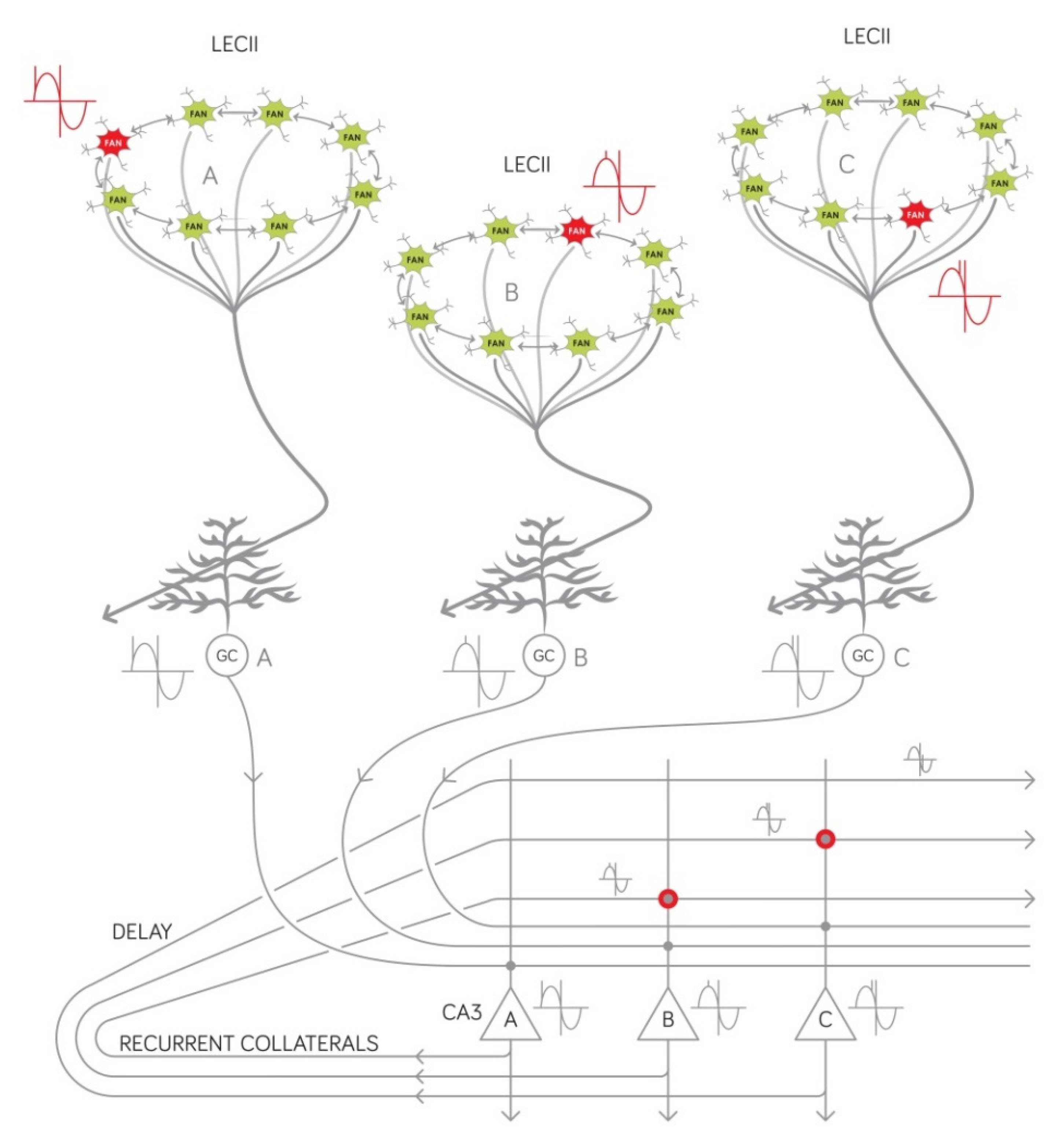
2.3. Our Circuit-Level Model Explains Why the Temporal Structure of Episodic Memories Deteriorate at the Onset of AD
3. Discussion
4. Methods
5. Conclusions
Funding
Conflicts of Interest
References
- Vogel, J.W.; Iturria-Medina, Y.; Strandberg, O.T.; Smith, R.; Levitis, E.; Evans, A.C.; Hansson, O.; Alzheimer’s Disease Neuroimaging Initiative; the Swedish BioFinder Study. Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 2020, 11, 2612. [Google Scholar] [CrossRef]
- Minati, L.; Edginton, T.; Bruzzone, M.G.; Giaccone, G. Reviews: Current Concepts in Alzheimer’s Disease: A Multidisciplinary Review. Am. J. Alzheimer’s Dis. Other Dement. 2009, 24, 95–121. [Google Scholar] [CrossRef]
- Kovács, K.A. Episodic Memories: How do the Hippocampus and the Entorhinal Ring Attractors Cooperate to Create Them? Front. Syst. Neurosci. 2020, 14, 559168. [Google Scholar] [CrossRef] [PubMed]
- Talamonti, D.; Koscik, R.; Johnson, S.; Bruno, D. Temporal contiguity and ageing: The role of memory organization in cognitive decline. J. Neuropsychol. 2021, 15, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Witter, M.P.; Doan, T.P.; Jacobsen, B.; Nilssen, E.S.; Ohara, S. Architecture of the Entorhinal Cortex A Review of Entorhinal Anatomy in Rodents with Some Comparative Notes. Front. Syst. Neurosci. 2017, 11, 46. [Google Scholar] [CrossRef]
- Ramos-Moreno, T.; Galazo, M.; Porrero, C.; Martínez-Cerdeño, V.; Clascá, F. Extracellular matrix molecules and synaptic plasticity: Immunomapping of intracellular and secreted Reelin in the adult rat brain. Eur. J. Neurosci. 2006, 23, 401–422. [Google Scholar] [CrossRef] [PubMed]
- Maloku, E.; Covelo, I.R.; Hanbauer, I.; Guidotti, A.; Kadriu, B.; Hu, Q.; Davis, J.M.; Costa, E. Lower number of cerebellar Purkinje neurons in psychosis is associated with reduced reelin expression. Proc. Natl. Acad. Sci. USA 2010, 107, 4407–4411. [Google Scholar] [CrossRef] [Green Version]
- Stranahan, A.M.; Erion, J.R.; Wosiski-Kuhn, M. Reelin signaling in development, maintenance, and plasticity of neural networks. Ageing Res. Rev. 2013, 12, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Pujadas, L.; Gruart, A.; Bosch, C.; Delgado, L.; Teixeira, C.M.; Rossi, D.; De Lecea, L.; Martínez, A.; Delgado-García, J.M.; Soriano, E. Reelin Regulates Postnatal Neurogenesis and Enhances Spine Hypertrophy and Long-Term Potentiation. J. Neurosci. 2010, 30, 4636–4649. [Google Scholar] [CrossRef] [Green Version]
- Stranahan, A.M.; Salas-Vega, S.; Jiam, N.T.; Gallagher, M. Interference with reelin signaling in the lateral entorhinal cortex impairs spatial memory. Neurobiol. Learn. Mem. 2011, 96, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Kulason, S.; Xu, E.; Tward, D.J.; Bakker, A.; Albert, M.; Younes, L.; Miller, M.I. Entorhinal and Transentorhinal Atrophy in Preclinical Alzheimer’s Disease. Front. Neurosci. 2020, 14, 804. [Google Scholar] [CrossRef]
