
Netrin-1: A Modulator of Macrophage Driven Acute and Chronic Inflammation


Abstract
1. Netrin-1 Acts as a Chemoattractant and Chemorepellent Guidance Cue
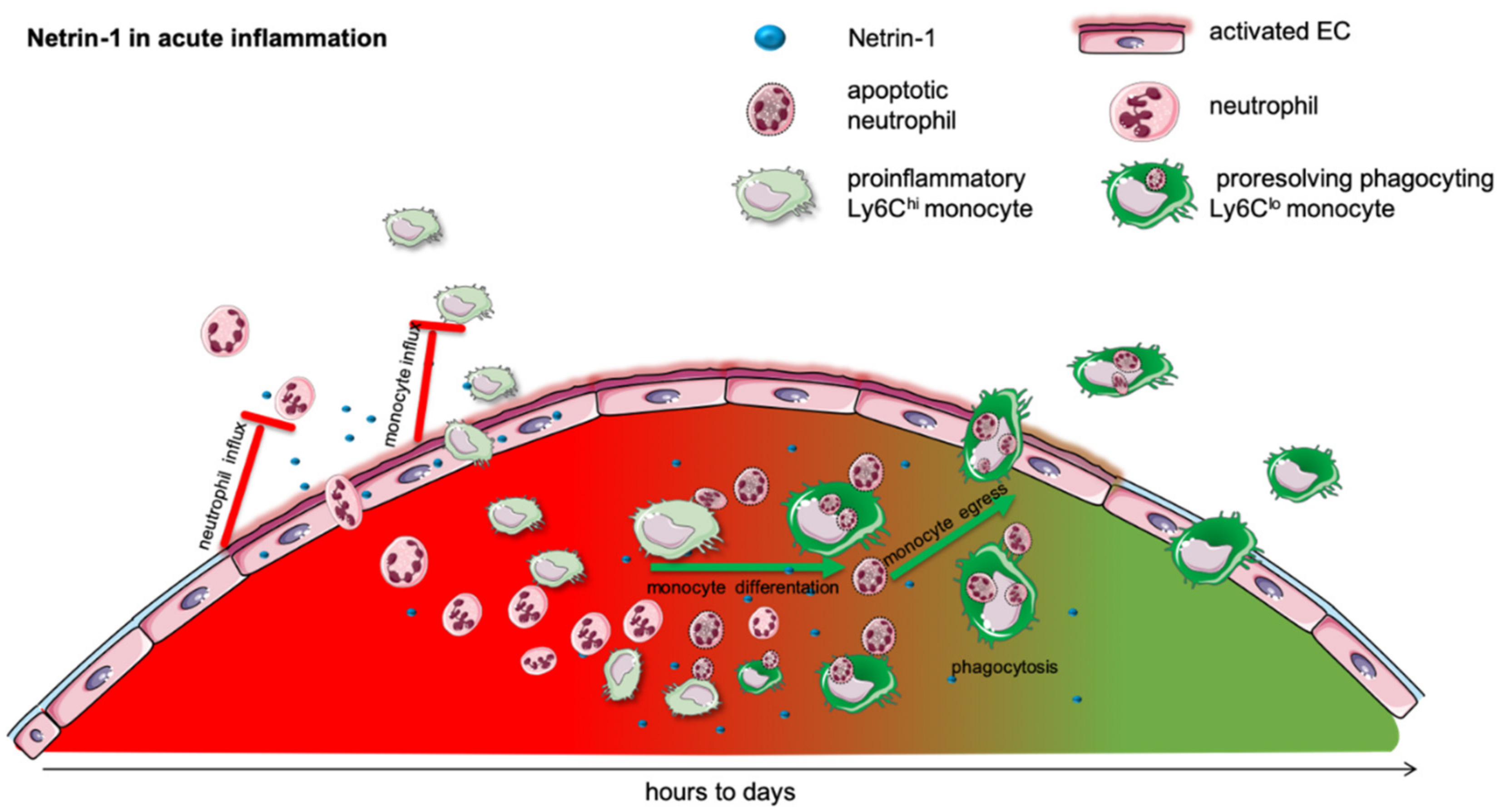
2. Acute Inflammation
2.1. Acute Lung Injury
2.2. Acute Abdominal Inflammation
2.2.1. Inflammatory Bowel Diseases
2.2.2. Acute Pancreatitis
2.2.3. Acute Peritonitis
2.3. Hypoxia and Ischemia–Reperfusion Injury
2.3.1. Hypoxia
2.3.2. Acute Ischemic Kidney Injury
2.3.3. Liver Ischemia and Reperfusion Injury
2.3.4. Acute Myocardial Infarction and Reperfusion Injury
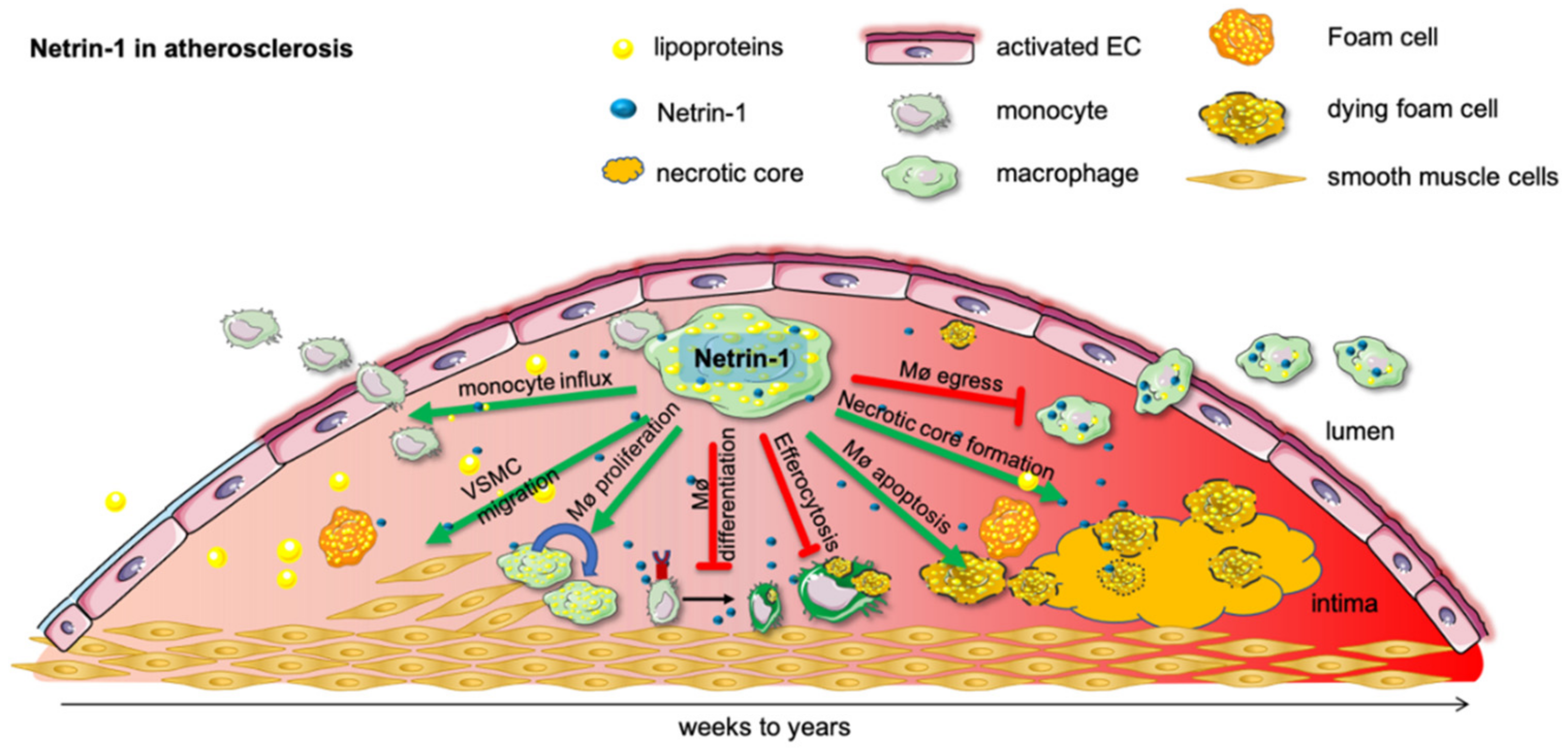
3. Chronic Low-Grade Inflammation
3.1. Netrin-1 in Atherosclerosis
3.2. Aortic Abdominal Aneurysma
3.3. Obesity
3.4. Rheumatoid Arthritis and Osteolysis
3.5. Diabetes
3.6. Chronic Inflammation of Lungs
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Barallobre, M.J.; Pascual, M.; Del Rio, J.A.; Soriano, E. The Netrin family of guidance factors: Emphasis on Netrin-1 signalling. Brain Res. Rev. 2005, 49, 22–47. [Google Scholar] [CrossRef]
- Cirulli, V.; Yebra, M. Netrins: Beyond the brain. Nat. Rev. Mol. Cell Biol. 2007, 8, 296–306. [Google Scholar] [CrossRef]
- Yurchenco, P.D.; Wadsworth, W.G. Assembly and tissue functions of early embryonic laminins and netrins. Curr. Opin. Cell Biol. 2004, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Serafini, T.; Kennedy, T.E.; Galko, M.J.; Mirzayan, C.; Jessell, T.M.; Tessier-Lavigne, M. The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell 1994, 78, 409–424. [Google Scholar] [CrossRef]
- Bruikman, C.S.; Zhang, H.; Kemper, A.M.; van Gils, J.M. Netrin Family: Role for Protein Isoforms in Cancer. J. Nucleic Acids 2019, 2019, 3947123. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wu, Z.; Renier, N.; Antipenko, A.; Tzvetkova-Robev, D.; Xu, Y.; Minchenko, M.; Nardi-Dei, V.; Rajashankar, K.R.; Himanen, J.; et al. Neural migration. Structures of netrin-1 bound to two receptors provide insight into its axon guidance mechanism. Science 2014, 344, 1275–1279. [Google Scholar] [CrossRef] [PubMed]
- Miloudi, K.; Binet, F.; Wilson, A.; Cerani, A.; Oubaha, M.; Menard, C.; Henriques, S.; Mawambo, G.; Dejda, A.; Nguyen, P.T.; et al. Truncated netrin-1 contributes to pathological vascular permeability in diabetic retinopathy. J. Clin. Investig. 2016, 126, 3006–3022. [Google Scholar] [CrossRef]
- Kruger, R.P.; Lee, J.; Li, W.; Guan, K.L. Mapping netrin receptor binding reveals domains of Unc5 regulating its tyrosine phosphorylation. J. Neurosci. 2004, 24, 10826–10834. [Google Scholar] [CrossRef]
- Finci, L.; Zhang, Y.; Meijers, R.; Wang, J.-H. Signaling mechanism of the netrin-1 receptor DCC in axon guidance. Prog. Biophys. Mol. Biol. 2015, 118, 153–160. [Google Scholar] [CrossRef]
- Mehlen, P.; Rabizadeh, S.; Snipas, S.J.; Assa-Munt, N.; Salvesen, G.S.; Bredesen, D.E. The DCC gene product induces apoptosis by a mechanism requiring receptor proteolysis. Nature 1998, 395, 801–804. [Google Scholar] [CrossRef]
- Paradisi, A.; Mehlen, P. Netrin-1, a missing link between chronic inflammation and tumor progression. Cell Cycle 2010, 9, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, H. Netrin-1 and its receptors in tumorigenesis. Nat. Rev. Cancer 2004, 4, 978–987. [Google Scholar] [CrossRef]
- Rosenberger, P.; Schwab, J.M.; Mirakaj, V.; Masekowsky, E.; Mager, A.; Morote-Garcia, J.C.; Unertl, K.; Eltzschig, H.K. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat. Immunol. 2009, 10, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Conrad, C.; Mills, T.W.; Berg, N.K.; Kim, B.; Ruan, W.; Lee, J.W.; Zhang, X.; Yuan, X.; Eltzschig, H.K. PMN-derived netrin-1 attenuates cardiac ischemia-reperfusion injury via myeloid ADORA2B signaling. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Corset, V.; Nguyen-Ba-Charvet, K.T.; Forcet, C.; Moyse, E.; Chedotal, A.; Mehlen, P. Netrin-1-mediated axon outgrowth and cAMP production requires interactionwith adenosine A2b receptor. Nature 2000, 407, 747–750. [Google Scholar] [CrossRef]
- Ranganathan, P.V.; Jayakumar, C.; Ramesh, G. Netrin-1-treated macrophages protect the kidney against ischemia-reperfusion injury and suppress inflammation by inducing M2 polarization. Am. J. Physiol. Renal Physiol. 2013, 304, F948–F957. [Google Scholar] [CrossRef] [PubMed]
- Gloerich, M.; Bos, J.L. Epac: Defining a new mechanism for cAMP action. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 355–375. [Google Scholar] [CrossRef]
- Lorenowicz, M.J.; Fernandez-Borja, M.; Hordijk, P.L. cAMP signaling in leukocyte transendothelial migration. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Yebra, M.; Montgomery, A.M.; Diaferia, G.R.; Kaido, T.; Silletti, S.; Perez, B.; Just, M.L.; Hildbrand, S.; Hurford, R.; Florkiewicz, E.; et al. Recognition of the neural chemoattractant Netrin-1 by integrins alpha6beta4 and alpha3beta1 regulates epithelial cell adhesion and migration. Dev. Cell 2003, 5, 695–707. [Google Scholar] [CrossRef]
