
Fibromyalgia: Pathogenesis, Mechanisms, Diagnosis and Treatment Options Update




Abstract
1. Fibromyalgia
2. Pathophysiology
2.1. Principal Processes Underlying FM
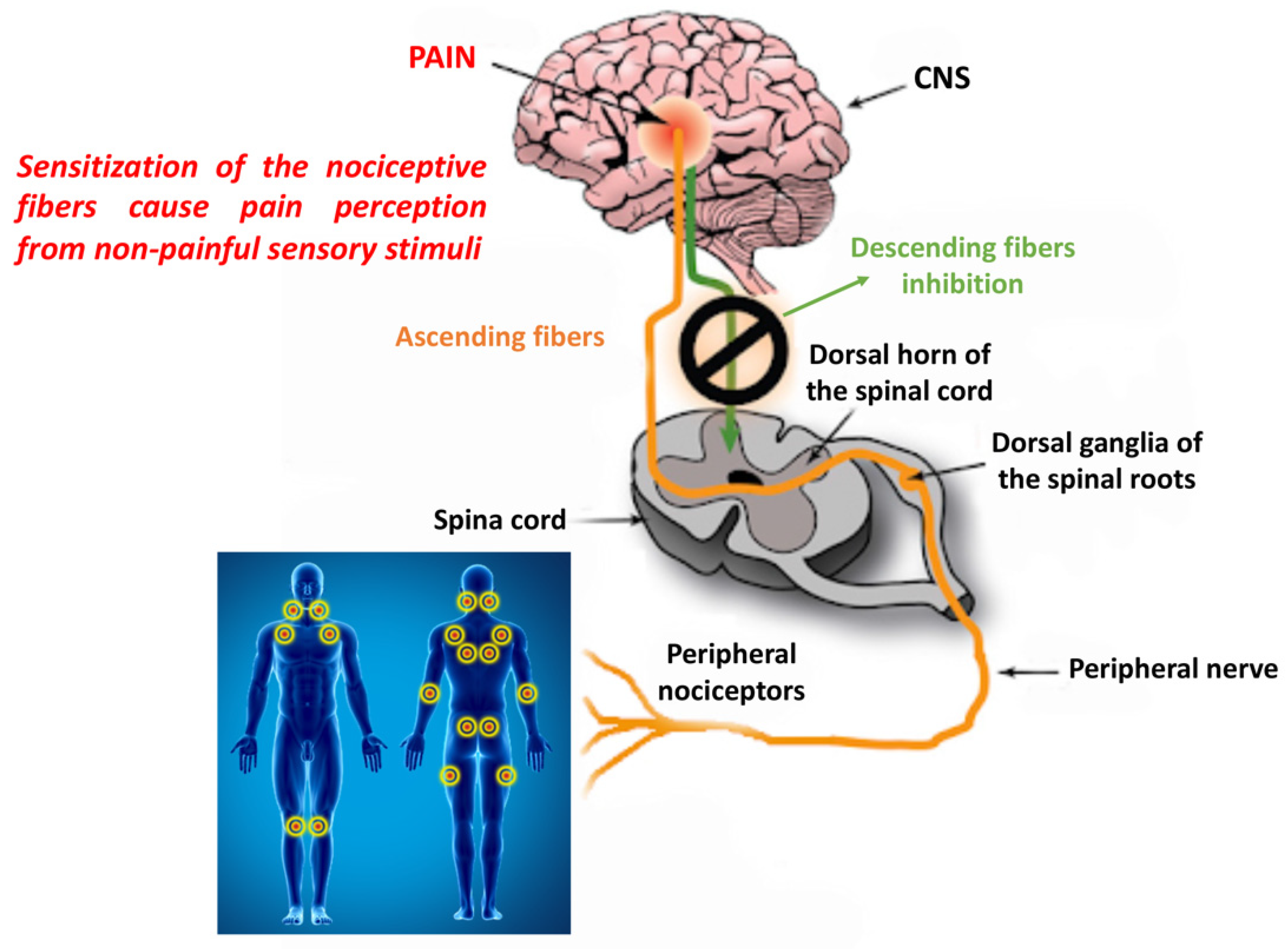
2.1.1. Peripheral and Central Sensitization
2.1.2. Inflammation and Immunity
2.1.3. Genetic Aspects
2.1.4. Endocrine Factors
2.1.5. Psychopathological Factors and Poor Sleep
3. Pain Amplification in FM
4. Diagnostic Biomarkers
4.1. Genetic Approach
4.2. Epigenetic Modifications
4.3. MicroRNAs as Novel Possible Biomarkers
4.4. Gene Expression
4.5. Mu-Opioid Receptor on B Lymphocytes as a Biomarker
5. Serological Markers
5.1. Autoantibodies
5.2. Neuropeptides
5.3. BDNF
5.4. Glutamate
5.5. Inflammatory Cytokines
5.6. Proteomic Approach
5.7. Metabolomic Approach
6. Antioxidants and Diet for Fibromyalgia Management
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gerdle, B.; Bjork, J.; Coster, L.; Henriksson, K.; Henriksson, C.; Bengtsson, A. Prevalence of widespread pain and associations with work status: A population study. BMC Musculoskelet. Disord. 2008, 9, 102. [Google Scholar] [CrossRef]
- Bennett, R.M.; Jones, J.; Turk, D.C.; Russell, I.J.; Matallana, L. An internet survey of 2596 people with fibromyalgia. BMC Musculoskelet. Disord. 2007, 8, 27. [Google Scholar]
- Bellato, E.; Marini, E.; Castoldi, F.; Barbasetti, N.; Mattei, L.; Bonasia, D.E.; Blonna, D. Fibromyalgia syndrome: Etiology, pathogenesis, diagnosis, and treatment. Pain Res. Treat. 2012, 2012, 426130. [Google Scholar] [CrossRef]
- Gowers, W.R. A Lecture on Lumbago: Its Lessons and Analogues: Delivered at the National Hospital for the Paralysed and Epileptic. Br. Med. J. 1904, 1, 117–121. [Google Scholar] [CrossRef]
- Graham, W. The fibrosits syndrome. Bull. Rheum. Dis. 1953, 3, 33–34. [Google Scholar] [PubMed]
- Smythe, H.A.; Moldofsky, H. Two contributions to understanding of the “fibrositis” syndrome. Bull. Rheum. Dis. 1977, 28, 928–931. [Google Scholar]
- Staud, R.; Vierck, C.J.; Cannon, R.L.; Mauderli, A.P.; Price, D.D. Abnormal sensitization and temporal summation of second pain (wind-up) in patients with fibromyalgia syndrome. Pain 2001, 91, 165–175. [Google Scholar] [CrossRef]
- Wolfe, F. New American College of Rheumatology criteria for fibromyalgia: A twenty-year journey. Arthritis Care Res. 2010, 62, 583–584. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, F.; Clauw, D.J.; Fitzcharles, M.A.; Goldenberg, D.L.; Katz, R.S.; Mease, P.; Russell, A.S.; Russell, I.J.; Winfield, J.B.; Yunus, M.B. The American College of Rheumatology preliminary diagnostic criteria for fibromyalgia and measurement of symptom severity. Arthritis Care Res. 2010, 62, 600–610. [Google Scholar] [CrossRef]
- Wolfe, F.; Clauw, D.J.; Fitzcharles, M.A.; Goldenberg, D.L.; Hauser, W.; Katz, R.L.; Mease, P.J.; Russell, A.S.; Russell, I.J.; Walitt, B. 2016 Revisions to the 2010/2011 fibromyalgia diagnostic criteria. Semin. Arthritis Rheum. 2016, 46, 319–329. [Google Scholar] [CrossRef]
- Wolfe, F.; Ross, K.; Anderson, J.; Russell, I.J.; Hebert, L. The prevalence and characteristics of fibromyalgia in the general population. Arthritis Rheum. 1995, 38, 19–28. [Google Scholar] [CrossRef]
- Bhargava, J.; Hurley, J.A. Fibromyalgia. In StatPearls; StatPearls Publishing: Treasure Island, Fl, USA, 2021. [Google Scholar]
- Meyer, H.P. Myofascial pain syndrome and its suggested role in the pathogenesis and treatment of fibromyalgia syndrome. Curr. Pain Headache Rep. 2002, 6, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Clauw, D.J. Fibromyalgia and related conditions. Mayo Clin. Proc. 2015, 90, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Malatji, B.G.; Mason, S.; Mienie, L.J.; Wevers, R.A.; Meyer, H.; van Reenen, M.; Reinecke, C.J. The GC-MS metabolomics signature in patients with fibromyalgia syndrome directs to dysbiosis as an aspect contributing factor of FMS pathophysiology. Metabolomics 2019, 15, 54. [Google Scholar] [CrossRef] [PubMed]
- Bradley, L.A. Pathophysiology of fibromyalgia. Am. J. Med. 2009, 122 (Suppl. S12), S22–S30. [Google Scholar] [CrossRef] [PubMed]
- Yunus, M.B.; Khan, M.A.; Rawlings, K.K.; Green, J.R.; Olson, J.M.; Shah, S. Genetic linkage analysis of multicase families with fibromyalgia syndrome. J. Rheumatol. 1999, 26, 408–412. [Google Scholar] [PubMed]
- Muir, W.W., 3rd; Woolf, C.J. Mechanisms of pain and their therapeutic implications. J. Am. Vet. Med. Assoc. 2001, 219, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Vierck, C.J., Jr. Mechanisms underlying development of spatially distributed chronic pain (fibromyalgia). Pain 2006, 124, 242–263. [Google Scholar] [CrossRef]
- Staud, R.; Nagel, S.; Robinson, M.E.; Price, D.D. Enhanced central pain processing of fibromyalgia patients is maintained by muscle afferent input: A randomized, double-blind, placebo-controlled study. Pain 2009, 145, 96–104. [Google Scholar] [CrossRef][Green Version]
- Brosschot, J.F. Cognitive-emotional sensitization and somatic health complaints. Scand. J. Psychol. 2002, 43, 113–121. [Google Scholar] [CrossRef]
- Overmier, J.B. Sensitization, conditioning, and learning: Can they help us understand somatization and disability? Scand. J. Psychol. 2002, 43, 105–112. [Google Scholar] [CrossRef]
- Jackson, P.L.; Meltzoff, A.N.; Decety, J. How do we perceive the pain of others? A window into the neural processes involved in empathy. Neuroimage 2005, 24, 771–779. [Google Scholar] [CrossRef]
- Collado, A.; Gomez, E.; Coscolla, R.; Sunyol, R.; Sole, E.; Rivera, J.; Altarriba, E.; Carbonell, J.; Castells, X. Work, family and social environment in patients with Fibromyalgia in Spain: An epidemiological study: EPIFFAC study. BMC Health Serv. Res. 2014, 14, 513. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, A.T.; Deitos, A.; Trinanes Pego, Y.; Fregni, F.; Carrillo-de-la-Pena, M.T. Defective Endogenous Pain Modulation in Fibromyalgia: A Meta-Analysis of Temporal Summation and Conditioned Pain Modulation Paradigms. J. Pain 2018, 19, 819–836. [Google Scholar] [CrossRef]
- Harris, R.E.; Clauw, D.J. Imaging central neurochemical alterations in chronic pain with proton magnetic resonance spectroscopy. Neurosci. Lett. 2012, 520, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Clauw, D.J. Fibromyalgia: A clinical review. JAMA 2014, 311, 1547–1555. [Google Scholar] [CrossRef]
- Sluka, K.A.; O’Donnell, J.M.; Danielson, J.; Rasmussen, L.A. Regular physical activity prevents development of chronic pain and activation of central neurons. J. Appl. Physiol. 2013, 114, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Bobinski, F.; Ferreira, T.A.A.; Cordova, M.M.; Dombrowski, P.A.; da Cunha, C.; Santo, C.; Poli, A.; Pires, R.G.W.; Martins-Silva, C.; Sluka, K.A.; et al. Role of brainstem serotonin in analgesia produced by low-intensity exercise on neuropathic pain after sciatic nerve injury in mice. Pain 2015, 156, 2595–2606. [Google Scholar] [CrossRef]
- Yokoyama, T.; Lisi, T.L.; Moore, S.A.; Sluka, K.A. Muscle fatigue increases the probability of developing hyperalgesia in mice. J. Pain 2007, 8, 692–699. [Google Scholar] [CrossRef]
- Sluka, K.A.; Rasmussen, L.A. Fatiguing exercise enhances hyperalgesia to muscle inflammation. Pain 2010, 148, 188–197. [Google Scholar] [CrossRef]
- Gregory, N.S.; Gibson-Corley, K.; Frey-Law, L.; Sluka, K.A. Fatigue-enhanced hyperalgesia in response to muscle insult: Induction and development occur in a sex-dependent manner. Pain 2013, 154, 2668–2676. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Ryals, J.M.; Liu, H.; Liu, W.; Wright, D.E. Acidic saline-induced primary and secondary mechanical hyperalgesia in mice. J. Pain 2009, 10, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Song, J.; Mun, H.; Park, K.U. Effect of the combined use of tramadol and milnacipran on pain threshold in an animal model of fibromyalgia. Korean J. Intern. Med. 2009, 24, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Maeda, Y.; Audette, K.M.; Sluka, K.A. Pregabalin reduces muscle and cutaneous hyperalgesia in two models of chronic muscle pain in rats. J. Pain 2007, 8, 422–429. [Google Scholar] [CrossRef]
- Sharma, N.K.; Ryals, J.M.; Gajewski, B.J.; Wright, D.E. Aerobic exercise alters analgesia and neurotrophin-3 synthesis in an animal model of chronic widespread pain. Phys. Ther. 2010, 90, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Giesecke, T.; Gracely, R.H.; Grant, M.A.; Nachemson, A.; Petzke, F.; Williams, D.A.; Clauw, D.J. Evidence of augmented central pain processing in idiopathic chronic low back pain. Arthritis Rheum. 2004, 50, 613–623. [Google Scholar] [CrossRef]
- Gracely, R.H.; Geisser, M.E.; Giesecke, T.; Grant, M.A.; Petzke, F.; Williams, D.A.; Clauw, D.J. Pain catastrophizing and neural responses to pain among persons with fibromyalgia. Brain 2004, 127 Pt 4, 835–843. [Google Scholar] [CrossRef]
- Wager, T.D.; Atlas, L.Y.; Lindquist, M.A.; Roy, M.; Woo, C.W.; Kross, E. An fMRI-based neurologic signature of physical pain. N. Engl. J. Med. 2013, 368, 1388–1397. [Google Scholar] [CrossRef]
- Segerdahl, A.R.; Mezue, M.; Okell, T.W.; Farrar, J.T.; Tracey, I. The dorsal posterior insula subserves a fundamental role in human pain. Nat. Neurosci. 2015, 18, 499–500. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Tracey, I. Imaging pain: A potent means for investigating pain mechanisms in patients. Br. J. Anaesth. 2013, 111, 64–72. [Google Scholar] [CrossRef]
- Tracey, I. “Seeing” how our drugs work brings translational added value. Anesthesiology 2013, 119, 1247–1248. [Google Scholar] [CrossRef]
- Eippert, F.; Tracey, I. Pain and the PAG: Learning from painful mistakes. Nat. Neurosci. 2014, 17, 1438–1439. [Google Scholar] [CrossRef] [PubMed]
- Napadow, V.; Kim, J.; Clauw, D.J.; Harris, R.E. Decreased intrinsic brain connectivity is associated with reduced clinical pain in fibromyalgia. Arthritis Rheum. 2012, 64, 2398–2403. [Google Scholar] [CrossRef] [PubMed]