- Gómez-Isla, T.; Price, J.L.; McKeel, D.W., Jr.; Morris, J.C.; Growdon, J.H.; Hyman, B.T. Profound Loss of Layer II Entorhinal Cortex Neurons Occurs in Very Mild Alzheimer’s Disease. J. Neurosci. 1996, 16, 4491–4500. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Kobro-Flatmoen, A.; Nagelhus, A.; Witter, M.P. Reelin-immunoreactive neurons in entorhinal cortex layer II selectively express intracellular amyloid in early Alzheimer’s disease. Neurobiol. Dis. 2016, 93, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K. From the Entorhinal Region via the Prosubiculum to the Dentate Fascia: Alzheimer Disease-Related Neurofibrillary Changes in the Temporal Allocortex. J. Neuropathol. Exp. Neurol. 2020, 79, 163–175. [Google Scholar] [CrossRef]
- Berron, D.; Van Westen, D.; Ossenkoppele, R.; Strandberg, O.; Hansson, O. Medial temporal lobe connectivity and its associations with cognition in early Alzheimer’s disease. Brain 2020, 143, 1233–1248. [Google Scholar] [CrossRef]
- Sáez-Valero, J.; Costell, M.; Sjögren, M.; Andreasen, N.; Blennow, K.; Luque, J.M. Altered levels of cerebrospinal fluid reelin in frontotemporal dementia and Alzheimer’s disease. J. Neurosci. Res. 2003, 72, 132–136. [Google Scholar] [CrossRef]
- Botella-López, A.; Burgaya, F.; Gavín, R.; García-Ayllón, M.-S.; Gómez-Tortosa, E.; Peña-Casanova, J.; Ureña, J.M.; Del Rio, J.A.; Blesa, R.; Soriano, E.; et al. Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5573–5578. [Google Scholar] [CrossRef] [Green Version]
- Chin, J.; Massaro, C.M.; Palop, J.; Thwin, M.T.; Yu, G.-Q.; Bien-Ly, N.; Bender, A.; Mucke, L. Reelin Depletion in the Entorhinal Cortex of Human Amyloid Precursor Protein Transgenic Mice and Humans with Alzheimer’s Disease. J. Neurosci. 2007, 27, 2727–2733. [Google Scholar] [CrossRef] [PubMed]
- Botella-López, A.; Cuchillo-Ibáñez, I.; Cotrufo, T.; Mok, S.S.; Li, Q.-X.; Barquero, M.-S.; Dierssen, M.; Soriano, E.; Sáez-Valero, J. β-amyloid controls altered Reelin expression and processing in Alzheimer’s disease. Neurobiol. Dis. 2010, 37, 682–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herring, A.; Donath, A.; Steiner, K.M.; Widera, M.P.; Hamzehian, S.; Kanakis, D.; Kölble, K.; ElAli, A.; Hermann, D.M.; Paulus, W.; et al. Reelin Depletion is an Early Phenomenon of Alzheimer’s Pathology. J. Alzheimer’s Dis. 2012, 30, 963–979. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo-Ibañez, I.; Mata-Balaguer, T.; Balmaceda, V.; Arranz, J.J.; Nimpf, J.; Sáez-Valero, J. The β-amyloid peptide compromises Reelin signaling in Alzheimer’s disease. Sci. Rep. 2016, 6, 31646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lidón, L.; Urrea, L.; Llorens, F.; Gil, V.; Alvarez, I.; Diez-Fairen, M.; Aguilar, M.; Pastor, P.; Zerr, I.; Alcolea, D.; et al. Disease-Specific Changes in Reelin Protein and mRNA in Neurodegenerative Diseases. Cells 2020, 9, 1252. [Google Scholar] [CrossRef]