- Ly, N.P.; Komatsuzaki, K.; Fraser, I.P.; Tseng, A.A.; Prodhan, P.; Moore, K.J.; Kinane, T.B. Netrin-1-inhibits-leukocyte-migration in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 14729–14734. [Google Scholar] [CrossRef]
- Claro, V.; Ferro, A. Netrin-1: Focus on its role in cardiovascular physiology and atherosclerosis. JRSM Cardiovasc. Dis. 2020, 9, 2048004020959574. [Google Scholar] [CrossRef]
- Bradford, D.; Cole, S.J.; Cooper, H.M. Netrin-1: Diversity in development. Int. J. Biochem. Cell Biol. 2009, 41, 487–493. [Google Scholar] [CrossRef]
- Castets, M.; Mehlen, P. Netrin-1 role in angiogenesis: To be or not to be a pro-angiogenic factor? Cell Cycle 2010, 9, 1466–1471. [Google Scholar] [CrossRef]
- Mazelin, L.; Bernet, A.; Bonod-Bidaud, C.; Pays, L.; Arnaud, S.; Gespach, C.; Bredesen, D.E.; Scoazec, J.Y.; Mehlen, P. Netrin-1 controls colorectal tumorigenesis by regulating apoptosis. Nature 2004, 431, 80–84. [Google Scholar] [CrossRef]
- Han, Y.; Jiang, N.; Su, T.; Yang, Q.-C.; Yan, C.-C.; Ye, L.; Yuan, Q.; Zhu, P.-W.; Li, W.; Liu, Z.-G.; et al. Netrin-1 promotes epithelium repair in corneal injury. Int. J. Ophthalmol. 2020, 13, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M. Chemokines and leukocyte traffic. Nature 1998, 392, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Mirakaj, V.; Dalli, J.; Granja, T.; Rosenberger, P.; Serhan, C.N. Vagus nerve controls resolution and pro-resolving mediators of inflammation. J. Exp. Med. 2014, 211, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, M.; Kohler, D.; Korner, A.; Granja, T.; Straub, A.; Giera, M.; Mirakaj, V. The neuroimmune guidance cue netrin-1 controls resolution programs and promotes liver regeneration. Hepatology 2016, 63, 1689–1705. [Google Scholar] [CrossRef] [PubMed]
- Mirakaj, V.; Thix, C.A.; Laucher, S.; Mielke, C.; Morote-Garcia, J.C.; Schmit, M.A.; Henes, J.; Unertl, K.E.; Köhler, D.; Rosenberger, P. Netrin-1 dampens pulmonary inflammation during acute lung injury. Am. J. Respir. Crit. Care Med. 2010, 181, 815–824. [Google Scholar] [CrossRef]
- Mutz, C.; Mirakaj, V.; Vagts, D.A.; Westermann, P.; Waibler, K.; König, K.; Iber, T.; Nöldge-Schomburg, G.; Rosenberger, P. The neuronal guidance protein netrin-1 reduces alveolar inflammation in a porcine model of acute lung injury. Crit. Care 2010, 14, R189. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, Y.; Zhou, J.; Li, Y.; Gong, C.; Wang, X. Netrin-1 reduces lung ischemia-reperfusion injury by increasing the proportion of regulatory T cells. J. Int. Med. Res. 2020, 48, 300060520926415. [Google Scholar] [CrossRef]
- He, J.; Zhao, Y.; Deng, W.; Wang, D. Netrin-1 promotes epithelial sodium channel-mediated alveolar fluid clearance via activation of the adenosine 2B receptor in lipopolysaccharide-induced acute lung injury. Respiration 2014, 87, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.-L.; Lin, J.-A.; Chen, K.-Y.; Hsu, A.-C.; Wu, S.-Y.; Tai, Y.-T.; Lin, K.-H.; Chung, W.-C.; Li, M.-H. Netrin-1 Dampens Hypobaric Hypoxia-Induced Lung Injury in Mice. High Alt. Med. Biol. 2019, 20, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Aherne, C.M.; Collins, C.B.; Masterson, J.C.; Tizzano, M.; Boyle, T.A.; Westrich, J.A.; Parnes, J.A.; Furuta, G.T.; Rivera-Nieves, J.; Eltzschig, H.K. Neuronal guidance molecule netrin-1 attenuates inflammatory cell trafficking during acute experimental colitis. Gut 2012, 61, 695–705. [Google Scholar] [CrossRef]
- Mirakaj, V.; Gatidou, D.; Pötzsch, C.; König, K.; Rosenberger, P. Netrin-1 signaling dampens inflammatory peritonitis. J. Immunol. 2011, 186, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cai, Q.-P.; Shen, P.-J.; Yan, R.-L.; Wang, C.-M.; Yang, D.-J.; Fu, H.-B.; Chen, X.-Y. Netrin-1 protects against L-Arginine-induced acute pancreatitis in mice. PLoS ONE 2012, 7, e46201. [Google Scholar] [CrossRef]
- Wang, W.; Reeves, W.B.; Ramesh, G. Netrin-1 and kidney injury. I. Netrin-1 protects against ischemia-reperfusion injury of the kidney. Am. J. Physiol. Renal Physiol. 2008, 294, F739–F747. [Google Scholar] [CrossRef]
- Wang, W.; Reeves, W.B.; Pays, L.; Mehlen, P.; Ramesh, G. Netrin-1 overexpression protects kidney from ischemia reperfusion injury by suppressing apoptosis. Am. J. Pathol. 2009, 175, 1010–1018. [Google Scholar] [CrossRef]
- Tadagavadi, R.K.; Wang, W.; Ramesh, G. Netrin-1 regulates Th1/Th2/Th17 cytokine production and inflammation through UNC5B receptor and protects kidney against ischemia-reperfusion injury. J. Immunol. 2010, 185, 3750–3758. [Google Scholar] [CrossRef]
- Grenz, A.; Dalton, J.H.; Bauerle, J.D.; Badulak, A.; Ridyard, D.; Gandjeva, A.; Aherne, C.M.; Brodsky, K.S.; Kim, J.-H.; Tuder, R.M.; et al. Partial netrin-1 deficiency aggravates acute kidney injury. PLoS ONE 2011, 6, e14812. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, H. Netrin-1 prevents ischemia/reperfusion-induced myocardial infarction via a DCC/ERK1/2/eNOS s1177/NO/DCC feed-forward mechanism. J. Mol. Cell Cardiol. 2010, 48, 1060–1070. [Google Scholar] [CrossRef] [PubMed]