- Napadow, V.; LaCount, L.; Park, K.; As-Sanie, S.; Clauw, D.J.; Harris, R.E. Intrinsic brain connectivity in fibromyalgia is associated with chronic pain intensity. Arthritis Rheum. 2010, 62, 2545–2555. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.B.; Loitoile, R.; Kosek, E.; Petzke, F.; Carville, S.; Fransson, P.; Marcus, H.; Williams, S.C.; Choy, E.; Mainguy, Y.; et al. Patients with fibromyalgia display less functional connectivity in the brain’s pain inhibitory network. Mol. Pain 2012, 8, 32. [Google Scholar] [CrossRef]
- Harris, R.E.; Sundgren, P.C.; Craig, A.D.; Kirshenbaum, E.; Sen, A.; Napadow, V.; Clauw, D.J. Elevated insular glutamate in fibromyalgia is associated with experimental pain. Arthritis Rheum. 2009, 60, 3146–3152. [Google Scholar] [CrossRef]
- Fayed, N.; Garcia-Campayo, J.; Magallon, R.; Andres-Bergareche, H.; Luciano, J.V.; Andres, E.; Beltran, J. Localized 1H-NMR spectroscopy in patients with fibromyalgia: A controlled study of changes in cerebral glutamate/glutamine, inositol, choline, and N-acetylaspartate. Arthritis Res. 2010, 12, R134. [Google Scholar] [CrossRef]
- Harris, R.E. Elevated excitatory neurotransmitter levels in the fibromyalgia brain. Arthritis Res. 2010, 12, 141. [Google Scholar] [CrossRef]
- Harris, R.E.; Sundgren, P.C.; Pang, Y.; Hsu, M.; Petrou, M.; Kim, S.H.; McLean, S.A.; Gracely, R.H.; Clauw, D.J. Dynamic levels of glutamate within the insula are associated with improvements in multiple pain domains in fibromyalgia. Arthritis Rheum. 2008, 58, 903–907. [Google Scholar] [CrossRef]
- Harte, S.E.; Clauw, D.J.; Napadow, V.; Harris, R.E. Pressure Pain Sensitivity and Insular Combined Glutamate and Glutamine (Glx) Are Associated with Subsequent Clinical Response to Sham but Not Traditional Acupuncture in Patients Who Have Chronic Pain. Med. Acupunct. 2013, 25, 154–160. [Google Scholar] [CrossRef]
- Foerster, B.R.; Nascimento, T.D.; DeBoer, M.; Bender, M.A.; Rice, I.C.; Truong, D.Q.; Bikson, M.; Clauw, D.J.; Zubieta, J.K.; Harris, R.E.; et al. Excitatory and inhibitory brain metabolites as targets of motor cortex transcranial direct current stimulation therapy and predictors of its efficacy in fibromyalgia. Arthritis Rheumatol. 2015, 67, 576–581. [Google Scholar] [CrossRef]
- Harris, R.E.; Napadow, V.; Huggins, J.P.; Pauer, L.; Kim, J.; Hampson, J.; Sundgren, P.C.; Foerster, B.; Petrou, M.; Schmidt-Wilcke, T.; et al. Pregabalin rectifies aberrant brain chemistry, connectivity, and functional response in chronic pain patients. Anesthesiology 2013, 119, 1453–1464. [Google Scholar] [CrossRef]
- Staud, R.; Vierck, C.J.; Robinson, M.E.; Price, D.D. Effects of the N-methyl-D-aspartate receptor antagonist dextromethorphan on temporal summation of pain are similar in fibromyalgia patients and normal control subjects. J. Pain 2005, 6, 323–332. [Google Scholar] [CrossRef]
- Cohen, S.P.; Verdolin, M.H.; Chang, A.S.; Kurihara, C.; Morlando, B.J.; Mao, J. The intravenous ketamine test predicts subsequent response to an oral dextromethorphan treatment regimen in fibromyalgia patients. J. Pain 2006, 7, 391–398. [Google Scholar] [CrossRef]
- Olivan-Blazquez, B.; Herrera-Mercadal, P.; Puebla-Guedea, M.; Perez-Yus, M.C.; Andres, E.; Fayed, N.; Lopez-Del-Hoyo, Y.; Magallon, R.; Roca, M.; Garcia-Campayo, J. Efficacy of memantine in the treatment of fibromyalgia: A double-blind, randomised, controlled trial with 6-month follow-up. Pain 2014, 155, 2517–2525. [Google Scholar] [CrossRef]
- Holton, K.F.; Taren, D.L.; Thomson, C.A.; Bennett, R.M.; Jones, K.D. The effect of dietary glutamate on fibromyalgia and irritable bowel symptoms. Clin. Exp. Rheumatol. 2012, 30 (6 Suppl. 74), 10–17. [Google Scholar]
- Skyba, D.A.; Lisi, T.L.; Sluka, K.A. Excitatory amino acid concentrations increase in the spinal cord dorsal horn after repeated intramuscular injection of acidic saline. Pain 2005, 119, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, R.; Sluka, K.A. Increased glutamate and decreased glycine release in the rostral ventromedial medulla during induction of a pre-clinical model of chronic widespread muscle pain. Neurosci. Lett. 2009, 457, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Skyba, D.A.; King, E.W.; Sluka, K.A. Effects of NMDA and non-NMDA ionotropic glutamate receptor antagonists on the development and maintenance of hyperalgesia induced by repeated intramuscular injection of acidic saline. Pain 2002, 98, 69–78. [Google Scholar] [CrossRef]
- Da Silva, L.F.; Desantana, J.M.; Sluka, K.A. Activation of NMDA receptors in the brainstem, rostral ventromedial medulla, and nucleus reticularis gigantocellularis mediates mechanical hyperalgesia produced by repeated intramuscular injections of acidic saline in rats. J. Pain 2010, 11, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, L.F.S.; Walder, R.Y.; Davidson, B.L.; Wilson, S.P.; Sluka, K.A. Changes in expression of NMDA-NR1 receptor subunits in the rostral ventromedial medulla modulate pain behaviors. Pain 2010, 151, 155–161. [Google Scholar] [CrossRef]
- Lee, H.; Im, J.; Won, H.; Nam, W.; Kim, Y.O.; Lee, S.W.; Lee, S.; Cho, I.H.; Kim, H.K.; Kwon, J.T.; et al. Effects of tianeptine on symptoms of fibromyalgia via BDNF signaling in a fibromyalgia animal model. Korean J. Physiol. Pharm. 2017, 21, 361–370. [Google Scholar] [CrossRef]
- Messersmith, D.J.; Kim, D.J.; Iadarola, M.J. Transcription factor regulation of prodynorphin gene expression following rat hindpaw inflammation. Brain Res. Mol. Brain Res. 1998, 53, 260–269. [Google Scholar] [CrossRef]
- Anderson, L.E.; Seybold, V.S. Phosphorylated cAMP response element binding protein increases in neurokinin-1 receptor-immunoreactive neurons in rat spinal cord in response to formalin-induced nociception. Neurosci. Lett. 2000, 283, 29–32. [Google Scholar] [CrossRef]
- Wei, F.; Qiu, C.S.; Kim, S.J.; Muglia, L.; Maas, J.W.; Pineda, V.V.; Xu, H.M.; Chen, Z.F.; Storm, D.R.; Muglia, L.J.; et al. Genetic elimination of behavioral sensitization in mice lacking calmodulin-stimulated adenylyl cyclases. Neuron 2002, 36, 713–726. [Google Scholar] [CrossRef]
- Ma, W.; Quirion, R. Increased phosphorylation of cyclic AMP response element-binding protein (CREB) in the superficial dorsal horn neurons following partial sciatic nerve ligation. Pain 2001, 93, 295–301. [Google Scholar] [CrossRef]
- Miletic, G.; Pankratz, M.T.; Miletic, V. Increases in the phosphorylation of cyclic AMP response element binding protein (CREB) and decreases in the content of calcineurin accompany thermal hyperalgesia following chronic constriction injury in rats. Pain 2002, 99, 493–500. [Google Scholar] [CrossRef]
- Hoeger-Bement, M.K.; Sluka, K.A. Phosphorylation of CREB and mechanical hyperalgesia is reversed by blockade of the cAMP pathway in a time-dependent manner after repeated intramuscular acid injections. J. Neurosci. 2003, 23, 5437–5445. [Google Scholar] [CrossRef]
- Chen, W.K.; Liu, I.Y.; Chang, Y.T.; Chen, Y.C.; Chen, C.C.; Yen, C.T.; Shin, H.S.; Chen, C.C. Ca(v)3.2 T-type Ca2+ channel-dependent activation of ERK in paraventricular thalamus modulates acid-induced chronic muscle pain. J. Neurosci. 2010, 30, 10360–10368. [Google Scholar] [CrossRef]
- Cheng, S.J.; Chen, C.C.; Yang, H.W.; Chang, Y.T.; Bai, S.W.; Chen, C.C.; Yen, C.T.; Min, M.Y. Role of extracellular signal-regulated kinase in synaptic transmission and plasticity of a nociceptive input on capsular central amygdaloid neurons in normal and acid-induced muscle pain mice. J. Neurosci. 2011, 31, 2258–2270. [Google Scholar] [CrossRef]
- Oaklander, A.L.; Herzog, Z.D.; Downs, H.M.; Klein, M.M. Objective evidence that small-fiber polyneuropathy underlies some illnesses currently labeled as fibromyalgia. Pain 2013, 154, 2310–2316. [Google Scholar] [CrossRef]
- Uceyler, N.; Zeller, D.; Kahn, A.K.; Kewenig, S.; Kittel-Schneider, S.; Schmid, A.; Casanova-Molla, J.; Reiners, K.; Sommer, C. Small fibre pathology in patients with fibromyalgia syndrome. Brain 2013, 136 Pt 6, 1857–1867. [Google Scholar] [CrossRef]
- Caro, X.J.; Winter, E.F. Evidence of abnormal epidermal nerve fiber density in fibromyalgia: Clinical and immunologic implications. Arthritis Rheumatol. 2014, 66, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Doppler, K.; Rittner, H.L.; Deckart, M.; Sommer, C. Reduced dermal nerve fiber diameter in skin biopsies of patients with fibromyalgia. Pain 2015, 156, 2319–2325. [Google Scholar] [CrossRef] [PubMed]
- Serra, J.; Collado, A.; Sola, R.; Antonelli, F.; Torres, X.; Salgueiro, M.; Quiles, C.; Bostock, H. Hyperexcitable C nociceptors in fibromyalgia. Ann. Neurol. 2014, 75, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Staud, R.; Weyl, E.E.; Bartley, E.; Price, D.D.; Robinson, M.E. Analgesic and anti-hyperalgesic effects of muscle injections with lidocaine or saline in patients with fibromyalgia syndrome. Eur. J. Pain 2014, 18, 803–812. [Google Scholar] [CrossRef]
- Srikuea, R.; Symons, T.B.; Long, D.E.; Lee, J.D.; Shang, Y.; Chomentowski, P.J.; Yu, G.; Crofford, L.J.; Peterson, C.A. Association of fibromyalgia with altered skeletal muscle characteristics which may contribute to postexertional fatigue in postmenopausal women. Arthritis Rheumathol. 2013, 65, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Gurley, K.; Symons, B.; Long, D.; Srikuea, R.; Crofford, L.J.; Peterson, C.A.; Yu, G. Noninvasive optical characterization of muscle blood flow, oxygenation, and metabolism in women with fibromyalgia. Arthritis Res. 2012, 14, R236. [Google Scholar] [CrossRef] [PubMed]
- Dailey, D.L.; Keffala, V.J.; Sluka, K.A. Do cognitive and physical fatigue tasks enhance pain, cognitive fatigue, and physical fatigue in people with fibromyalgia? Arthritis Care Res. 2015, 67, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Law, L.A.F.; Sluka, K.A.; McMullen, T.; Lee, J.; Arendt-Nielsen, L.; Graven-Nielsen, T. Acidic buffer induced muscle pain evokes referred pain and mechanical hyperalgesia in humans. Pain 2008, 140, 254–264. [Google Scholar] [CrossRef]
- Abdelhamid, R.E.; Sluka, K.A. ASICs Mediate Pain and Inflammation in Musculoskeletal Diseases. Physiology 2015, 30, 449–459. [Google Scholar] [CrossRef]