- Yamanaka, H.; Kamimura, K.; Tanahashi, H.; Takahashi, K.; Asada, T.; Tabira, T. Genetic Risk Factors in Japanese Alzheimer’s Disease Patients: α1-ACT, VLDLR, and ApoE. Neurobiol. Aging 1998, 19, S43–S46. [Google Scholar] [CrossRef]
- Seripa, D.; Matera, M.G.; Franceschi, M.; Daniele, A.; Bizzarro, A.; Rinaldi, M.; Panza, F.; Fazio, V.M.; Gravina, C.; D’Onofrio, G.; et al. The RELN Locus in Alzheimer’s Disease. J. Alzheimer’s Dis. 2008, 14, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Tao, Y.; He, Q.; Song, F.; Saffen, D. Functional enrichment analysis of three Alzheimer’s disease genome-wide association studies identities DAB1 as a novel candidate liability/protective gene. Biochem. Biophys. Res. Commun. 2015, 463, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Hinrich, A.J.; Jodelka, F.M.; Chang, J.L.; Brutman, D.; Bruno, A.M.; Briggs, C.A.; James, B.D.; Stutzmann, G.E.; Bennett, D.A.; Miller, S.A.; et al. Therapeutic correction of ApoER2 splicing in Alzheimer’s disease mice using antisense oligonucleotides. EMBO Mol. Med. 2016, 8, 328–345. [Google Scholar] [CrossRef] [Green Version]
- Pujadas, L.; Rossi, D.; Andres, R.; Teixeira, C.; Serra-Vidal, B.; Parcerisas, A.; Maldonado, R.; Giralt, E.; Carulla, N.; Soriano, E. Reelin delays amyloid-beta fibril formation and rescues cognitive deficits in a model of Alzheimer’s disease. Nat. Commun. 2014, 5, 3443. [Google Scholar] [CrossRef]
- Rossi, D.; Gruart, A.; Contreras-Murillo, G.; Muhaisen, A.; Ávila, J.; Delgado-García, J.M.; Pujadas, L.; Soriano, E. Reelin reverts biochemical, physiological and cognitive alterations in mouse models of Tauopathy. Prog. Neurobiol. 2020, 186, 101743. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Renfro, A.; Quattrocchi, C.C.; Sheldon, M.; D’Arcangelo, G. Reelin Promotes Hippocampal Dendrite Development through the VLDLR/ApoER2-Dab1 Pathway. Neuron 2004, 41, 71–84. [Google Scholar] [CrossRef] [Green Version]
- Niu, S.; Yabut, O.; D’Arcangelo, G. The Reelin Signaling Pathway Promotes Dendritic Spine Development in Hippocampal Neurons. J. Neurosci. 2008, 28, 10339–10348. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.M.; Kron, M.M.; Masachs, N.; Zhang, H.; Lagace, D.C.; Martinez, A.; Reillo, I.; Duan, X.; Bosch, C.; Pujadas, L.; et al. Cell-Autonomous Inactivation of the Reelin Pathway Impairs Adult Neurogenesis in the Hippocampus. J. Neurosci. 2012, 32, 12051–12065. [Google Scholar] [CrossRef] [Green Version]
- Bosch, C.; Masachs, N.; Exposito-Alonso, D.; Martínez, A.; Teixeira, C.M.; Fernaud, I.; Pujadas, L.; Ulloa, F.; Comella, J.; DeFelipe, J.; et al. Reelin Regulates the Maturation of Dendritic Spines, Synaptogenesis and Glial Ensheathment of Newborn Granule Cells. Cereb. Cortex 2016, 26, 4282–4298. [Google Scholar] [CrossRef] [PubMed]
- Kobro-Flatmoen, A.; Lagartos-Donate, M.J.; Aman, Y.; Edison, P.; Witter, M.P.; Fang, E.F. Re-emphasizing early Alzheimer’s disease pathology starting in select entorhinal neurons, with a special focus on mitophagy. Ageing Res. Rev. 2021, 67, 101307. [Google Scholar] [CrossRef]