- Bouhidel, J.O.; Wang, P.; Li, Q.; Cai, H. Pharmacological postconditioning treatment of myocardial infarction with netrin-1. Front. Biosci. 2014, 19, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Bouhidel, J.O.; Wang, P.; Siu, K.L.; Li, H.; Youn, J.Y.; Cai, H. Netrin-1 improves post-injury cardiac function in vivo via DCC/NO-dependent preservation of mitochondrial integrity, while attenuating autophagy. Biochim. Biophys. Acta 2015, 1852, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Xing, H.; Mao, A.; Jiang, H.; Cheng, L.; Liu, Y.; Quan, X.; Li, L. Netrin-1 attenuates cardiac ischemia reperfusion injury and generates alternatively activated macrophages. Inflammation 2014, 37, 573–580. [Google Scholar] [CrossRef]
- Ke, T.; Wu, Y.; Li, L.; Liu, Y.; Yao, X.; Zhang, J.; Kong, D.; Li, C. Netrin-1 ameliorates myocardial infarction-induced myocardial injury: Mechanisms of action in rats and diabetic mice. Hum. Gene Ther. 2014, 25, 787–797. [Google Scholar] [CrossRef]
- Siu, K.L.; Lotz, C.; Ping, P.; Cai, H. Netrin-1 abrogates ischemia/reperfusion-induced cardiac mitochondrial dysfunction via nitric oxide-dependent attenuation of NOX4 activation and recoupling of NOS. J. Mol. Cell. Cardiol. 2015, 78, 174–185. [Google Scholar] [CrossRef]
- Rubenfeld, G.D.; Caldwell, E.; Peabody, E.; Weaver, J.; Martin, D.P.; Neff, M.; Stern, E.J.; Hudson, L.D. Incidence and outcomes of acute lung injury. N. Engl. J. Med. 2005, 353, 1685–1693. [Google Scholar] [CrossRef]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef]
- Paradisi, A.; Maisse, C.; Bernet, A.; Coissieux, M.M.; Maccarrone, M.; Scoazec, J.Y.; Mehlen, P. NF-kappaB regulates netrin-1 expression and affects the conditional tumor suppressive activity of the netrin-1 receptors. Gastroenterology 2008, 135, 1248–1257. [Google Scholar] [CrossRef]
- Ramkhelawon, B.; Hennessy, E.J.; Ménager, M.; Ray, T.D.; Sheedy, F.J.; Hutchison, S.; Wanschel, A.; Oldebeken, S.; Geoffrion, M.; Spiro, W.; et al. Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat. Med. 2014, 20, 377–384. [Google Scholar] [CrossRef]
- Liu, J.; Du, J.; Cheng, X.; Zhang, X.; Li, Y.; Fu, X.; Chen, X. Effect of Netrin-1 Anti-Inflammatory Factor on Acute Lung Injury in Sepsis Rats. Med. Sci. Monit. 2019, 25, 7928–7935. [Google Scholar] [CrossRef]
- Berg, N.K.; Li, J.; Kim, B.; Mills, T.; Pei, G.; Zhao, Z.; Li, X.; Zhang, X.; Ruan, W.; Eltzschig, H.K.; et al. Hypoxia-inducible factor-dependent induction of myeloid-derived netrin-1 attenuates natural killer cell infiltration during endotoxin-induced lung injury. FASEB J. 2021, 35, e21334. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef]
- Kuhl, A.A.; Erben, U.; Kredel, L.I.; Siegmund, B. Diversity of Intestinal Macrophages in Inflammatory Bowel Diseases. Front. Immunol. 2015, 6, 613. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Lissner, D.; Schumann, M.; Batra, A.; Kredel, L.I.; Kuhl, A.A.; Erben, U.; May, C.; Schulzke, J.D.; Siegmund, B. Monocyte and M1 Macrophage-induced Barrier Defect Contributes to Chronic Intestinal Inflammation in IBD. Inflamm. Bowel Dis. 2015, 21, 1297–1305. [Google Scholar] [CrossRef]
- Demetter, P.; de Vos, M.; van Huysse, J.A.; Baeten, D.; Ferdinande, L.; Peeters, H.; Mielants, H.; Veys, E.M.; de Keyser, F.; Cuvelier, C.A. Colon mucosa of patients both with spondyloarthritis and Crohn’s disease is enriched with macrophages expressing the scavenger receptor CD163. Ann. Rheum. Dis. 2005, 64, 321–324. [Google Scholar] [CrossRef]
- Franze, E.; Caruso, R.; Stolfi, C.; Sarra, M.; Cupi, M.L.; Caprioli, F.; Monteleone, I.; Zorzi, F.; de Nitto, D.; Colantoni, A.; et al. Lesional accumulation of CD163-expressing cells in the gut of patients with inflammatory bowel disease. PLoS ONE 2013, 8, e69839. [Google Scholar] [CrossRef] [PubMed]
- Aherne, C.M.; Collins, C.B.; Eltzschig, H.K. Netrin-1 guides inflammatory cell migration to control mucosal immune responses during intestinal inflammation. Tissue Barriers 2013, 1, e24957. [Google Scholar] [CrossRef] [PubMed]
- Paradisi, A.; Maisse, C.; Coissieux, M.-M.; Gadot, N.; Lépinasse, F.; Delloye-Bourgeois, C.; Delcros, J.-G.; Svrcek, M.; Neufert, C.; Fléjou, J.-F.; et al. Netrin-1 up-regulation in inflammatory bowel diseases is required for colorectal cancer progression. Proc. Natl. Acad. Sci. USA. 2009, 106, 17146–17151. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, D.C. Clinical practice. Acute pancreatitis. N. Engl. J. Med. 2006, 354, 2142–2150. [Google Scholar] [CrossRef]
- Shrivastava, P.; Bhatia, M. Essential role of monocytes and macrophages in the progression of acute pancreatitis. World J. Gastroenterol. 2010, 16, 3995–4002. [Google Scholar] [CrossRef]
- Gea-Sorlí, S.; Closa, D. Role of macrophages in the progression of acute pancreatitis. World J. Gastrointest. Pharmacol. Ther. 2010, 1, 107–111. [Google Scholar] [CrossRef]
- Karlsson, S.; Varpula, M.; Ruokonen, E.; Pettila, V.; Parviainen, I.; Ala-Kokko, T.I.; Kolho, E.; Rintala, E.M. Incidence, treatment, and outcome of severe sepsis in ICU-treated adults in Finland: The Finnsepsis study. Intensive Care Med. 2007, 33, 435–443. [Google Scholar] [CrossRef]