- Sluka, K.A.; Gregory, N.S. The dichotomized role for acid sensing ion channels in musculoskeletal pain and inflammation. Neuropharmacology 2015, 94, 58–63. [Google Scholar] [CrossRef]
- Deval, E.; Gasull, X.; Noel, J.; Salinas, M.; Baron, A.; Diochot, S.; Lingueglia, E. Acid-sensing ion channels (ASICs): Pharmacology and implication in pain. Pharmacol. Ther. 2010, 128, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Molliver, D.C.; Immke, D.C.; Fierro, L.; Pare, M.; Rice, F.L.; McCleskey, E.W. ASIC3, an acid-sensing ion channel, is expressed in metaboreceptive sensory neurons. Mol. Pain 2005, 1, 35. [Google Scholar] [CrossRef] [PubMed]
- Walder, R.Y.; Gautam, M.; Wilson, S.P.; Benson, C.J.; Sluka, K.A. Selective targeting of ASIC3 using artificial miRNAs inhibits primary and secondary hyperalgesia after muscle inflammation. Pain 2011, 152, 2348–2356. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, J.; Spencer, R.H.; Garsky, V.M.; Liang, A.; Leitl, M.D.; Cato, M.J.; Cook, S.P.; Kane, S.; Urban, M.O. Reversal of acid-induced and inflammatory pain by the selective ASIC3 inhibitor, APETx2. Br. J. Pharm. 2010, 161, 950–960. [Google Scholar] [CrossRef]
- Chen, W.N.; Chen, C.C. Acid mediates a prolonged antinociception via substance P signaling in acid-induced chronic widespread pain. Mol. Pain 2014, 10, 30. [Google Scholar] [CrossRef]
- Gregory, N.S.; Brito, R.G.; Fusaro, M.; Sluka, K.A. ASIC3 Is Required for Development of Fatigue-Induced Hyperalgesia. Mol. Neurobiol. 2016, 53, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Sluka, K.A.; Price, M.P.; Breese, N.M.; Stucky, C.L.; Wemmie, J.A.; Welsh, M.J. Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. Pain 2003, 106, 229–239. [Google Scholar] [CrossRef]
- Gautam, M.; Benson, C.J.; Ranier, J.D.; Light, A.R.; Sluka, K.A. ASICs Do Not Play a Role in Maintaining Hyperalgesia Induced by Repeated Intramuscular Acid Injections. Pain Res. Treat. 2012, 2012, 817347. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Katanosaka, K.; Yasui, M.; Hayashi, K.; Yamashita, M.; Wakatsuki, K.; Kiyama, H.; Yamanaka, A.; Mizumura, K. Peripheral and spinal mechanisms of nociception in a rat reserpine-induced pain model. Pain 2015, 156, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Navratilova, E.; Porreca, F. Substance P and Inflammatory Pain: Getting It Wrong and Right Simultaneously. Neuron 2019, 101, 353–355. [Google Scholar] [CrossRef]
- De Felipe, C.; Herrero, J.F.; O’Brien, J.A.; Palmer, J.A.; Doyle, C.A.; Smith, A.J.; Laird, J.M.; Belmonte, C.; Cervero, F.; Hunt, S.P. Altered nociception, analgesia and aggression in mice lacking the receptor for substance P. Nature 1998, 392, 394–397. [Google Scholar] [CrossRef]
- Zimmer, A.; Zimmer, A.M.; Baffi, J.; Usdin, T.; Reynolds, K.; Konig, M.; Palkovits, M.; Mezey, E. Hypoalgesia in mice with a targeted deletion of the tachykinin 1 gene. Proc. Natl. Acad. Sci. USA 1998, 95, 2630–2635. [Google Scholar] [CrossRef] [PubMed]
- Hill, R. NK1 (substance P) receptor antagonists--why are they not analgesic in humans? Trends Pharm. Sci 2000, 21, 244–246. [Google Scholar] [CrossRef]
- Lin, C.C.; Chen, W.N.; Chen, C.J.; Lin, Y.W.; Zimmer, A.; Chen, C.C. An antinociceptive role for substance P in acid-induced chronic muscle pain. Proc. Natl. Acad. Sci. USA 2012, 109, E76–E83. [Google Scholar] [CrossRef] [PubMed]
- Nugraha, B.; Karst, M.; Engeli, S.; Gutenbrunner, C. Brain-derived neurotrophic factor and exercise in fibromyalgia syndrome patients: A mini review. Rheumatol. Int. 2012, 32, 2593–2599. [Google Scholar] [CrossRef] [PubMed]
- Giovengo, S.L.; Russell, I.J.; Larson, A.A. Increased concentrations of nerve growth factor in cerebrospinal fluid of patients with fibromyalgia. J. Rheumatol. 1999, 26, 1564–1569. [Google Scholar]
- Baumeister, D.; Eich, W.; Saft, S.; Geisel, O.; Hellweg, R.; Finn, A.; Svensson, C.I.; Tesarz, J. No evidence for altered plasma NGF and BDNF levels in fibromyalgia patients. Sci. Rep. 2019, 9, 13667. [Google Scholar] [CrossRef] [PubMed]
- Slade, G.D.; Conrad, M.S.; Diatchenko, L.; Rashid, N.U.; Zhong, S.; Smith, S.; Rhodes, J.; Medvedev, A.; Makarov, S.; Maixner, W.; et al. Cytokine biomarkers and chronic pain: Association of genes, transcription, and circulating proteins with temporomandibular disorders and widespread palpation tenderness. Pain 2011, 152, 2802–2812. [Google Scholar] [CrossRef]
- Sturgill, J.; McGee, E.; Menzies, V. Unique cytokine signature in the plasma of patients with fibromyalgia. J. Immunol. Res. 2014, 2014, 938576. [Google Scholar] [CrossRef]
- Mendieta, D.; De la Cruz-Aguilera, D.L.; Barrera-Villalpando, M.I.; Becerril-Villanueva, E.; Arreola, R.; Hernandez-Ferreira, E.; Perez-Tapia, S.M.; Perez-Sanchez, G.; Garces-Alvarez, M.E.; Aguirre-Cruz, L.; et al. IL-8 and IL-6 primarily mediate the inflammatory response in fibromyalgia patients. J. Neuroimmunol. 2016, 290, 22–25. [Google Scholar] [CrossRef]
- Littlejohn, G.; Guymer, E. Neurogenic inflammation in fibromyalgia. Semin. Immunopathol. 2018, 40, 291–300. [Google Scholar] [CrossRef]
- Bote, M.E.; Garcia, J.J.; Hinchado, M.D.; Ortega, E. An exploratory study of the effect of regular aquatic exercise on the function of neutrophils from women with fibromyalgia: Role of IL-8 and noradrenaline. Brain Behav. Immunol. 2014, 39, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Kadetoff, D.; Lampa, J.; Westman, M.; Andersson, M.; Kosek, E. Evidence of central inflammation in fibromyalgia-increased cerebrospinal fluid interleukin-8 levels. J. Neuroimmunol. 2012, 242, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Coskun Benlidayi, I. Role of inflammation in the pathogenesis and treatment of fibromyalgia. Rheumatol. Int. 2019, 39, 781–791. [Google Scholar] [CrossRef]
- Kosek, E.; Altawil, R.; Kadetoff, D.; Finn, A.; Westman, M.; Le Maitre, E.; Andersson, M.; Jensen-Urstad, M.; Lampa, J. Evidence of different mediators of central inflammation in dysfunctional and inflammatory pain--interleukin-8 in fibromyalgia and interleukin-1 beta in rheumatoid arthritis. J. Neuroimmunol. 2015, 280, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Bote, M.E.; Garcia, J.J.; Hinchado, M.D.; Ortega, E. Inflammatory/stress feedback dysregulation in women with fibromyalgia. Neuroimmunomodulation 2012, 19, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Imamura, M.; Targino, R.A.; Hsing, W.T.; Imamura, S.; Azevedo, R.S.; Boas, L.S.; Tozetto-Mendoza, T.R.; Alfieri, F.M.; Filippo, T.R.; Battistella, L.R. Concentration of cytokines in patients with osteoarthritis of the knee and fibromyalgia. Clin. Interv. Aging 2014, 9, 939–944. [Google Scholar]
- Ranzolin, A.; Duarte, A.L.; Bredemeier, M.; da Costa Neto, C.A.; Ascoli, B.M.; Wollenhaupt-Aguiar, B.; Kapczinski, F.; Xavier, R.M. Evaluation of cytokines, oxidative stress markers and brain-derived neurotrophic factor in patients with fibromyalgia–A controlled cross-sectional study. Cytokine 2016, 84, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, F.; Frydas, I.; Ronconi, G.; Kritas, S.K.; Tettamanti, L.; Caraffa, A.C.; DOvidio, C.; Younes, A.; Gallenga, C.E.; Conti, P. Low-grade chronic inflammation mediated by mast cells in fibromyalgia: Role of IL-37. J. Biol. Regul. Homeost. Agents 2018, 32, 195–198. [Google Scholar]
- Cicuttini, F.M.; Wluka, A.E. Not just loading and age: The dynamics of osteoarthritis, obesity and inflammation. Med. J. Aust. 2016, 204, 47. [Google Scholar] [CrossRef]
- Rossi, H.L.; Luu, A.K.; DeVilbiss, J.L.; Recober, A. Obesity increases nociceptive activation of the trigeminal system. Eur. J. Pain 2013, 17, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Rossi, H.L.; Luu, A.K.; Kothari, S.D.; Kuburas, A.; Neubert, J.K.; Caudle, R.M.; Recober, A. Effects of diet-induced obesity on motivation and pain behavior in an operant assay. Neuroscience 2013, 235, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Smart, P.A.; Waylonis, G.W.; Hackshaw, K.V. Immunologic profile of patients with fibromyalgia. Am. J. Phys. Med. Rehabil. 1997, 76, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Batista, E.D.; Andretta, A.; de Miranda, R.C.; Nehring, J.; Dos Santos Paiva, E.; Schieferdecker, M.E. Food intake assessment and quality of life in women with fibromyalgia. Rev. Bras. Reumatol. Engl. Ed. 2016, 56, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Rokyta, R.; Holecek, V.; Pekarkova, I.; Krejcova, J.; Racek, J.; Trefil, L.; Yamamotova, A. Free radicals after painful stimulation are influenced by antioxidants and analgesics. Neuro Endocrinol. Lett. 2003, 24, 304–309. [Google Scholar] [PubMed]
- Mogil, J.S. Pain genetics: Past, present and future. Trends Genet. 2012, 28, 258–266. [Google Scholar] [CrossRef]
- Oertel, B.; Lotsch, J. Genetic mutations that prevent pain: Implications for future pain medication. Pharmacogenomics 2008, 9, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Arnold, L.M.; Fan, J.; Russell, I.J.; Yunus, M.B.; Khan, M.A.; Kushner, I.; Olson, J.M.; Iyengar, S.K. The fibromyalgia family study: A genome-wide linkage scan study. Arthritis Rheum. 2013, 65, 1122–1128. [Google Scholar] [CrossRef]
- Mergener, M.; Becker, R.M.; dos Santos, A.F.; dos Santos, G.A.; de Andrade, F.M. Influence of the interaction between environmental quality and T102C SNP in the HTR2A gene on fibromyalgia susceptibility. Rev. Bras. Reum. 2011, 51, 594–602. [Google Scholar]
- Mickle, A.D.; Shepherd, A.J.; Mohapatra, D.P. Sensory TRP channels: The key transducers of nociception and pain. Prog. Mol. Biol. Transl. Sci. 2015, 131, 73–118. [Google Scholar]
- Lee, Y.H.; Choi, S.J.; Ji, J.D.; Song, G.G. Candidate gene studies of fibromyalgia: A systematic review and meta-analysis. Rheumatol. Int. 2012, 32, 417–426. [Google Scholar] [CrossRef]
- Docampo, E.; Escaramis, G.; Gratacos, M.; Villatoro, S.; Puig, A.; Kogevinas, M.; Collado, A.; Carbonell, J.; Rivera, J.; Vidal, J.; et al. Genome-wide analysis of single nucleotide polymorphisms and copy number variants in fibromyalgia suggest a role for the central nervous system. Pain 2014, 155, 1102–1109. [Google Scholar] [CrossRef]