- Hiesberger, T.; Trommsdorff, M.; Howell, B.; Goffinet, A.; Mumby, M.C.; Cooper, J.A.; Herz, J. Direct Binding of Reelin to VLDL Receptor and ApoE Receptor 2 Induces Tyrosine Phosphorylation of Disabled-1 and Modulates Tau Phosphorylation. Neuron 1999, 24, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Ohkubo, N.; Lee, Y.; Morishima, A.; Terashima, T.; Kikkawa, S.; Tohyama, M.; Sakanaka, M.; Tanaka, J.; Maeda, N.; Vitek, M.P.; et al. Apolipoprotein E and Reelin ligands modulate tau phosphorylation through an Apolipoprotein E receptor/disabled-1/glycogen synthase kinase-3β cascade. FASEB J. 2002, 17, 295–297. [Google Scholar] [CrossRef] [Green Version]
- Xian, X.; Pohlkamp, T.; Durakoglugil, M.S.; Wong, C.H.; Beck, J.K.; Lane-Donovan, C.; Plattner, F.; Herz, J. Reversal of ApoE4-induced recycling block as a novel prevention approach for Alzheimer’s disease. eLife 2018, 7, 40048. [Google Scholar] [CrossRef]
- Vandrey, B.; Garden, D.L.; Ambrozova, V.; McClure, C.; Nolan, M.F.; Ainge, J.A. Fan Cells in Layer 2 of the Lateral Entorhinal Cortex Are Critical for Episodic-like Memory. Curr. Biol. 2020, 30, 169–175.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, B. Prodromal Alzheimer’s disease’: A more useful concept than mild cognitive impairment? Curr. Opin. Neurobiol. 2000, 13, 367–369. [Google Scholar] [CrossRef]
- Hainmueller, T.; Bartos, M. Dentate gyrus circuits for encoding, retrieval and discrimination of episodic memories. Nat. Rev. Neurosci. 2020, 21, 153–168. [Google Scholar] [CrossRef]
- Imayoshi, I.; Sakamoto, M.; Ohtsuka, T.; Takao, K.; Miyakawa, T.; Yamaguchi, M.; Mori, K.; Ikeda, T.; Itohara, S.; Kageyama, R. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat. Neurosci. 2008, 11, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Kitamura, T.; Saitoh, Y.; Ohkawa, N.; Kondo, T.; Inokuchi, K. Adult Neurogenesis Conserves Hippocampal Memory Capacity. J. Neurosci. 2018, 38, 6854–6863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Frankland, P.W.; Miller, F.D. Regenerating your senses: Multiple roles for neurogenesis in the adult brain. Nat. Neurosci. 2008, 11, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Sibbe, M.; Kuner, E.; Althof, D.; Frotscher, M. Stem- and Progenitor Cell Proliferation in the Dentate Gyrus of the Reeler Mouse. PLoS ONE 2015, 10, e0119643. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Brunne, B.; Zhao, S.; Chai, X.; Virgilio, F.A.; Lau, J.; Failla, A.V.; Zobiak, B.; Sibbe, M.; Westbrook, G.L.; et al. Trajectory Analysis Unveils Reelin’s Role in the Directed Migration of Granule Cells in the Dentate Gyrus. J. Neurosci. 2018, 38, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Spalding, K.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Boström, E.; Westerlund, I.; Vial, C.; Buchholz, B.; et al. Dynamics of Hippocampal Neurogenesis in Adult Humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef] [Green Version]