- Pieracci, F.M.; Barie, P.S. Management of severe sepsis of abdominal origin. Scand. J. Surg. 2007, 96, 184–196. [Google Scholar] [CrossRef]
- Tridente, A.; Clarke, G.M.; Walden, A.; McKechnie, S.; Hutton, P.; Mills, G.H.; Gordon, A.C.; Holloway, P.A.; Chiche, J.D.; Bion, J.; et al. Patients with faecal peritonitis admitted to European intensive care units: An epidemiological survey of the GenOSept cohort. Intensive Care Med. 2014, 40, 202–210. [Google Scholar] [CrossRef]
- Hackett, P.H.; Roach, R.C. High-altitude illness. N. Engl. J. Med. 2001, 345, 107–114. [Google Scholar] [CrossRef]
- Eckle, T.; Faigle, M.; Grenz, A.; Laucher, S.; Thompson, L.F.; Eltzschig, H.K. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood 2008, 111, 2024–2035. [Google Scholar] [CrossRef]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef]
- Anand, R.J.; Gribar, S.C.; Li, J.; Kohler, J.W.; Branca, M.F.; Dubowski, T.; Sodhi, C.P.; Hackam, D.J. Hypoxia causes an increase in phagocytosis by macrophages in a HIF-1alpha-dependent manner. J. Leukoc. Biol. 2007, 82, 1257–1265. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and Inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef]
- Taylor, C.T.; Doherty, G.; Fallon, P.G.; Cummins, E.P. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J. Clin. Investig. 2016, 126, 3716–3724. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Ramkhelawon, B.; Yang, Y.; van Gils, J.M.; Hewing, B.; Rayner, K.J.; Parathath, S.; Guo, L.; Oldebeken, S.; Feig, J.L.; Fisher, E.A.; et al. Hypoxia induces netrin-1 and Unc5b in atherosclerotic plaques: Mechanism for macrophage retention and survival. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1180–1188. [Google Scholar] [CrossRef]
- Bystrom, J.; Evans, I.; Newson, J.; Stables, M.; Toor, I.; van Rooijen, N.; Crawford, M.; Colville-Nash, P.; Farrow, S.; Gilroy, D.W. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood 2008, 112, 4117–4127. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Wu, W.; Mishra, P.K.; Chen, F.; Millman, A.; Csoka, B.; Koscso, B.; Eltzschig, H.K.; Hasko, G.; Gause, W.C. A2B adenosine receptor induces protective antihelminth type 2 immune responses. Cell Host Microbe 2014, 15, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Luan, H.; Chai, H.; Yan, L.; Zhang, J.; Wang, Q.; Cao, L. Netrin-1 interference potentiates epithelial-to-mesenchymal transition through the PI3K/AKT pathway under the hypoxic microenvironment conditions of non-small cell lung cancer. Int. J. Oncol. 2019, 54, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Ranganathan, P.; Mohamed, R.; Jayakumar, C.; Ramesh, G. Guidance cue netrin-1 and the regulation of inflammation in acute and chronic kidney disease. Mediat. Inflamm. 2014, 2014, 525891. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, C.; Mohamed, R.; Ranganathan, P.V.; Ramesh, G. Intracellular kinases mediate increased translation and secretion of netrin-1 from renal tubular epithelial cells. PLoS ONE 2011, 6, e26776. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Jayakumar, C.; Navankasattusas, S.; Li, D.Y.; Kim, I.; Ramesh, G. UNC5B receptor deletion exacerbates tissue injury in response to AKI. J. Am. Soc. Nephrol. 2014, 25, 239–249. [Google Scholar] [CrossRef]
- Ramesh, G.; Krawczeski, C.D.; Woo, J.G.; Wang, Y.; Devarajan, P. Urinary netrin-1 is an early predictive biomarker of acute kidney injury after cardiac surgery. Clin. J. Am. Soc. Nephrol. 2010, 5, 395–401. [Google Scholar] [CrossRef]
- Barbosa Evora, P.R.; Celotto, A.C.; Sumarelli Albuquerque, A.A.; Martinez Évora, P. Circulatory Shock, Ischemia-Reperfusion Injury, Systemic Inflammatory Response Syndrome (SIRS), and Multiple Organ Failure. In Vasoplegic Endothelial Dysfunction: Circulatory Shock and Methylene Blue; Barbosa Evora, P.R., Celotto, A.C., Sumarelli Albuquerque, A.A., Martinez Évora, P., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 29–34. ISBN 978-3-030-74096-2. [Google Scholar]
- Jaeschke, H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G15–G26. [Google Scholar] [CrossRef]
- Lentsch, A.B.; Kato, A.; Yoshidome, H.; McMasters, K.M.; Edwards, M.J. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology 2000, 32, 169–173. [Google Scholar] [CrossRef]
- Ibanez, B.; James, S.; Agewall, S.; Antunes, M.J.; Bucciarelli-Ducci, C.; Bueno, H.; Caforio, A.L.P.; Crea, F.; Goudevenos, J.A.; Halvorsen, S.; et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2017. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef]
- Daliang, Z.; Lifang, Y.; Hong, F.; Lingling, Z.; Lin, W.; Dapeng, L.; Tianshu, Z.; Weimin, L. Netrin-1 plays a role in the effect of moderate exercise on myocardial fibrosis in rats. PLoS ONE 2019, 14, e0199802. [Google Scholar] [CrossRef]
- Mutlu, H.; Akilli, N.; Cander, B.; Köylü, Ö.; Gul, M.; Köylü, R. Effect of Serum Netrin-1 Levels on Diagnosis and Prognosis in Patients Admitted to the Emergency Service for Acute Coronary Syndrome. Cureus 2020, 12, e7741. [Google Scholar] [CrossRef] [PubMed]