- Wood, P.B.; Schweinhardt, P.; Jaeger, E.; Dagher, A.; Hakyemez, H.; Rabiner, E.A.; Bushnell, M.C.; Chizh, B.A. Fibromyalgia patients show an abnormal dopamine response to pain. Eur. J. Neurosci. 2007, 25, 3576–3582. [Google Scholar] [CrossRef]
- Gold, S.J.; Ni, Y.G.; Dohlman, H.G.; Nestler, E.J. Regulators of G-protein signaling (RGS) proteins: Region-specific expression of nine subtypes in rat brain. J. Neurosci. 1997, 17, 8024–8037. [Google Scholar] [CrossRef]
- Lu, A.T.; Ogdie, M.N.; Jarvelin, M.R.; Moilanen, I.K.; Loo, S.K.; McCracken, J.T.; McGough, J.J.; Yang, M.H.; Peltonen, L.; Nelson, S.F.; et al. Association of the cannabinoid receptor gene (CNR1) with ADHD and post-traumatic stress disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008, 147B, 1488–1494. [Google Scholar] [CrossRef]
- Park, J.M.; Choi, M.G.; Cho, Y.K.; Lee, I.S.; Kim, S.W.; Choi, K.Y.; Chung, I.S. Cannabinoid receptor 1 gene polymorphism and irritable bowel syndrome in the Korean population: A hypothesis-generating study. J. Clin. Gastroenterol. 2011, 45, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Bleakman, D.; Alt, A.; Nisenbaum, E.S. Glutamate receptors and pain. Semin. Cell Dev. Biol. 2006, 17, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Crofford, L.J.; Pillemer, S.R.; Kalogeras, K.T.; Cash, J.M.; Michelson, D.; Kling, M.A.; Sternberg, E.M.; Gold, P.W.; Chrousos, G.P.; Wilder, R.L. Hypothalamic-pituitary-adrenal axis perturbations in patients with fibromyalgia. Arthritis Rheum. 1994, 37, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- McCain, G.A.; Tilbe, K.S. Diurnal hormone variation in fibromyalgia syndrome: A comparison with rheumatoid arthritis. J. Rheumatol. Suppl. 1989, 19, 154–157. [Google Scholar]
- McLean, S.A.; Williams, D.A.; Harris, R.E.; Kop, W.J.; Groner, K.H.; Ambrose, K.; Lyden, A.K.; Gracely, R.H.; Crofford, L.J.; Geisser, M.E.; et al. Momentary relationship between cortisol secretion and symptoms in patients with fibromyalgia. Arthritis Rheum. 2005, 52, 3660–3669. [Google Scholar] [CrossRef]
- Weissbecker, I.; Floyd, A.; Dedert, E.; Salmon, P.; Sephton, S. Childhood trauma and diurnal cortisol disruption in fibromyalgia syndrome. Psychoneuroendocrinology 2006, 31, 312–324. [Google Scholar] [CrossRef]
- Crofford, L.J.; Young, E.A.; Engleberg, N.C.; Korszun, A.; Brucksch, C.B.; McClure, L.A.; Brown, M.B.; Demitrack, M.A. Basal circadian and pulsatile ACTH and cortisol secretion in patients with fibromyalgia and/or chronic fatigue syndrome. Brain Behav. Immun. 2004, 18, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Abeles, A.M.; Pillinger, M.H.; Solitar, B.M.; Abeles, M. Narrative review: The pathophysiology of fibromyalgia. Ann. Intern. Med. 2007, 146, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Rea, K.; Dinan, T.G.; Cryan, J.F. The microbiome: A key regulator of stress and neuroinflammation. Neurobiol. Stress 2016, 4, 23–33. [Google Scholar] [CrossRef]
- McLean, S.A.; Williams, D.A.; Stein, P.K.; Harris, R.E.; Lyden, A.K.; Whalen, G.; Park, K.M.; Liberzon, I.; Sen, A.; Gracely, R.H.; et al. Cerebrospinal fluid corticotropin-releasing factor concentration is associated with pain but not fatigue symptoms in patients with fibromyalgia. Neuropsychopharmacology 2006, 31, 2776–2782. [Google Scholar] [CrossRef]
- Wingenfeld, K.; Heim, C.; Schmidt, I.; Wagner, D.; Meinlschmidt, G.; Hellhammer, D.H. HPA axis reactivity and lymphocyte glucocorticoid sensitivity in fibromyalgia syndrome and chronic pelvic pain. Psychosom. Med. 2008, 70, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.M.; Cook, D.M.; Clark, S.R.; Burckhardt, C.S.; Campbell, S.M. Hypothalamic-pituitary-insulin-like growth factor-I axis dysfunction in patients with fibromyalgia. J. Rheumatol. 1997, 24, 1384–1389. [Google Scholar]
- Paul-Savoie, E.; Marchand, S.; Morin, M.; Bourgault, P.; Brissette, N.; Rattanavong, V.; Cloutier, C.; Bissonnette, A.; Potvin, S. Is the deficit in pain inhibition in fibromyalgia influenced by sleep impairments? Open Rheumatol. J. 2012, 6, 296–302. [Google Scholar] [CrossRef]
- Okifuji, A.; Turk, D.C. Sex hormones and pain in regularly menstruating women with fibromyalgia syndrome. J. Pain 2006, 7, 851–859. [Google Scholar] [CrossRef]
- Koca, T.; Kocyigit, B.; Seyithanoglu, M.; Berk, E. The Importance of G-protein Coupled Estrogen Receptor in Patients with Fibromyalgia. Arch. Rheumatol. 2019, 34, 419–425. [Google Scholar] [CrossRef]
- Epstein, S.A.; Kay, G.; Clauw, D.; Heaton, R.; Klein, D.; Krupp, L.; Kuck, J.; Leslie, V.; Masur, D.; Wagner, M.; et al. Psychiatric disorders in patients with fibromyalgia. A multicenter investigation. Psychosomatics 1999, 40, 57–63. [Google Scholar] [CrossRef]
- Leino, P.; Magni, G. Depressive and distress symptoms as predictors of low back pain, neck-shoulder pain, and other musculoskeletal morbidity: A 10-year follow-up of metal industry employees. Pain 1993, 53, 89–94. [Google Scholar] [CrossRef]
- Giesecke, T.; Gracely, R.H.; Williams, D.A.; Geisser, M.E.; Petzke, F.W.; Clauw, D.J. The relationship between depression, clinical pain, and experimental pain in a chronic pain cohort. Arthritis Rheum. 2005, 52, 1577–1584. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, P.G.; Balden, E.; Tomkins, G.; Santoro, J.; Kroenke, K.; Jackson, J.L. Treatment of fibromyalgia with antidepressants: A meta-analysis. J. Gen. Intern. Med. 2000, 15, 659–666. [Google Scholar] [CrossRef]
- Jackson, J.L.; O’Malley, P.G.; Tomkins, G.; Balden, E.; Santoro, J.; Kroenke, K. Treatment of functional gastrointestinal disorders with antidepressant medications: A meta-analysis. Am. J. Med. 2000, 108, 65–72. [Google Scholar] [CrossRef]
- Tomkins, G.E.; Jackson, J.L.; O’Malley, P.G.; Balden, E.; Santoro, J.E. Treatment of chronic headache with antidepressants: A meta-analysis. Am. J. Med. 2001, 111, 54–63. [Google Scholar] [CrossRef]
- Moret, C.; Briley, M. Antidepressants in the treatment of fibromyalgia. Neuropsychiatr. Dis. Treat. 2006, 2, 537–548. [Google Scholar] [CrossRef]
- Jennings, E.M.; Okine, B.N.; Roche, M.; Finn, D.P. Stress-induced hyperalgesia. Prog. Neurobiol. 2014, 121, 1–18. [Google Scholar] [CrossRef]
- Michaux, G.P.N.; Magerl, W.; Anton, F.; Treede, R.D. Experimental characterization of the effects of acute stresslike doses of hydrocortisone in human neurogenic hyperalgesia models. Pain 2012, 153, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, L.K.; Michaux, G.P.; Richter, S.; Schachinger, H.; Anton, F. Increased basal mechanical pain sensitivity but decreased perceptual wind-up in a human model of relative hypocortisolism. Pain 2010, 149, 539–546. [Google Scholar] [CrossRef]
- Suarez-Roca, H.; Silva, J.A.; Arcaya, J.L.; Quintero, L.; Maixner, W.; Pinerua-Shuhaibar, L. Role of mu-opioid and NMDA receptors in the development and maintenance of repeated swim stress-induced thermal hyperalgesia. Behav. Brain Res. 2006, 167, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Nasu, T.; Taguchi, T.; Mizumura, K. Persistent deep mechanical hyperalgesia induced by repeated cold stress in rats. Eur. J. Pain 2010, 14, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Nishiyori, M.; Uchida, H.; Nagai, J.; Araki, K.; Mukae, T.; Kishioka, S.; Ueda, H. Permanent relief from intermittent cold stress-induced fibromyalgia-like abnormal pain by repeated intrathecal administration of antidepressants. Mol. Pain 2011, 7, 69. [Google Scholar] [CrossRef]
- Khasar, S.G.; Dina, O.A.; Green, P.G.; Levine, J.D. Sound stress-induced long-term enhancement of mechanical hyperalgesia in rats is maintained by sympathoadrenal catecholamines. J. Pain 2009, 10, 1073–1077. [Google Scholar] [CrossRef]
- Sluka, K.A.; Danielson, J.; Rasmussen, L.; DaSilva, L.F. Exercise-induced pain requires NMDA receptor activation in the medullary raphe nuclei. Med. Sci. Sports Exerc. 2012, 44, 420–427. [Google Scholar] [CrossRef]
- Quintero, J.E.; Dooley, D.J.; Pomerleau, F.; Huettl, P.; Gerhardt, G.A. Amperometric measurement of glutamate release modulation by gabapentin and pregabalin in rat neocortical slices: Role of voltage-sensitive Ca2+ alpha2delta-1 subunit. J. Pharm. Exp. 2011, 338, 240–245. [Google Scholar] [CrossRef]
- Suarez-Roca, H.; Leal, L.; Silva, J.A.; Pinerua-Shuhaibar, L.; Quintero, L. Reduced GABA neurotransmission underlies hyperalgesia induced by repeated forced swimming stress. Behav. Brain Res. 2008, 189, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Itoh, E.; Kawabata, A. Changes in CNS levels of serotonin and its metabolite in SART-stressed (repeatedly cold-stressed) rats. Jpn. J. Pharm. 1991, 56, 101–104. [Google Scholar] [CrossRef]
- Finan, P.H.; Goodin, B.R.; Smith, M.T. The association of sleep and pain: An update and a path forward. J. Pain 2013, 14, 1539–1552. [Google Scholar] [CrossRef]
- Mundal, I.; Grawe, R.W.; Bjorngaard, J.H.; Linaker, O.M.; Fors, E.A. Prevalence and long-term predictors of persistent chronic widespread pain in the general population in an 11-year prospective study: The HUNT study. BMC Musculoskelet. Disord. 2014, 15, 213. [Google Scholar] [CrossRef]
- Haack, M.; Sanchez, E.; Mullington, J.M. Elevated inflammatory markers in response to prolonged sleep restriction are associated with increased pain experience in healthy volunteers. Sleep 2007, 30, 1145–1152. [Google Scholar] [CrossRef]
- Irwin, M.R.; Olmstead, R.; Carrillo, C.; Sadeghi, N.; Fitzgerald, J.D.; Ranganath, V.K.; Nicassio, P.M. Sleep loss exacerbates fatigue, depression, and pain in rheumatoid arthritis. Sleep 2012, 35, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.T.; Edwards, R.R.; McCann, U.D.; Haythornthwaite, J.A. The effects of sleep deprivation on pain inhibition and spontaneous pain in women. Sleep 2007, 30, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Parrino, L.; Grassi, A.; Milioli, G. Cyclic alternating pattern in polysomnography: What is it and what does it mean? Curr. Opin. Pulm. Med. 2014, 20, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, M.; Sarzi-Puttini, P.; Atzeni, F.; Capsoni, F.; Andreoli, A.; Pecis, M.; Colombo, S.; Carrabba, M.; Sergi, M. Cyclic alternating pattern: A new marker of sleep alteration in patients with fibromyalgia? J. Rheumatol. 2004, 31, 1193–1199. [Google Scholar] [PubMed]