- Vivar, C.; Potter, M.C.; Choi, J.; Lee, J.-Y.; Stringer, T.P.; Callaway, E.M.; Gage, F.H.; Suh, H.; Van Praag, H. Monosynaptic inputs to new neurons in the dentate gyrus. Nat. Commun. 2012, 3, 1107. [Google Scholar] [CrossRef] [Green Version]
- Toni, N.; Schinder, A.F. Maturation and Functional Integration of New Granule Cells into the Adult Hippocampus. Cold Spring Harb. Perspect. Biol. 2016, 8, a018903. [Google Scholar] [CrossRef] [Green Version]
- Schroeter, M.L.; Stein, T.; Maslowski, N.; Neumann, J. Neural correlates of Alzheimer’s disease and mild cognitive impairment: A systematic and quantitative meta-analysis involving 1351 patients. NeuroImage 2009, 47, 1196–1206. [Google Scholar] [CrossRef] [Green Version]
- Salat, D.; Tuch, D.; van der Kouwe, A.; Greve, D.; Pappu, V.; Lee, S.; Hevelone, N.; Zaleta, A.; Growdon, J.; Corkin, S.; et al. White matter pathology isolates the hippocampal formation in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 244–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paredes, M.F.; Sorrells, S.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Does Adult Neurogenesis Persist in the Human Hippocampus? Cell Stem Cell 2018, 23, 780–781. [Google Scholar] [CrossRef] [Green Version]
- Sorrells, S.; Paredes, M.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 2018, 555, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Jiménez, E.P.; Terreros-Roncal, J.; Flor-García, M.; Rábano, A.; Llorens-Martín, M. Evidences for Adult Hippocampal Neurogenesis in Humans. J. Neurosci. 2021, 41, 2541–2553. [Google Scholar] [CrossRef]
- Manganas, L.N.; Zhang, X.; Li, Y.; Hazel, R.D.; Smith, S.D.; Wagshul, M.E.; Henn, F.; Benveniste, H.; Djurić, P.M.; Enikolopov, G.; et al. Magnetic Resonance Spectroscopy Identifies Neural Progenitor Cells in the Live Human Brain. Science 2007, 318, 980–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curiel, R.E.; Crocco, E.; Acevedo, A.; Duara, R.; Agron, J. A New Scale for the Evaluation of Proactive and Retroactive Interference in Mild Cognitive Impairment and Early Alzheimer’s Disease. J. Aging Sci. 2013, 1, 1000102. [Google Scholar] [CrossRef] [Green Version]
- Kitaigorodsky, M.; Crocco, E.; Curiel-Cid, R.E.; Leal, G.; Zheng, D.; Eustache, M.K.; Greig-Custo, M.T.; Barker, W.; Duara, R.; Loewenstein, D.A. The relationship of semantic intrusions to different etiological subtypes of MCI and cognitively healthy older adults. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2021, 13, e12192. [Google Scholar] [CrossRef]
- Schlesiger, M.; Cannova, C.C.; Boublil, B.L.; Hales, J.B.; Mankin, E.A.; Brandon, M.P.; Leutgeb, J.; Leibold, C.; Leutgeb, S. The medial entorhinal cortex is necessary for temporal organization of hippocampal neuronal activity. Nat. Neurosci. 2015, 18, 1123–1132. [Google Scholar] [CrossRef]