- Leocádio, P.; Menta, P.; Dias, M.; Fraga, J.; Goulart, A.; Santos, I.; Lotufo, P.; Bensenor, I.; Alvarez-Leite, J. Níveis Elevados de Netrina-1 e IL-1β em Mulheres Idosas com SCA: Pior Prognóstico no Acompanhamento de Dois Anos. Arq. Bras. Cardiol. 2020, 114, 507–514. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.-J.; Rama, N.; Imbach, J.; Fiore, S.; Ducarouge, B.; Neves, D.; Chen, H.-W.; Bernard, D.; Yang, P.-C.; Bernet, A.; et al. Cancer-Associated Fibroblasts Produce Netrin-1 to Control Cancer Cell Plasticity. Cancer Res. 2019, 79, 3651–3661. [Google Scholar] [CrossRef] [PubMed]
- Bruikman, C.S.; Vreeken, D.; Zhang, H.; van Gils, M.J.; Peter, J.; van Zonneveld, A.J.; Hovingh, G.K.; van Gils, J.M. The identification and function of a Netrin-1 mutation in a pedigree with premature atherosclerosis. Atherosclerosis 2020, 301, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, J.; Chen, L.; Yuan, Z.; Qin, X.; Wu, Q.; Shen, D.; He, H.; Yu, C. The role of UNC5b in ox-LDL inhibiting migration of RAW264.7 macrophages and the involvement of CCR7. Biochem. Biophys. Res. Commun. 2018, 505, 637–643. [Google Scholar] [CrossRef]
- van Gils, J.M.; Derby, M.C.; Fernandes, L.R.; Ramkhelawon, B.; Ray, T.D.; Rayner, K.J.; Parathath, S.; Distel, E.; Feig, J.L.; Alvarez-Leite, J.I.; et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat. Immunol. 2012, 13, 136–143. [Google Scholar] [CrossRef]
- Schlegel, M.; Sharma, M.; Brown, E.J.; Newman, A.A.C.; Cyr, Y.; Afonso, M.S.; Corr, E.M.; Koelwyn, G.J.; van Solingen, C.; Guzman, J.; et al. Silencing Myeloid Netrin-1 Induces Inflammation Resolution and Plaque Regression. Circ. Res. 2021, 129, 530–546. [Google Scholar] [CrossRef]
- Sharma, M.; Schlegel, M.; Brown, E.J.; Sansbury, B.E.; Weinstock, A.; Afonso, M.S.; Corr, E.M.; van Solingen, C.; Shanley, L.C.; Peled, D.; et al. Netrin-1 Alters Adipose Tissue Macrophage Fate and Function in Obesity. Immunometabolism 2019, 1. [Google Scholar] [CrossRef]
- Zhang, X.; Qin, J.; Wang, X.; Guo, X.; Liu, J.; Wang, X.; Wu, X.; Lu, X.; Li, W.; Liu, X. Netrin-1 improves adipose-derived stem cell proliferation, migration, and treatment effect in type 2 diabetic mice with sciatic denervation. Stem Cell Res. Ther. 2018, 9, 285. [Google Scholar] [CrossRef]
- Jiao, X.; Zhang, D.; Hong, Q.; Yan, L.; Han, Q.; Shao, F.; Cai, G.; Chen, X.; Zhu, H. Netrin-1 works with UNC5B to regulate angiogenesis in diabetic kidney disease. Front. Med. 2020, 14, 293–304. [Google Scholar] [CrossRef]
- Maruyama, K.; Kawasaki, T.; Hamaguchi, M.; Hashimoto, M.; Furu, M.; Ito, H.; Fujii, T.; Takemura, N.; Karuppuchamy, T.; Kondo, T.; et al. Bone-protective Functions of Netrin 1 Protein. J. Biol. Chem. 2016, 291, 23854–23868. [Google Scholar] [CrossRef]
- Mediero, A.; Ramkhelawon, B.; Wilder, T.; Purdue, P.E.; Goldring, S.R.; Dewan, M.Z.; Loomis, C.; Moore, K.J.; Cronstein, B.N. Netrin-1 is highly expressed and required in inflammatory infiltrates in wear particle-induced osteolysis. Ann. Rheum. Dis. 2016, 75, 1706–1713. [Google Scholar] [CrossRef]
- Wang, L.; Gao, Z.; Zhang, J.; Huo, Y.; Xu, Q.; Qiu, Y. Netrin-1 regulates ERK1/2 signaling pathway and autophagy activation in wear particle-induced osteoclastogenesis. Cell Biol. Int. 2021, 45, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Huffman, M.D.; Moran, A.E.; Feigin, V.; Mensah, G.A.; Naghavi, M.; Murray, C.J. Global and regional patterns in cardiovascular mortality from 1990 to 2013. Circulation 2015, 132, 1667–1678. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef]
- Kasikara, C.; Doran, A.C.; Cai, B.; Tabas, I. The role of non-resolving inflammation in atherosclerosis. J. Clin. Investig. 2018, 128, 2713–2723. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Li, X.; Xiong, K.; Song, Z.; Tian, J.; Wen, Y.; Sun, A.; Deng, X. The Entry and Egress of Monocytes in Atherosclerosis: A Biochemical and Biomechanical Driven Process. Cardiovasc. Ther. 2021, 2021, 6642927. [Google Scholar] [CrossRef] [PubMed]
- Bruikman, C.S.; van Gils, J.M. Netrin-1 in coronary artery disease (CAD): Mechanism of action and potential as a therapeutic target. Expert Opin. Ther. Targets 2019, 23, 729–731. [Google Scholar] [CrossRef]
- Shatrov, V.A.; Sumbayev, V.V.; Zhou, J.; Brüne, B. Oxidized low-density lipoprotein (oxLDL) triggers hypoxia-inducible factor-1alpha (HIF-1alpha) accumulation via redox-dependent mechanisms. Blood 2003, 101, 4847–4849. [Google Scholar] [CrossRef]
- Steinberg, D. Low density lipoprotein oxidation and its pathobiological significance. J. Biol. Chem. 1997, 272, 20963–20966. [Google Scholar] [CrossRef]
- Rosenfeld, M.E. Macrophage proliferation in atherosclerosis: An historical perspective. Arterioscler. Thromb. Vasc. Biol. 2014, 34, e21–e22. [Google Scholar] [CrossRef]