- Tiede, W.; Magerl, W.; Baumgartner, U.; Durrer, B.; Ehlert, U.; Treede, R.D. Sleep restriction attenuates amplitudes and attentional modulation of pain-related evoked potentials, but augments pain ratings in healthy volunteers. Pain 2010, 148, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Moldofsky, H.; Scarisbrick, P.; England, R.; Smythe, H. Musculosketal symptoms and non-REM sleep disturbance in patients with “fibrositis syndrome” and healthy subjects. Psychosom. Med. 1975, 37, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Gottshall, J.L.; Adams, Z.M.; Forgacs, P.B.; Schiff, N.D. Daytime Central Thalamic Deep Brain Stimulation Modulates Sleep Dynamics in the Severely Injured Brain: Mechanistic Insights and a Novel Framework for Alpha-Delta Sleep Generation. Front. Neurol. 2019, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, S.; Klerman, E.B.; Adler, G.K.; Kopell, N.J. Thalamic mechanisms underlying alpha-delta sleep with implications for fibromyalgia. J. Neurophysiol. 2015, 114, 1923–1930. [Google Scholar] [CrossRef]
- Roizenblatt, S.; Moldofsky, H.; Benedito-Silva, A.A.; Tufik, S. Alpha sleep characteristics in fibromyalgia. Arthritis Rheum. 2001, 44, 222–230. [Google Scholar] [CrossRef]
- Steriade, M. Corticothalamic resonance, states of vigilance and mentation. Neuroscience 2000, 101, 243–276. [Google Scholar] [CrossRef]
- Steriade, M.; Deschenes, M. The thalamus as a neuronal oscillator. Brain Res. 1984, 320, 1–63. [Google Scholar] [CrossRef]
- Steriade, M.; McCormick, D.A.; Sejnowski, T.J. Thalamocortical oscillations in the sleeping and aroused brain. Science 1993, 262, 679–685. [Google Scholar] [CrossRef]
- Steriade, M.; Amzica, F. Coalescence of sleep rhythms and their chronology in corticothalamic networks. Sleep Res. Online 1998, 1, 1–10. [Google Scholar] [PubMed]
- Principe, J.C.; Smith, J.R. Sleep spindle characteristics as a function of age. Sleep 1982, 5, 73–84. [Google Scholar]
- Landis, C.A.; Lentz, M.J.; Rothermel, J.; Buchwald, D.; Shaver, J.L. Decreased sleep spindles and spindle activity in midlife women with fibromyalgia and pain. Sleep 2004, 27, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.M.; Xiao, Z.; Robinson, E.J.; Jami, A.S.; Zhang, J.; Zhou, H.; Henin, S.E.; Liu, A.A.; Osorio, R.S.; Wang, J.; et al. A deep learning approach for real-time detection of sleep spindles. J. Neural Eng. 2019, 16, 036004. [Google Scholar] [CrossRef]
- Caravan, B.; Hu, L.; Veyg, D.; Kulkarni, P.; Zhang, Q.; Chen, Z.S.; Wang, J. Sleep spindles as a diagnostic and therapeutic target for chronic pain. Mol. Pain 2020, 16, 1744806920902350. [Google Scholar] [CrossRef] [PubMed]
- Granovsky, Y.; Matre, D.; Sokolik, A.; Lorenz, J.; Casey, K.L. Thermoreceptive innervation of human glabrous and hairy skin: A contact heat evoked potential analysis. Pain 2005, 115, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Grayston, R.; Czanner, G.; Elhadd, K.; Goebel, A.; Frank, B.; Uceyler, N.; Malik, R.A.; Alam, U. A systematic review and meta-analysis of the prevalence of small fiber pathology in fibromyalgia: Implications for a new paradigm in fibromyalgia etiopathogenesis. Semin. Arthritis Rheum. 2019, 48, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Uceyler, N.; Sommer, C. Fibromyalgia syndrome: A disease of the small nerve fibers? Z. Rheumatol. 2015, 74, 490–492, 494–495. [Google Scholar] [PubMed]
- Yam, M.F.; Loh, Y.C.; Tan, C.S.; Khadijah Adam, S.; Abdul Manan, N.; Basir, R. General Pathways of Pain Sensation and the Major Neurotransmitters Involved in Pain Regulation. Int. J. Mol. Sci. 2018, 19, 2164. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; von Hehn, C.A.; Woolf, C.J. Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat. Neurosci. 2012, 15, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, L.S.; Eddinger, K.A.; Woller, S.A.; Yaksh, T.L. Origins of antidromic activity in sensory afferent fibers and neurogenic inflammation. Semin. Immunopathol. 2018, 40, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Lerma, C.; Martinez, A.; Ruiz, N.; Vargas, A.; Infante, O.; Martinez-Lavin, M. Nocturnal heart rate variability parameters as potential fibromyalgia biomarker: Correlation with symptoms severity. Arthritis Res. 2011, 13, R185. [Google Scholar] [CrossRef]
- Lerma, C.; Martinez-Martinez, L.A.; Ruiz, N.; Vargas, A.; Infante, O.; Martinez-Lavin, M. Fibromyalgia beyond reductionism. Heart rhythm fractal analysis to assess autonomic nervous system resilience. Scand. J. Rheumatol. 2016, 45, 151–157. [Google Scholar] [CrossRef]
- Dawson, L.F.; Phillips, J.K.; Finch, P.M.; Inglis, J.J.; Drummond, P.D. Expression of alpha1-adrenoceptors on peripheral nociceptive neurons. Neuroscience 2011, 175, 300–314. [Google Scholar] [CrossRef]
- Maestroni, G.J. Sympathetic nervous system influence on the innate immune response. Ann. N. Y. Acad. Sci. 2006, 1069, 195–207. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, D.H.; Oh, D.H.; Clauw, D.J. Characteristic electron microscopic findings in the skin of patients with fibromyalgia--preliminary study. Clin. Rheumatol. 2008, 27, 407–411. [Google Scholar] [CrossRef]
- Leinders, M.; Doppler, K.; Klein, T.; Deckart, M.; Rittner, H.; Sommer, C.; Uceyler, N. Increased cutaneous miR-let-7d expression correlates with small nerve fiber pathology in patients with fibromyalgia syndrome. Pain 2016, 157, 2493–2503. [Google Scholar] [CrossRef]
- Harte, S.E.; Clauw, D.J.; Hayes, J.M.; Feldman, E.L.; St Charles, I.C.; Watson, C.J. Reduced intraepidermal nerve fiber density after a sustained increase in insular glutamate: A proof-of-concept study examining the pathogenesis of small fiber pathology in fibromyalgia. Pain Rep. 2017, 2, e590. [Google Scholar] [CrossRef]
- McLean, S.A.; Clauw, D.J. Biomedical models of fibromyalgia. Disabil. Rehabil. 2005, 27, 659–665. [Google Scholar] [CrossRef]
- Schrepf, A.; Moser, S.; Harte, S.E.; Basu, N.; Kaplan, C.; Kolarik, E.; Tsodikov, A.; Brummett, C.M.; Clauw, D.J. Top down or bottom up? An observational investigation of improvement in fibromyalgia symptoms following hip and knee replacement. Rheumatology 2020, 59, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Hans, G.; Dickenson, A.H. Peripheral input and its importance for central sensitization. Ann. Neurol. 2013, 74, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, F.; Smythe, H.A.; Yunus, M.B.; Bennett, R.M.; Bombardier, C.; Goldenberg, D.L.; Tugwell, P.; Campbell, S.M.; Abeles, M.; Clark, P.; et al. The American College of Rheumatology 1990 Criteria for the Classification of Fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum. 1990, 33, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Julien, N.; Goffaux, P.; Arsenault, P.; Marchand, S. Widespread pain in fibromyalgia is related to a deficit of endogenous pain inhibition. Pain 2005, 114, 295–302. [Google Scholar] [CrossRef]
- Sluka, K.A.; Clauw, D.J. Neurobiology of fibromyalgia and chronic widespread pain. Neuroscience 2016, 338, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Ablin, J.N.; Buskila, D. Update on the genetics of the fibromyalgia syndrome. Best Pract. Res. Clin. Rheumatol. 2015, 29, 20–28. [Google Scholar] [CrossRef]
- D’Agnelli, S.; Arendt-Nielsen, L.; Gerra, M.C.; Zatorri, K.; Boggiani, L.; Baciarello, M.; Bignami, E. Fibromyalgia: Genetics and epigenetics insights may provide the basis for the development of diagnostic biomarkers. Mol. Pain 2019, 15, 1744806918819944. [Google Scholar] [CrossRef] [PubMed]
- Tour, J.; Lofgren, M.; Mannerkorpi, K.; Gerdle, B.; Larsson, A.; Palstam, A.; Bileviciute-Ljungar, I.; Bjersing, J.; Martin, I.; Ernberg, M.; et al. Gene-to-gene interactions regulate endogenous pain modulation in fibromyalgia patients and healthy controls-antagonistic effects between opioid and serotonin-related genes. Pain 2017, 158, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Offenbaecher, M.; Bondy, B.; de Jonge, S.; Glatzeder, K.; Kruger, M.; Schoeps, P.; Ackenheil, M. Possible association of fibromyalgia with a polymorphism in the serotonin transporter gene regulatory region. Arthritis Rheum. 1999, 42, 2482–2488. [Google Scholar] [CrossRef]
- Cohen, H.; Buskila, D.; Neumann, L.; Ebstein, R.P. Confirmation of an association between fibromyalgia and serotonin transporter promoter region (5- HTTLPR) polymorphism, and relationship to anxiety-related personality traits. Arthritis Rheum. 2002, 46, 845–847. [Google Scholar] [CrossRef]
- Gursoy, S. Absence of association of the serotonin transporter gene polymorphism with the mentally healthy subset of fibromyalgia patients. Clin. Rheumatol. 2002, 21, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Jauand, M.; Sitges, C.; Rodriguez, V.; Picornell, A.; Ramon, M.; Buskila, D.; Montoya, P. Pain sensitivity in fibromyalgia is associated with catechol-O-methyltransferase (COMT) gene. Eur. J. Pain 2013, 17, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Inanir, A.; Karakus, N.; Ates, O.; Sezer, S.; Bozkurt, N.; Inanir, S.; Yigit, S. Clinical symptoms in fibromyalgia are associated to catechol-O-methyltransferase (COMT) gene Val158Met polymorphism. Xenobiotica 2014, 44, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.; Neumann, L.; Glazer, Y.; Ebstein, R.P.; Buskila, D. The relationship between a common catechol-O-methyltransferase (COMT) polymorphism val(158) met and fibromyalgia. Clin. Exp. Rheumatol. 2009, 27 (5 Suppl. 56), S51–S56. [Google Scholar]
- Lee, Y.H.; Kim, J.H.; Song, G.G. Association between the COMT Val158Met polymorphism and fibromyalgia susceptibility and fibromyalgia impact questionnaire score: A meta-analysis. Rheumatol. Int. 2015, 35, 159–166. [Google Scholar] [CrossRef]