- O’Keefe, J.; Recce, M.L. Phase relationship between hippocampal place units and the EEG theta rhythm. Hippocampus 1993, 3, 317–330. [Google Scholar] [CrossRef]
- Montchal, M.E.; Reagh, Z.M.; Yassa, M.A. Precise temporal memories are supported by the lateral entorhinal cortex in humans. Nat. Neurosci. 2019, 22, 284–288. [Google Scholar] [CrossRef]
- Bruno, D.; Koscik, R.L.; Woodard, J.L.; Pomara, N.; Johnson, S.C. The recency ratio as predictor of early MCI. Int. Psychogeriatr. 2018, 30, 1883–1888. [Google Scholar] [CrossRef] [PubMed]
- Gillis, M.M.; Quinn, K.M.; Phillips, P.A.; Hampstead, B.M. Impaired retention is responsible for temporal order memory deficits in mild cognitive impairment. Acta Psychol. 2013, 143, 88–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Type of Relationship | Year of Publication | Molecule Involved | Finding |
|---|---|---|---|
| Change of reelin expression in AD patients | 2003 | protein | increase in the cerebrospinal fluid [17] |
| 2006 | protein, mRNA | increase in the frontal cortex [18] | |
| 2007 | protein | decrease in the entorhinal cortex [19] | |
| 2010 | protein | increase in the frontal cortex [20] | |
| 2012 | protein, mRNA | decrease in the entorhinal cortex [21] | |
| 2016 | protein | increase in the frontal cortex and the hippocampus [22] | |
| 2020 | mRNA | increase in the frontal cortex [23] | |
| Established genetic links with molecules of the reelin signaling pathway in human subjects with AD | 1998 | VLDLR | [24] |
| 2008 | reelin | [25] | |
| 2015 | DAB1 | [26] | |
| 2016 | ApoER2 | [27] | |
| Rescue by overexpression in disease model animals | 2014 | reelin | novel object recognition rescued in the AD model mice strain J20 [28] |
| 2020 | reelin | passive avoidance rescued in τ-overexpressing mice [29] | |
| Processes related to hippocampal neurogenesis supported by molecules of the reelin signaling pathway | 2004 | reelin | dendritogenesis [30] |
| 2008 | reelin | dendritic spine development [31] | |
| 2012 | DAB1 | neurogenesis [32] | |
| 2016 | reelin | synaptogenesis [33] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, K.A. Relevance of a Novel Circuit-Level Model of Episodic Memories to Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 462. https://doi.org/10.3390/ijms23010462
Kovács KA. Relevance of a Novel Circuit-Level Model of Episodic Memories to Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(1):462. https://doi.org/10.3390/ijms23010462
Chicago/Turabian StyleKovács, Krisztián A. 2022. "Relevance of a Novel Circuit-Level Model of Episodic Memories to Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 1: 462. https://doi.org/10.3390/ijms23010462
APA StyleKovács, K. A. (2022). Relevance of a Novel Circuit-Level Model of Episodic Memories to Alzheimer’s Disease. International Journal of Molecular Sciences, 23(1), 462. https://doi.org/10.3390/ijms23010462