- Nadkarni, S.K.; Bilenca, A.; Bouma, B.E.; Tearney, G.J. Measurement of fibrous cap thickness in atherosclerotic plaques by spatiotemporal analysis of laser speckle images. J. Biomed. Opt. 2006, 11, 21006. [Google Scholar] [CrossRef][Green Version]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- van Gils, J.M.; Ramkhelawon, B.; Fernandes, L.; Stewart, M.C.; Guo, L.; Seibert, T.; Menezes, G.B.; Cara, D.C.; Chow, C.; Kinane, T.B.; et al. Endothelial expression of guidance cues in vessel wall homeostasis dysregulation under proatherosclerotic conditions. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Passacquale, G.; Phinikaridou, A.; Warboys, C.; Cooper, M.; Lavin, B.; Alfieri, A.; Andia, M.E.; Botnar, R.M.; Ferro, A. Aspirin-induced histone acetylation in endothelial cells enhances synthesis of the secreted isoform of netrin-1 thus inhibiting monocyte vascular infiltration. Br. J. Pharmacol. 2015, 172, 3548–3564. [Google Scholar] [CrossRef] [PubMed]
- Llodra, J.; Angeli, V.; Liu, J.; Trogan, E.; Fisher, E.A.; Randolph, G.J. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc. Natl. Acad. Sci. USA 2004, 101, 11779–11784. [Google Scholar] [CrossRef] [PubMed]
- Trogan, E.; Feig, J.E.; Dogan, S.; Rothblat, G.H.; Angeli, V.; Tacke, F.; Randolph, G.J.; Fisher, E.A. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3781–3786. [Google Scholar] [CrossRef] [PubMed]
- Bruikman, C.S.; Vreeken, D.; Hoogeveen, R.M.; Bom, M.J.; Danad, I.; Pinto-Sietsma, S.-J.; van Zonneveld, A.J.; Knaapen, P.; Hovingh, G.K.; Stroes, E.S.G.; et al. Netrin-1 and the Grade of Atherosclerosis Are Inversely Correlated in Humans. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Alebrahim, D.; Nayak, M.; Ward, A.; Ursomanno, P.; Shams, R.; Corsica, A.; Sleiman, R.; Fils, K.H.; Silvestro, M.; Boytard, L.; et al. Mapping Semaphorins and Netrins in the Pathogenesis of Human Thoracic Aortic Aneurysms. Int. J. Mol. Sci. 2019, 20, 2100. [Google Scholar] [CrossRef]
- Hadi, T.; Boytard, L.; Silvestro, M.; Alebrahim, D.; Jacob, S.; Feinstein, J.; Barone, K.; Spiro, W.; Hutchison, S.; Simon, R.; et al. Macrophage-derived netrin-1 promotes abdominal aortic aneurysm formation by activating MMP3 in vascular smooth muscle cells. Nat. Commun. 2018, 9, 5022. [Google Scholar] [CrossRef] [PubMed]
- Finucane, M.M.; Stevens, G.A.; Cowan, M.J.; Danaei, G.; Lin, J.K.; Paciorek, C.J.; Singh, G.M.; Gutierrez, H.R.; Lu, Y.; Bahalim, A.N.; et al. National, regional, and global trends in body-mass index since 1980: Systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet 2011, 377, 557–567. [Google Scholar] [CrossRef]
- Ogden, C.L.; Yanovski, S.Z.; Carroll, M.D.; Flegal, K.M. The epidemiology of obesity. Gastroenterology 2007, 132, 2087–2102. [Google Scholar] [CrossRef] [PubMed]
- Berrington de Gonzalez, A.; Hartge, P.; Cerhan, J.R.; Flint, A.J.; Hannan, L.; MacInnis, R.J.; Moore, S.C.; Tobias, G.S.; Anton-Culver, H.; Freeman, L.B.; et al. Body-mass index and mortality among 1.46 million white adults. N. Engl. J. Med. 2010, 363, 2211–2219. [Google Scholar] [CrossRef] [PubMed]
- Flegal, K.M.; Graubard, B.I.; Williamson, D.F.; Gail, M.H. Cause-specific excess deaths associated with underweight, overweight, and obesity. JAMA 2007, 298, 2028–2037. [Google Scholar] [CrossRef]
- Zheng, W.; McLerran, D.F.; Rolland, B.; Zhang, X.; Inoue, M.; Matsuo, K.; He, J.; Gupta, P.C.; Ramadas, K.; Tsugane, S.; et al. Association between body-mass index and risk of death in more than 1 million Asians. N. Engl. J. Med. 2011, 364, 719–729. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W., Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Investig. 2006, 116, 115–124. [Google Scholar] [CrossRef]
- Sanders, T.R.; Kim, D.W.; Glendining, K.A.; Jasoni, C.L. Maternal obesity and IL-6 lead to aberrant developmental gene expression and deregulated neurite growth in the fetal arcuate nucleus. Endocrinology 2014, 155, 2566–2577. [Google Scholar] [CrossRef]
- Yim, J.; Kim, G.; Lee, B.-W.; Kang, E.S.; Cha, B.-S.; Kim, J.-H.; Cho, J.W.; Lee, S.-G.; Lee, Y.-H. Relationship Between Circulating Netrin-1 Concentration, Impaired Fasting Glucose, and Newly Diagnosed Type 2 Diabetes. Front. Endocrinol. 2018, 9, 691. [Google Scholar] [CrossRef]
- Mediero, A.; Wilder, T.; Ramkhelawon, B.; Moore, K.J.; Cronstein, B.N. Netrin-1 and its receptor Unc5b are novel targets for the treatment of inflammatory arthritis. FASEB J. 2016, 30, 3835–3844. [Google Scholar] [CrossRef]
- Zhu, S.; Zhu, J.; Zhen, G.; Hu, Y.; An, S.; Li, Y.; Zheng, Q.; Chen, Z.; Yang, Y.; Wan, M.; et al. Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain. J. Clin. Investig. 2019, 129, 1076–1093. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Ke, X.; Wang, Y.; Feng, X.; Li, Q.; Zhang, Y.; Zhu, J.; Li, Q. The level of netrin-1 is decreased in newly diagnosed type 2 diabetes mellitus patients. BMC Endocr. Disord. 2016, 16, 33. [Google Scholar] [CrossRef] [PubMed]
- Toque, H.A.; Fernandez-Flores, A.; Mohamed, R.; Caldwell, R.B.; Ramesh, G.; Caldwell, R.W. Netrin-1 is a novel regulator of vascular endothelial function in diabetes. PLoS ONE 2017, 12, e0186734. [Google Scholar] [CrossRef]
- Yang, Y.H.C.; Szabat, M.; Bragagnini, C.; Kott, K.; Helgason, C.D.; Hoffman, B.G.; Johnson, J.D. Paracrine signalling loops in adult human and mouse pancreatic islets: Netrins modulate beta cell apoptosis signalling via dependence receptors. Diabetologia 2011, 54, 828–842. [Google Scholar] [CrossRef] [PubMed]