- Smith, S.B.; Maixner, D.W.; Fillingim, R.B.; Slade, G.; Gracely, R.H.; Ambrose, K.; Zaykin, D.V.; Hyde, C.; John, S.; Tan, K.; et al. Large candidate gene association study reveals genetic risk factors and therapeutic targets for fibromyalgia. Arthritis Rheum. 2012, 64, 584–593. [Google Scholar] [CrossRef]
- Solak, O.; Erdogan, M.O.; Yildiz, H.; Ulasli, A.M.; Yaman, F.; Terzi, E.S.; Ulu, S.; Dundar, U.; Solak, M. Assessment of opioid receptor mu1 gene A118G polymorphism and its association with pain intensity in patients with fibromyalgia. Rheumatol. Int. 2014, 34, 1257–1261. [Google Scholar] [CrossRef]
- Arnold, L.M.; Bennett, R.M.; Crofford, L.J.; Dean, L.E.; Clauw, D.J.; Goldenberg, D.L.; Fitzcharles, M.A.; Paiva, E.S.; Staud, R.; Sarzi-Puttini, P.; et al. AAPT Diagnostic Criteria for Fibromyalgia. J. Pain 2019, 20, 611–628. [Google Scholar] [CrossRef]
- Park, D.J.; Lee, S.S. New insights into the genetics of fibromyalgia. Korean J. Intern. Med. 2017, 32, 984–995. [Google Scholar] [CrossRef]
- Garcia Rodriguez, D.F.; Abud Mendoza, C. Physiopathology of fibromyalgia. Reum. Clin. 2020, 16, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Dolcino, M.; Tinazzi, E.; Puccetti, A.; Lunardi, C. Gene Expression Profiling in Fibromyalgia Indicates an Autoimmune Origin of the Disease and Opens New Avenues for Targeted Therapy. J. Clin. Med. 2020, 9, 1814. [Google Scholar] [CrossRef] [PubMed]
- Park, D.J.; Kim, S.H.; Nah, S.S.; Lee, J.H.; Kim, S.K.; Lee, Y.A.; Hong, S.J.; Kim, H.S.; Lee, H.S.; Kim, H.A.; et al. Polymorphisms of the TRPV2 and TRPV3 genes associated with fibromyalgia in a Korean population. Rheumatology 2016, 55, 1518–1527. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, D.; Cesareo, E.; Mariani, S.; Raskovic, D.; Ientile, R.; Curro, M.; Korkina, L.; De Luca, C. Xenobiotic sensor- and metabolism-related gene variants in environmental sensitivity-related illnesses: A survey on the Italian population. Oxid. Med. Cell Longev. 2013, 2013, 831969. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Russell, I.J.; Liu, Y.G. A brain-derived neurotrophic factor polymorphism Val66Met identifies fibromyalgia syndrome subgroup with higher body mass index and C-reactive protein. Rheumatol. Int. 2012, 32, 2479–2485. [Google Scholar] [CrossRef] [PubMed]
- Haas, L.; Portela, L.V.; Bohmer, A.E.; Oses, J.P.; Lara, D.R. Increased plasma levels of brain derived neurotrophic factor (BDNF) in patients with fibromyalgia. Neurochem. Res. 2010, 35, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Park, D.J.; Kim, S.H.; Nah, S.S.; Lee, J.H.; Kim, S.K.; Lee, Y.A.; Hong, S.J.; Kim, H.S.; Lee, H.S.; Kim, H.A.; et al. Association between brain-derived neurotrophic factor gene polymorphisms and fibromyalgia in a Korean population: A multicenter study. Arthritis Res. 2018, 20, 220. [Google Scholar] [CrossRef] [PubMed]
- Nugraha, B.; Anwar, S.L.; Gutenbrunner, C.; Korallus, C. Polymorphisms of brain-derived neurotrophic factor genes are associated with anxiety and body mass index in fibromyalgia syndrome patients. BMC Res. Notes 2020, 13, 402. [Google Scholar] [CrossRef]
- Polli, A.; Ghosh, M.; Bakusic, J.; Ickmans, K.; Monteyne, D.; Velkeniers, B.; Bekaert, B.; Godderis, L.; Nijs, J. DNA Methylation and Brain-Derived Neurotrophic Factor Expression Account for Symptoms and Widespread Hyperalgesia in Patients with Chronic Fatigue Syndrome and Comorbid Fibromyalgia. Arthritis Rheumatol. 2020, 72, 1936–1944. [Google Scholar] [CrossRef]
- Menzies, V.; Lyon, D.E.; Archer, K.J.; Zhou, Q.; Brumelle, J.; Jones, K.H.; Gao, G.; York, T.P.; Jackson-Cook, C. Epigenetic alterations and an increased frequency of micronuclei in women with fibromyalgia. Nurs. Res. Pract. 2013, 2013, 795784. [Google Scholar] [CrossRef]
- Burri, A.; Marinova, Z.; Robinson, M.D.; Kuhnel, B.; Waldenberger, M.; Wahl, S.; Kunze, S.; Gieger, C.; Livshits, G.; Williams, F. Are Epigenetic Factors Implicated in Chronic Widespread Pain? PLoS ONE 2016, 11, e0165548. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.D.; Gelbart, T.; Whisenant, T.C.; Waalen, J.; Mondala, T.S.; Ikle, D.N.; Salomon, D.R.; Bennett, R.M.; Kurian, S.M. Genome-wide expression profiling in the peripheral blood of patients with fibromyalgia. Clin. Exp. Rheumatol. 2016, 34 (2 Suppl. 96), S89–S98. [Google Scholar] [PubMed]
- Buskila, D.; Cohen, H.; Neumann, L.; Ebstein, R.P. An association between fibromyalgia and the dopamine D4 receptor exon III repeat polymorphism and relationship to novelty seeking personality traits. Mol. Psychiatry 2004, 9, 730–731. [Google Scholar] [CrossRef]
- Gursoy, S.; Erdal, E.; Herken, H.; Madenci, E.; Alasehirli, B.; Erdal, N. Significance of catechol-O-methyltransferase gene polymorphism in fibromyalgia syndrome. Rheumatol. Int. 2003, 23, 104–107. [Google Scholar] [CrossRef]
- Finan, P.H.; Zautra, A.J.; Davis, M.C.; Lemery-Chalfant, K.; Covault, J.; Tennen, H. Genetic influences on the dynamics of pain and affect in fibromyalgia. Health Psychol. 2010, 29, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Alarcon, G.; Fragoso, J.M.; Cruz-Robles, D.; Vargas, A.; Martinez, A.; Lao-Villadoniga, J.I.; Garcia-Fructuoso, F.; Vallejo, M.; Martinez-Lavin, M. Association of adrenergic receptor gene polymorphisms with different fibromyalgia syndrome domains. Arthritis Rheum. 2009, 60, 2169–2173. [Google Scholar] [CrossRef]
- Szyf, M.; Bick, J. DNA methylation: A mechanism for embedding early life experiences in the genome. Child. Dev. 2013, 84, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Denk, F.; McMahon, S.B. Chronic pain: Emerging evidence for the involvement of epigenetics. Neuron 2012, 73, 435–444. [Google Scholar] [CrossRef]
- Wang, F.; Stefano, G.B.; Kream, R.M. Epigenetic modification of DRG neuronal gene expression subsequent to nerve injury: Etiological contribution to complex regional pain syndromes (Part II). Med. Sci. Monit. 2014, 20, 1188–1200. [Google Scholar] [PubMed]
- Seo, S.; Grzenda, A.; Lomberk, G.; Ou, X.M.; Cruciani, R.A.; Urrutia, R. Epigenetics: A promising paradigm for better understanding and managing pain. J. Pain 2013, 14, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Villen, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef]
- Andersen, H.H.; Duroux, M.; Gazerani, P. MicroRNAs as modulators and biomarkers of inflammatory and neuropathic pain conditions. Neurobiol. Dis. 2014, 71, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Bjersing, J.L.; Lundborg, C.; Bokarewa, M.I.; Mannerkorpi, K. Profile of cerebrospinal microRNAs in fibromyalgia. PLoS ONE 2013, 8, e78762. [Google Scholar] [CrossRef]
- Masotti, A.; Baldassarre, A.; Guzzo, M.P.; Iannuccelli, C.; Barbato, C.; Di Franco, M. Circulating microRNA Profiles as Liquid Biopsies for the Characterization and Diagnosis of Fibromyalgia Syndrome. Mol. Neurobiol. 2017, 54, 7129–7136. [Google Scholar] [CrossRef]
- Nakamura, T.; Schwander, S.K.; Donnelly, R.; Ortega, F.; Togo, F.; Broderick, G.; Yamamoto, Y.; Cherniack, N.S.; Rapoport, D.; Natelson, B.H. Cytokines across the night in chronic fatigue syndrome with and without fibromyalgia. Clin. Vaccine Immunol. 2010, 17, 582–587. [Google Scholar] [CrossRef]
- Montana, M.C.; Gereau, R.W. Metabotropic glutamate receptors as targets for analgesia: Antagonism, activation, and allosteric modulation. Curr. Pharm. Biotechnol. 2011, 12, 1681–1688. [Google Scholar] [CrossRef]
- Raffaeli, W.; Malafoglia, V.; Bonci, A.; Tenti, M.; Ilari, S.; Gremigni, P.; Iannuccelli, C.; Gioia, C.; Di Franco, M.; Mollace, V.; et al. Identification of MOR-Positive B Cell as Possible Innovative Biomarker (Mu Lympho-Marker) for Chronic Pain Diagnosis in Patients with Fibromyalgia and Osteoarthritis Diseases. Int. J. Mol. Sci. 2020, 21, 1499. [Google Scholar] [CrossRef]
- Applbaum, E.; Lichtbroun, A. Novel Sjogren’s autoantibodies found in fibromyalgia patients with sicca and/or xerostomia. Autoimmun. Rev. 2019, 18, 199–202. [Google Scholar] [CrossRef]
- Nishikai, M.; Tomomatsu, S.; Hankins, R.W.; Takagi, S.; Miyachi, K.; Kosaka, S.; Akiya, K. Autoantibodies to a 68/48 kDa protein in chronic fatigue syndrome and primary fibromyalgia: A possible marker for hypersomnia and cognitive disorders. Rheumatology 2001, 40, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Arora, N.; Gupta, A.; Reddy, S.B. Antinuclear Antibody and Subserology Testing in the Evaluation of Fibromyalgia: A Teachable Moment. JAMA Intern. Med. 2017, 177, 1369–1370. [Google Scholar] [CrossRef] [PubMed]
- Kotter, I.; Neuscheler, D.; Gunaydin, I.; Wernet, D.; Klein, R. Is there a predisposition for the development of autoimmune diseases in patients with fibromyalgia? Retrospective analysis with long term follow-up. Rheumatol. Int. 2007, 27, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.; Wittrup, I.H.; Wiik, A.; Bliddal, H.; Friis, A.S.; McLaughlin, J.K.; Danneskiold-Samsoe, B.; Olsen, J.H. Antipolymer antibodies in Danish fibromyalgia patients. Clin. Exp. Rheumatol. 2004, 22, 227–229. [Google Scholar] [PubMed]
- Di Franco, M.; Iannuccelli, C.; Alessandri, C.; Paradiso, M.; Riccieri, V.; Libri, F.; Valesini, G. Autonomic dysfunction and neuropeptide Y in fibromyalgia. Clin. Exp. Rheumatol. 2009, 27 (5 Suppl. 56), S75–S78. [Google Scholar]
- Staines, D.R. Is fibromyalgia an autoimmune disorder of endogenous vasoactive neuropeptides? Med. Hypotheses 2004, 62, 665–669. [Google Scholar] [CrossRef]
- Tsilioni, I.; Russell, I.J.; Stewart, J.M.; Gleason, R.M.; Theoharides, T.C. Neuropeptides CRH, SP, HK-1, and Inflammatory Cytokines IL-6 and TNF Are Increased in Serum of Patients with Fibromyalgia Syndrome, Implicating Mast Cells. J. Pharm. Exp. 2016, 356, 664–672. [Google Scholar] [CrossRef]
- Laske, C.; Stransky, E.; Eschweiler, G.W.; Klein, R.; Wittorf, A.; Leyhe, T.; Richartz, E.; Kohler, N.; Bartels, M.; Buchkremer, G.; et al. Increased BDNF serum concentration in fibromyalgia with or without depression or antidepressants. J. Psychiatr. Res. 2007, 41, 600–605. [Google Scholar] [CrossRef]