- Binet, F.; Mawambo, G.; Sitaras, N.; Tetreault, N.; Lapalme, E.; Favret, S.; Cerani, A.; Leboeuf, D.; Tremblay, S.; Rezende, F.; et al. Neuronal ER stress impedes myeloid-cell-induced vascular regeneration through IRE1α degradation of netrin-1. Cell Metab. 2013, 17, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Tak, E.; Ridyard, D.; Badulak, A.; Giebler, A.; Shabeka, U.; Werner, T.; Clambey, E.; Moldovan, R.; Zimmerman, M.A.; Eltzschig, H.K.; et al. Protective role for netrin-1 during diabetic nephropathy. J. Mol. Med. 2013, 91, 1071–1080. [Google Scholar] [CrossRef]
- Gao, S.; Zhang, X.; Qin, Y.; Xu, S.; Zhang, J.; Wang, Z.; Wang, W.; Kong, D.; Li, C. Dual actions of Netrin-1 on islet insulin secretion and immune modulation. Clin. Sci. 2016, 130, 1901–1911. [Google Scholar] [CrossRef]
- Gao, R.; Peng, X.; Perry, C.; Sun, H.; Ntokou, A.; Ryu, C.; Gomez, J.L.; Reeves, B.C.; Walia, A.; Kaminski, N.; et al. Macrophage-derived netrin-1 drives adrenergic nerve-associated lung fibrosis. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Kerget, B.; Afşin, D.E.; Kerget, F.; Koçak, A.O.; Aksakal, A.; Kızıltunç, A.; Araz, Ö.; Yılmazel Uçar, E.; Akgün, M. KOAH akut alevlenme hastalarında plasma netrin-1 düzeyinin inflamatuar denge ve komorbiditeler üzerine etkisi. Tuberk. Toraks 2021, 69, 30–38. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Acute Inflammation | ||||
|---|---|---|---|---|
| Model | Receptor | Netrin-1 Expression | Outcome | Ref. |
| Lung Inflammation | ||||
| S. aureus pneumonia in mice | Unc5b | ⇓ | Netrin-1 expression is attenuated in S. aureus septicemia. | [20] |
| LPS and ventilator induced lung injury in mice | Adora2b | ⇓ | Netrin-1 expression is attenuated by inflammation, and netrin-1 limits neutrophil influx into the lung. | [29] |
| LPS induced lung injury in pigs | not defined | Intravenous and inhalative netrin-1 mitigated pulmonary inflammation and lung damage. | [30] | |
| Mouse model with LIRI | Adora2b | ⇓ | Positive correlation between netrin-1 expression and Treg cell population. | [31] |
| An ALI model was established by intratracheal instillation of LPS in C57BL/J mice | Adora2b | ⇓ | Receptor binding has the potential to enhance ENaC-dependent alveolar fluid clearance by supplementation of netrin-1. | [32] |
| Hypobaric hypoxia-induced lung injury in mice | UNC5HB | ⇓ | Pretreatment of netrin-1 dampen ALI and inhibits neutrophil migration. | [33] |
| Abdominal Inflammation | ||||
| Colitis | ||||
| DSS-induced colitis | Adora2b | ⇑ in colon | Netrin-1 is induced during acute colitis limits neutrophil influx into the colonic epithelium. | [34] |
| Peritonitis | ||||
| ZyA-induced peritonitis | Adora2b | ⇓ | Netrin-1 expression is reduced during peritonitis. Exogenous netrin-1 application attenuates inflammation. | [35] |
| ZyA-induced peritonitis | Adora2b | Netrin-1 synergistically interacts with RvD2 in an anti-inflammatory and pro-resolving fashion. | [27] | |
| Pancreatitis | ||||
| L-arginine-induced acute pancreatitis | not defined | ⇓ | Netrin-1 administration reduced pancreas and lung injury. | [36] |
| Acute Kidney Injury | ||||
| Ischemia–reperfusion-induced AKI | not defined | ⇑ protein levels in renal tissue mRNA in renal tissue protein and mRNA levels in intestine | Netrin-1 is induced in renal tissue following IR. Ntn1 administration attenuates renal failure and kidney inflammation. | [37] |
| Ischemia–reperfusion-induced AKI | not defined | not defined | Netrin-1 overexpression attenuates renal failure by decreased apoptosis and increased tubular epithelium proliferation. | [38] |
| Ischemia–reperfusion-induced AKI | Unc5b | ⇓ mRNA in renal tissue | Netrin-1 overexpression attenuates renal failure and systemic inflammation. | [39] |
| Ischemia–reperfusion-induced AKI | not defined | ⇑ renal tissue | Netrin-1 attenuates renal failure and systemic inflammation. | [40] |
| Ischemia–reperfusion-induced AKI | not defined | not defined | Netrin-1 attenuates renal failure and promotes M2 polarization through PPARγ. | [16] |
| Hypoxia | ||||
| In vivo hypoxia | Adora2b | ⇑ in lung and colon via HIF-1α | Netrin-1 attenuates systemic inflammation and neutrophil recruitment. | [13] |
| Liver-Ischemia Reperfusion | ||||
| Adora2b | ⇓ in liver | Netrin-1 attenuates neutrophil recruitment, local and systemic inflammation and promotes resolution and tissue regeneration. | [28] | |
| Myocardial Infarct | ||||
| Normothermic ischemia reperfusion Langendorff perfusion | DCC | not defined | Netrin-1 pre- and postconditioning decrease infarct size and attenuate myocardiocyte apoptosis through ERK1/2 and NO induction. | [41,42,43] |
| Heterotopic cardiac transplant with 8 h of cold ischemia | ⇓ | Netrin-1 protects from reperfusion injury by limiting leukocyte influx, cardiomyocyte apoptosis, and M2 macrophage polarization through PPARg. | [44] | |
| Chronic LAD coronary ligation | not defined | not defined | Intracardial application of netrin-1-transduced mesenchymal stem cells attenuated infarction size and prevented cardiac hypertrophic remodeling. | [45] |
| Normothermic ischemia reperfusion Langendorff perfusion | not defined | not defined | Netrin-1 preconditioning attenuates infarcts size by preventing superoxide and NADPH oxidase production, preventing mitochondrial dysfunction. | [46] |
| Chronic Inflammation | ||||
|---|---|---|---|---|
| Model | Receptor | Netrin-1 Expression | Outcome | Ref. |
| Atherosclerosis | ||||
| Analysis of p.R590L variant of netrin-1 (mutNetrin-1) | UNC5B DCC Neogenin | Decrease in binding capacity to UNC5B and DCC and an increase in binding capacity to neogenin. Stimulation of monocyte adhesion and expression of IL-6, CCL2, and ICAM-1. Diminishing macrophages and smooth muscle cell migration. | [97] | |
| Induction of Raw264.7 macrophages with oxLDL | UNC5B | ⇑ | Downregulation of CCR7 expression and inhibition of macrophage migration. | [98] |
| Bone marrow transplantation of Ntn-1−/−-deficient cells in LDLR−/− KO mice on western diet in plaque progression | UNC5B Neogenin | ⇑ | Hematopoetic netrin-1 KO prevents atherosclerosis development by mitigating MØ and VSMC influx and facilitating MØ egress | [99] |
| Monocyte- and macrophage-specific tamoxifen-inducible CX3CR1-driven cre recombinase netrin-1 floxed mice (Ntn1fl/fl Cx3cr1creERT2+) in plaque regression | UNC5B | Reduced plaque size and complexity in aortic wall, inflammation resolution, IL-10 production, and efferocytosis by myeloid Ntn1 deletion. | [100] | |
| Obesity | ||||
| Mouse model of diet-induced obesity | Unc5B | ⇑ in obese, but not lean adipose tissue | Expression of netrin-1 and its receptor are regulated by saturated fatty acid. Macrophages with a reduced migratory capacity. Restored by blocking netrin-1. | [52] |
| Hematopoietic deletion of Ntn1 | ⇓ | Relief of adipose tissue macrophage emigration, reduction of inflammation, and improvement of insulin sensitivity. | [52] | |
| Mouse model with myeloid- specific deletion of netrin-1 (Ntn1fl/fl LysMCre+/−; Ntn1 mac) | not defined | Ntn1 mac mice: modest decrease in HFD-induced adiposity and adipocyte size. Ntn1 mac macrophages: reduced expression of genes involved in arachidonic acid metabolism and decreases in proinflammatory eicosanoids. Myeloid-specific deletion of netrin-1 caused a decrease of ATMs, particularly the resident macrophage subset. Macrophages reprogram the ATM phenotype, leading to reduced adipose inflammation and improved lipid handling and metabolic function. | [101] | |
| Diabetes | ||||
| Adipose-derived stem cells modified by netrin-1 gene (NTN-1) in vitro, condition of high glucose | ⇑ | Proliferation, migration, adhesion, and inhibition of the apoptosis of ADSCs. | [102] | |
| Injected adipose-derived stem cells modified by netrin-1 gene (NTN-1) in vivo (sciatic denervated mice with type 2 diabetes mellitus) | ⇑ | Capillaries and endothelium were formed by differentiation of N-ADSCs, higher density of microvessels. Upregulation of AKT/PI3K/eNOS/P-38/NF-κB signaling pathways. | [102] | |
| Exogenous netrin-1 in UNC5B-depleted human renal glomerular endothelial cells (HRGECs) | UNC5B | not defined | Inhibition of cell migration and tubulogenesis. Association with SRC pathway deactivation UNC5B antagonizes netrin-1 and that UNC5B upregulation for the enhancement of angiogenesis. | [103] |
| Destructive Joint Disease / Osteoarthritis | ||||
| Female mice with or without knockout of netrin-1 or receptor to detect differences in expression and effect on bone structure. | UNC5B | ⇑ | Activation of SHP1, inhibition of multinucleation of osteoclasts and preventing bone erosion in autoimmune arthritis. | [104] |
| Mouse model with implanted ultrahigh-molecular-weight-polyethylene particles (UHMWPE) over the calvaria and weekly injection of antibodies for netrin-1 and its receptor | UNC5B | not defined | Reduced particle-induced bone pitting, inflammatory processes, and TRAP (tartrate-resistant acid phosphatase)-positive osteoclasts. | [105] |
| RAW 264.7 mouse monocyte macrophages and air pouch model of bone resorption | UNC5B | not defined | Effect of netrin-1 via the ERK1/2 signaling pathway on osteoclast development by promoting autophagy. | [106] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziegon, L.; Schlegel, M. Netrin-1: A Modulator of Macrophage Driven Acute and Chronic Inflammation. Int. J. Mol. Sci. 2022, 23, 275. https://doi.org/10.3390/ijms23010275
Ziegon L, Schlegel M. Netrin-1: A Modulator of Macrophage Driven Acute and Chronic Inflammation. International Journal of Molecular Sciences. 2022; 23(1):275. https://doi.org/10.3390/ijms23010275
Chicago/Turabian StyleZiegon, Laura, and Martin Schlegel. 2022. "Netrin-1: A Modulator of Macrophage Driven Acute and Chronic Inflammation" International Journal of Molecular Sciences 23, no. 1: 275. https://doi.org/10.3390/ijms23010275
APA StyleZiegon, L., & Schlegel, M. (2022). Netrin-1: A Modulator of Macrophage Driven Acute and Chronic Inflammation. International Journal of Molecular Sciences, 23(1), 275. https://doi.org/10.3390/ijms23010275