- Pyke, T.L.; Osmotherly, P.G.; Baines, S. Measuring Glutamate Levels in the Brains of Fibromyalgia Patients and a Potential Role for Glutamate in the Pathophysiology of Fibromyalgia Symptoms: A Systematic Review. Clin. J. Pain 2017, 33, 944–954. [Google Scholar] [CrossRef]
- Clos-Garcia, M.; Andres-Marin, N.; Fernandez-Eulate, G.; Abecia, L.; Lavin, J.L.; van Liempd, S.; Cabrera, D.; Royo, F.; Valero, A.; Errazquin, N.; et al. Gut microbiome and serum metabolome analyses identify molecular biomarkers and altered glutamate metabolism in fibromyalgia. EBioMedicine 2019, 46, 499–511. [Google Scholar] [CrossRef]
- Ernberg, M.; Christidis, N.; Ghafouri, B.; Bileviciute-Ljungar, I.; Lofgren, M.; Bjersing, J.; Palstam, A.; Larsson, A.; Mannerkorpi, K.; Gerdle, B.; et al. Plasma Cytokine Levels in Fibromyalgia and Their Response to 15 Weeks of Progressive Resistance Exercise or Relaxation Therapy. Mediat. Inflamm. 2018, 2018, 3985154. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Linker-Israeli, M.; Hallegua, D.; Silverman, S.; Silver, D.; Weisman, M.H. Cytokines play an aetiopathogenetic role in fibromyalgia: A hypothesis and pilot study. Rheumatology 2001, 40, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Han, C.L.; Sheng, Y.C.; Wang, S.Y.; Chen, Y.H.; Kang, J.H. Serum proteome profiles revealed dysregulated proteins and mechanisms associated with fibromyalgia syndrome in women. Sci. Rep. 2020, 10, 12347. [Google Scholar] [CrossRef] [PubMed]
- Menzies, V.; Starkweather, A.; Yao, Y.; Thacker, L.R., 2nd; Garrett, T.J.; Swift-Scanlan, T.; Kelly, D.L.; Patel, P.; Lyon, D.E. Metabolomic Differentials in Women with and Without Fibromyalgia. Clin. Transl. Sci. 2020, 13, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Menzies, V.; Starkweather, A.; Yao, Y.; Kelly, D.L.; Garrett, T.J.; Yang, G.; Booker, S.; Swift-Scanlan, T.; Mahmud, I.; Lyon, D.E. Exploring Associations Between Metabolites and Symptoms of Fatigue, Depression and Pain in Women with Fibromyalgia. Biol. Res. Nurs. 2021, 23, 119–126. [Google Scholar] [CrossRef]
- Caboni, P.; Liori, B.; Kumar, A.; Santoru, M.L.; Asthana, S.; Pieroni, E.; Fais, A.; Era, B.; Cacace, E.; Ruggiero, V.; et al. Metabolomics analysis and modeling suggest a lysophosphocholines-PAF receptor interaction in fibromyalgia. PLoS ONE 2014, 9, e107626. [Google Scholar] [CrossRef]
- Malatji, B.G.; Meyer, H.; Mason, S.; Engelke, U.F.H.; Wevers, R.A.; van Reenen, M.; Reinecke, C.J. A diagnostic biomarker profile for fibromyalgia syndrome based on an NMR metabolomics study of selected patients and controls. BMC Neurol. 2017, 17, 88. [Google Scholar] [CrossRef] [PubMed]
- Hackshaw, K.V.; Rodriguez-Saona, L.; Plans, M.; Bell, L.N.; Buffington, C.A. A bloodspot-based diagnostic test for fibromyalgia syndrome and related disorders. Analyst 2013, 138, 4453–4462. [Google Scholar] [CrossRef] [PubMed]
- Hackshaw, K.V.; Aykas, D.P.; Sigurdson, G.T.; Plans, M.; Madiai, F.; Yu, L.; Buffington, C.A.T.; Giusti, M.M.; Rodriguez-Saona, L. Metabolic fingerprinting for diagnosis of fibromyalgia and other rheumatologic disorders. J. Biol. Chem. 2019, 294, 2555–2568. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, F. “Silicone related symptoms” are common in patients with fibromyalgia: No evidence for a new disease. J. Rheumatol. 1999, 26, 1172–1175. [Google Scholar]
- Iannuccelli, C.; Di Franco, M.; Alessandri, C.; Guzzo, M.P.; Croia, C.; Di Sabato, F.; Foti, M.; Valesini, G. Pathophysiology of fibromyalgia: A comparison with the tension-type headache, a localized pain syndrome. Ann. N. Y. Acad. Sci 2010, 1193, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Berg, P.A. High incidence of antibodies to 5-hydroxytryptamine, gangliosides and phospholipids in patients with chronic fatigue and fibromyalgia syndrome and their relatives: Evidence for a clinical entity of both disorders. Eur. J. Med. Res. 1995, 1, 21–26. [Google Scholar] [PubMed]
- Werle, E.; Fischer, H.P.; Muller, A.; Fiehn, W.; Eich, W. Antibodies against serotonin have no diagnostic relevance in patients with fibromyalgia syndrome. J. Rheumatol. 2001, 28, 595–600. [Google Scholar] [PubMed]
- Bazzichi, L.; Rossi, A.; Giacomelli, C.; Bombardieri, S. Exploring the abyss of fibromyalgia biomarkers. Clin. Exp. Rheumatol. 2010, 28 (6 Suppl. 63), S125–S130. [Google Scholar]
- Pamuk, O.N.; Cakir, N. The frequency of thyroid antibodies in fibromyalgia patients and their relationship with symptoms. Clin. Rheumatol. 2007, 26, 55–59. [Google Scholar] [CrossRef]
- Bazzichi, L.; Rossi, A.; Zirafa, C.; Monzani, F.; Tognini, S.; Dardano, A.; Santini, F.; Tonacchera, M.; De Servi, M.; Giacomelli, C.; et al. Thyroid autoimmunity may represent a predisposition for the development of fibromyalgia? Rheumatol. Int. 2012, 32, 335–341. [Google Scholar] [CrossRef]
- Ciregia, F.; Giacomelli, C.; Giusti, L.; Bazzichi, L.; Lucacchini, A. Diagnosis of Fibromyalgia Syndrome:Potential Biomarkers and Proteomic Approach. In New Insights into Fibromyalgia; Eilke, W.S., Ed.; Intech: London, UK, 2012. [Google Scholar]
- Maes, M.; Verkerk, R.; Delmeire, L.; Van Gastel, A.; van Hunsel, F.; Scharpe, S. Serotonergic markers and lowered plasma branched-chain-amino acid concentrations in fibromyalgia. Psychiatry Res. 2000, 97, 11–20. [Google Scholar] [CrossRef]
- Bazzichi, L.; Palego, L.; Giannaccini, G.; Rossi, A.; De Feo, F.; Giacomelli, C.; Betti, L.; Giusti, L.; Mascia, G.; Bombardieri, S.; et al. Altered amino acid homeostasis in subjects affected by fibromyalgia. Clin. Biochem. 2009, 42, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Anderberg, U.M.; Liu, Z.; Berglund, L.; Nyberg, F. Elevated plasma levels of neuropeptide Y in female fibromyalgia patients. Eur. J. Pain 1999, 3, 19–30. [Google Scholar] [CrossRef]
- Evengard, B.; Nilsson, C.G.; Lindh, G.; Lindquist, L.; Eneroth, P.; Fredrikson, S.; Terenius, L.; Henriksson, K.G. Chronic fatigue syndrome differs from fibromyalgia. No evidence for elevated substance P levels in cerebrospinal fluid of patients with chronic fatigue syndrome. Pain 1998, 78, 153–155. [Google Scholar] [CrossRef]
- Andersen, M.L.; Nascimento, D.C.; Machado, R.B.; Roizenblatt, S.; Moldofsky, H.; Tufik, S. Sleep disturbance induced by substance P in mice. Behav. Brain Res. 2006, 167, 212–218. [Google Scholar] [CrossRef]
- Sarchielli, P.; Mancini, M.L.; Floridi, A.; Coppola, F.; Rossi, C.; Nardi, K.; Acciarresi, M.; Pini, L.A.; Calabresi, P. Increased levels of neurotrophins are not specific for chronic migraine: Evidence from primary fibromyalgia syndrome. J. Pain 2007, 8, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Valdes, M.; Collado, A.; Bargallo, N.; Vazquez, M.; Rami, L.; Gomez, E.; Salamero, M. Increased glutamate/glutamine compounds in the brains of patients with fibromyalgia: A magnetic resonance spectroscopy study. Arthritis Rheum. 2010, 62, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Bazzichi, L.; Ciregia, F.; Giusti, L.; Baldini, C.; Giannaccini, G.; Giacomelli, C.; Sernissi, F.; Bombardieri, S.; Lucacchini, A. Detection of potential markers of primary fibromyalgia syndrome in human saliva. Proteom. Clin. Appl. 2009, 3, 1296–1304. [Google Scholar] [CrossRef]
- Bagis, S.; Tamer, L.; Sahin, G.; Bilgin, R.; Guler, H.; Ercan, B.; Erdogan, C. Free radicals and antioxidants in primary fibromyalgia: An oxidative stress disorder? Rheumatol. Int. 2005, 25, 188–190. [Google Scholar] [CrossRef]
- Miranda-Díaz, A.G.; Rodríguez-Lara, S.Q. The Role of Oxidants/Antioxidants, Mitochondrial. Dysfunction, and Autophagy in Fibromyalgia. In Discussions of Unusual Topics in Fibromyalgia; Wilke, W.S., Ed.; Inthech: London, UK, 2018. [Google Scholar]
- Quintans-Junior, L.J.; Brito, R.G.; Quintans, J.S.S.; Santos, P.L.; Camargo, Z.T.; Barreto, P.A.; Arrigoni-Blank, M.F.; Lucca-Junior, W.; Scotti, L.; Scotti, M.T.; et al. Nanoemulsion Thermoreversible Pluronic F127-Based Hydrogel Containing Hyptis pectinata (Lamiaceae) Leaf Essential Oil Produced a Lasting Anti-hyperalgesic Effect in Chronic Noninflammatory Widespread Pain in Mice. Mol. Neurobiol. 2018, 55, 1665–1675. [Google Scholar] [CrossRef]
- Lister, R.E. An open, pilot study to evaluate the potential benefits of coenzyme Q10 combined with Ginkgo biloba extract in fibromyalgia syndrome. J. Int. Med. Res. 2002, 30, 195–199. [Google Scholar] [CrossRef]
- Oliveira, M.G.; Brito, R.G.; Santos, P.L.; Araujo-Filho, H.G.; Quintans, J.S.; Menezes, P.P.; Serafini, M.R.; Carvalho, Y.M.; Silva, J.C.; Almeida, J.R.; et al. Alpha-Terpineol, a monoterpene alcohol, complexed with beta-cyclodextrin exerts antihyperalgesic effect in animal model for fibromyalgia aided with docking study. Chem. Biol. Interact. 2016, 254, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Quintans-Junior, L.J.; Araujo, A.A.; Brito, R.G.; Santos, P.L.; Quintans, J.S.; Menezes, P.P.; Serafini, M.R.; Silva, G.F.; Carvalho, F.M.; Brogden, N.K.; et al. Beta-caryophyllene, a dietary cannabinoid, complexed with beta-cyclodextrin produced anti-hyperalgesic effect involving the inhibition of Fos expression in superficial dorsal horn. Life Sci. 2016, 149, 34–41. [Google Scholar] [CrossRef]
- Casanueva, B.; Rodero, B.; Quintial, C.; Llorca, J.; Gonzalez-Gay, M.A. Short-term efficacy of topical capsaicin therapy in severely affected fibromyalgia patients. Rheumatol. Int. 2013, 33, 2665–2670. [Google Scholar] [CrossRef] [PubMed]
- Fusco, R.; Siracusa, R.; D’Amico, R.; Peritore, A.F.; Cordaro, M.; Gugliandolo, E.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. Melatonin Plus Folic Acid Treatment Ameliorates Reserpine-Induced Fibromyalgia: An Evaluation of Pain, Oxidative Stress, and Inflammation. Antioxidants 2019, 8, 628. [Google Scholar] [CrossRef] [PubMed]
- de Zanette, S.A.; Vercelino, R.; Laste, G.; Rozisky, J.R.; Schwertner, A.; Machado, C.B.; Xavier, F.; de Souza, I.C.; Deitos, A.; Torres, I.L.; et al. Melatonin analgesia is associated with improvement of the descending endogenous pain-modulating system in fibromyalgia: A phase II, randomized, double-dummy, controlled trial. BMC Pharm. Toxicol. 2014, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.A.; Al, K., II; Jasim, N.A.; Gorial, F.I. Adjuvant use of melatonin for treatment of fibromyalgia. J. Pineal Res. 2011, 50, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Cordero, M.D.; Cotan, D.; del-Pozo-Martin, Y.; Carrion, A.M.; de Miguel, M.; Bullon, P.; Sanchez-Alcazar, J.A. Oral coenzyme Q10 supplementation improves clinical symptoms and recovers pathologic alterations in blood mononuclear cells in a fibromyalgia patient. Nutrition 2012, 28, 1200–1203. [Google Scholar] [CrossRef] [PubMed]
- Cordero, M.D.; Cano-Garcia, F.J.; Alcocer-Gomez, E.; De Miguel, M.; Sanchez-Alcazar, J.A. Oxidative stress correlates with headache symptoms in fibromyalgia: Coenzyme Q(1)(0) effect on clinical improvement. PLoS ONE 2012, 7, e35677. [Google Scholar] [CrossRef]
- Sawaddiruk, P.; Apaijai, N.; Paiboonworachat, S.; Kaewchur, T.; Kasitanon, N.; Jaiwongkam, T.; Kerdphoo, S.; Chattipakorn, N.; Chattipakorn, S.C. Coenzyme Q10 supplementation alleviates pain in pregabalin-treated fibromyalgia patients via reducing brain activity and mitochondrial dysfunction. Free Radic. Res. 2019, 53, 901–909. [Google Scholar] [CrossRef]
- Joustra, M.L.; Minovic, I.; Janssens, K.A.M.; Bakker, S.J.L.; Rosmalen, J.G.M. Vitamin and mineral status in chronic fatigue syndrome and fibromyalgia syndrome: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0176631. [Google Scholar] [CrossRef]
- Ellis, S.D.; Kelly, S.T.; Shurlock, J.H.; Hepburn, A.L.N. The role of vitamin D testing and replacement in fibromyalgia: A systematic literature review. BMC Rheumatol. 2018, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Gugliandolo, E.; Peritore, A.F.; Piras, C.; Cuzzocrea, S.; Crupi, R. Palmitoylethanolamide and Related ALIAmides: Prohomeostatic Lipid Compounds for Animal Health and Wellbeing. Vet. Sci. 2020, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Peritore, A.F.; Siracusa, R.; Crupi, R.; Cuzzocrea, S. Therapeutic Efficacy of Palmitoylethanolamide and Its New Formulations in Synergy with Different Antioxidant Molecules Present in Diets. Nutrients 2019, 11, 2175. [Google Scholar] [CrossRef]
- D’Amico, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. ALIAmides Update: Palmitoylethanolamide and Its Formulations on Management of Peripheral Neuropathic Pain. Int. J. Mol. Sci. 2020, 21, 5330. [Google Scholar] [CrossRef] [PubMed]
- Impellizzeri, D.; Bruschetta, G.; Cordaro, M.; Crupi, R.; Siracusa, R.; Esposito, E.; Cuzzocrea, S. Micronized/ultramicronized palmitoylethanolamide displays superior oral efficacy compared to nonmicronized palmitoylethanolamide in a rat model of inflammatory pain. J. Neuroinflam. 2014, 11, 136. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Peritore, A.F.; Cordaro, M.; Gugliandolo, E.; Siracusa, R.; Crupi, R.; D’Amico, R.; Fusco, R.; Evangelista, M.; Cuzzocrea, S.; et al. The neuroprotective effects of micronized PEA (PEA-m) formulation on diabetic peripheral neuropathy in mice. FASEB J. 2019, 33, 11364–11380. [Google Scholar] [CrossRef] [PubMed]
- Bartolucci, M.L.; Marini, I.; Bortolotti, F.; Impellizzeri, D.; Di Paola, R.; Bruschetta, G.; Crupi, R.; Portelli, M.; Militi, A.; Oteri, G.; et al. Micronized palmitoylethanolamide reduces joint pain and glial cell activation. Inflamm. Res. 2018, 67, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, R.; Fusco, R.; Cordaro, M.; Peritore, A.F.; D’Amico, R.; Gugliandolo, E.; Crupi, R.; Genovese, T.; Evangelista, M.; Di Paola, R.; et al. The Protective Effects of Pre- and Post-Administration of Micronized Palmitoylethanolamide Formulation on Postoperative Pain in Rats. Int. J. Mol. Sci. 2020, 21, 7700. [Google Scholar] [CrossRef]
- Gatti, A.; Lazzari, M.; Gianfelice, V.; Di Paolo, A.; Sabato, E.; Sabato, A.F. Palmitoylethanolamide in the treatment of chronic pain caused by different etiopathogenesis. Pain Med. 2012, 13, 1121–1130. [Google Scholar] [CrossRef]
- Truini, A.; Biasiotta, A.; Di Stefano, G.; La Cesa, S.; Leone, C.; Cartoni, C.; Federico, V.; Petrucci, M.T.; Cruccu, G. Palmitoylethanolamide restores myelinated-fibre function in patients with chemotherapy-induced painful neuropathy. CNS Neurol. Disord. Drug Targets 2011, 10, 916–920. [Google Scholar] [CrossRef]
- Schifilliti, C.; Cucinotta, L.; Fedele, V.; Ingegnosi, C.; Luca, S.; Leotta, C. Micronized palmitoylethanolamide reduces the symptoms of neuropathic pain in diabetic patients. Pain Res. Treat. 2014, 2014, 849623. [Google Scholar] [CrossRef] [PubMed]
- Passavanti, M.B.; Fiore, M.; Sansone, P.; Aurilio, C.; Pota, V.; Barbarisi, M.; Fierro, D.; Pace, M.C. The beneficial use of ultramicronized palmitoylethanolamide as add-on therapy to Tapentadol in the treatment of low back pain: A pilot study comparing prospective and retrospective observational arms. BMC Anesth. 2017, 17, 171. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, V.; Martini, A.; Bellamoli, P.; Donadello, K.; Schievano, C.; Balzo, G.D.; Sarzi-Puttini, P.; Parolini, M.; Polati, E. Ultramicronized Palmitoylethanolamide (um-PEA) as Add-on Treatment in Fibromyalgia Syndrome (FMS): Retrospective Observational Study on 407 Patients. CNS Neurol. Disord. Drug Targets 2019, 18, 326–333. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Type of Study | References |
|---|---|---|
| 5-HTT | Human | [203,204,205,206] |
| COMT | Human | [207,208,209,210] |
| TAAR1 | Human | [211] |
| Opioid receptor μ1 gene A118G | Human | [212] |
| RGS4 | Human | [121,211] |
| CNR1 | Human | [211,213] |
| GRIA4 | Human | [211,213,214] |
| SLC64A4 | Human | [215,216] |
| TRPV2 | Human | [214,215,217] |
| MYT1L | Human | [214] |
| NRXN3 | Human | [125,215] |
| CYP450 | Human | [218] |
| BDNF | Human | [219,220,221,222,223] |
| NAT15 | Human | [224] |
| HDAC4 | Human | [224] |
| PRKCA | Human | [224,225] |
| RTN1 | Human | [224] |
| PRKG1 | Human | [224] |
| SLC1A5 | Human | [226] |
| SLC25A22 | Human | [226] |
| GRM6 | Human | [226] |
| Markers | Type of Study | References |
|---|---|---|
| Classic autoantibodies (SS-A/Ro, SS-B/La, ANA, and RF) Specific autoantibodies (SP-1, CA6, PSP) | Human | [244,245,246,247,248] |
| Neuropeptides | Human | [249,250,251] |
| BDNF | Human | [220,252] |
| Glutamate | Human | [253,254] |
| Inflammatory cytokines | Human | [102,255,256] |
| Proteomic analysis | Human | [257] |
| Metabolomic analysis | Human | [258,259,260,261,262,263] |
| Compound | Effects | References |
|---|---|---|
| Melatonin | In an animal study, melatonin was able to improve behavioral defects, oxidative and nitrosative stress, mast cell infiltration and activation of microglia in a reserpine-induced FM model. | [287] |
| In a clinical trial, the exogenous administration of 10 mg of melatonin once every 24 h increased endogenous pain inhibition, assessed on a numerical scale (0–10). The combination of amitriptyline and melatonin provided better results than amitriptyline alone, as calculated by the visual analog pain scale, in subjects with FM. | [288] | |
| A randomized trial found that melatonin alone or in combination with fluoxetine was beneficial for the treatment of FM. Using melatonin (3 or 5 mg/day) in combination with 20 mg/day fluoxetine caused a significant reduction in both total and individual components of the Fibromyalgia Impact Questionnaire score compared to the pretreatment values. | [289] | |
| Coenzyme Q10 | CoQ10 treatment showed effects on clinical symptoms, blood mononuclear cells and markers of mitochondrial and oxidative stress in women with FM. | [290] |
| The results of this clinical study suggest that CoQ10 supplementation plays a role in the modulation of mitochondrial dysfunction and oxidative stress that induce headaches in individuals with FM. | [291] | |
| In a clinical study, CoQ10 supplementation was shown to provide additional benefits for relieving pain sensation in FM patients treated with pregabalin, possibly by improving mitochondrial function, reducing inflammation and decreasing brain activity. | [292] | |
| Vitamins D and E | A clinical study found that women with FM had a lower qualitative and quantitative intake than control subjects. In particular, an association has been found between vitamin D deficiency and FM. However, its role in FM pathophysiology and the clinical relevance of its identification and treatment requires further clarification. Only vitamin E appears to be related to quality of life and pain sensation. | [117,293,294] |
| Palmitoylethanolamide (PEA) | PEA is a major anti-inflammatory, analgesic and neuroprotective mediator in central and peripheral organs and systems and acts on several molecular targets. | [295,296] |
| PEA is emerging as a candidate biomarker due to its anti-inflammatory and anti-hyperalgesic effects via the downregulation of mast cell activation. Preclinical and clinical studies support the idea that PEA merits further consideration as a therapeutic approach for controlling inflammatory responses, pain, related peripheral neuropathic pain and symptoms of FM. | [297,298,299,300,301,302,303,304,305,306] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siracusa, R.; Paola, R.D.; Cuzzocrea, S.; Impellizzeri, D. Fibromyalgia: Pathogenesis, Mechanisms, Diagnosis and Treatment Options Update. Int. J. Mol. Sci. 2021, 22, 3891. https://doi.org/10.3390/ijms22083891
Siracusa R, Paola RD, Cuzzocrea S, Impellizzeri D. Fibromyalgia: Pathogenesis, Mechanisms, Diagnosis and Treatment Options Update. International Journal of Molecular Sciences. 2021; 22(8):3891. https://doi.org/10.3390/ijms22083891
Chicago/Turabian StyleSiracusa, Rosalba, Rosanna Di Paola, Salvatore Cuzzocrea, and Daniela Impellizzeri. 2021. "Fibromyalgia: Pathogenesis, Mechanisms, Diagnosis and Treatment Options Update" International Journal of Molecular Sciences 22, no. 8: 3891. https://doi.org/10.3390/ijms22083891
APA StyleSiracusa, R., Paola, R. D., Cuzzocrea, S., & Impellizzeri, D. (2021). Fibromyalgia: Pathogenesis, Mechanisms, Diagnosis and Treatment Options Update. International Journal of Molecular Sciences, 22(8), 3891. https://doi.org/10.3390/ijms22083891