
Hereditary Prostate Cancer: Genes Related, Target Therapy and Prevention


 ,
,
Abstract
1. Introduction
2. Epidemiology of Hereditary Prostate Cancer
3. Genes Involved in the Predisposition to Hereditary Prostate Cancer
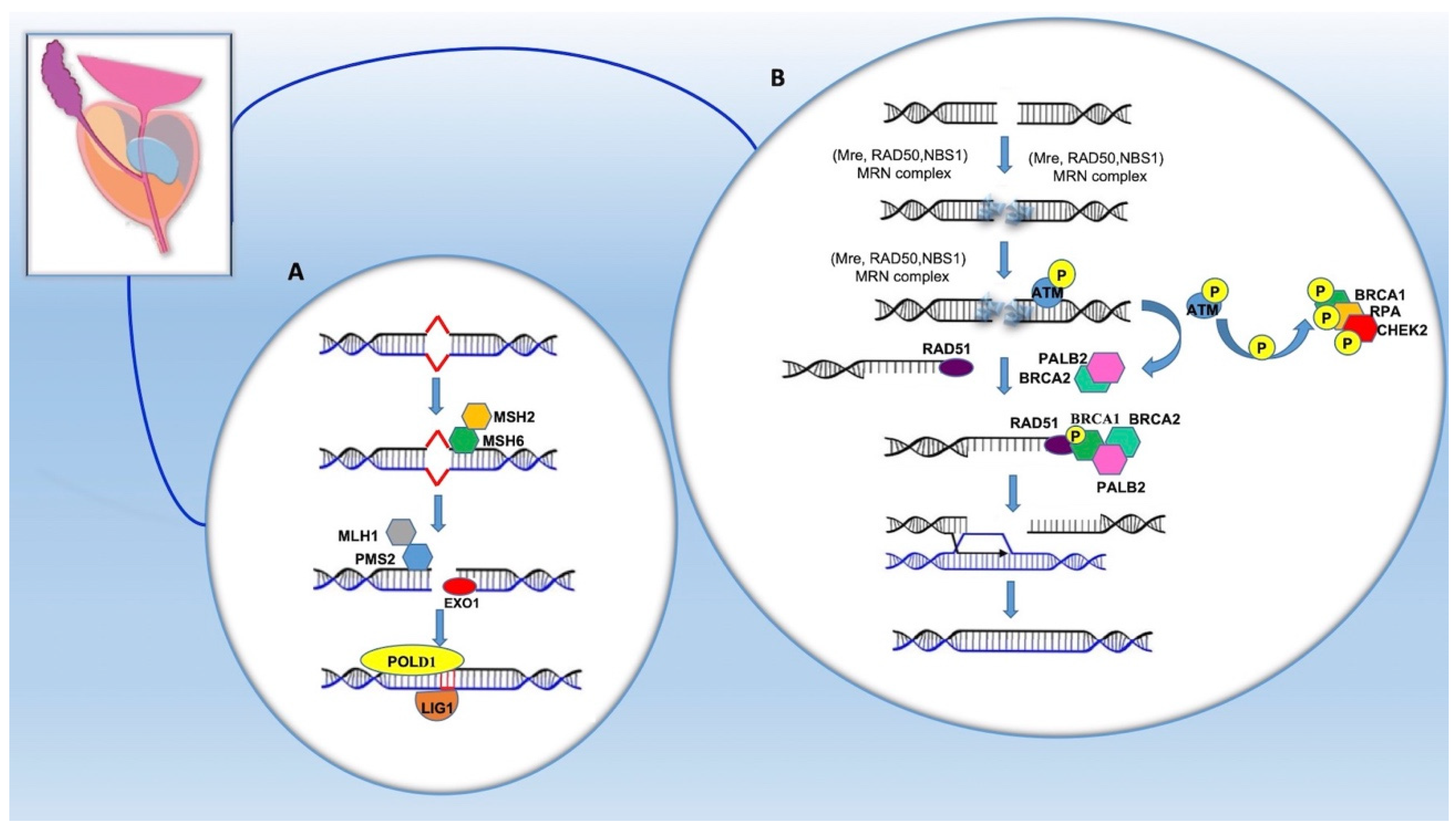
4. Pathogenetic Mechanisms of PCa Onset
5. Mutations and Genotype–Phenotype Correlation
6. Therapeutic Target
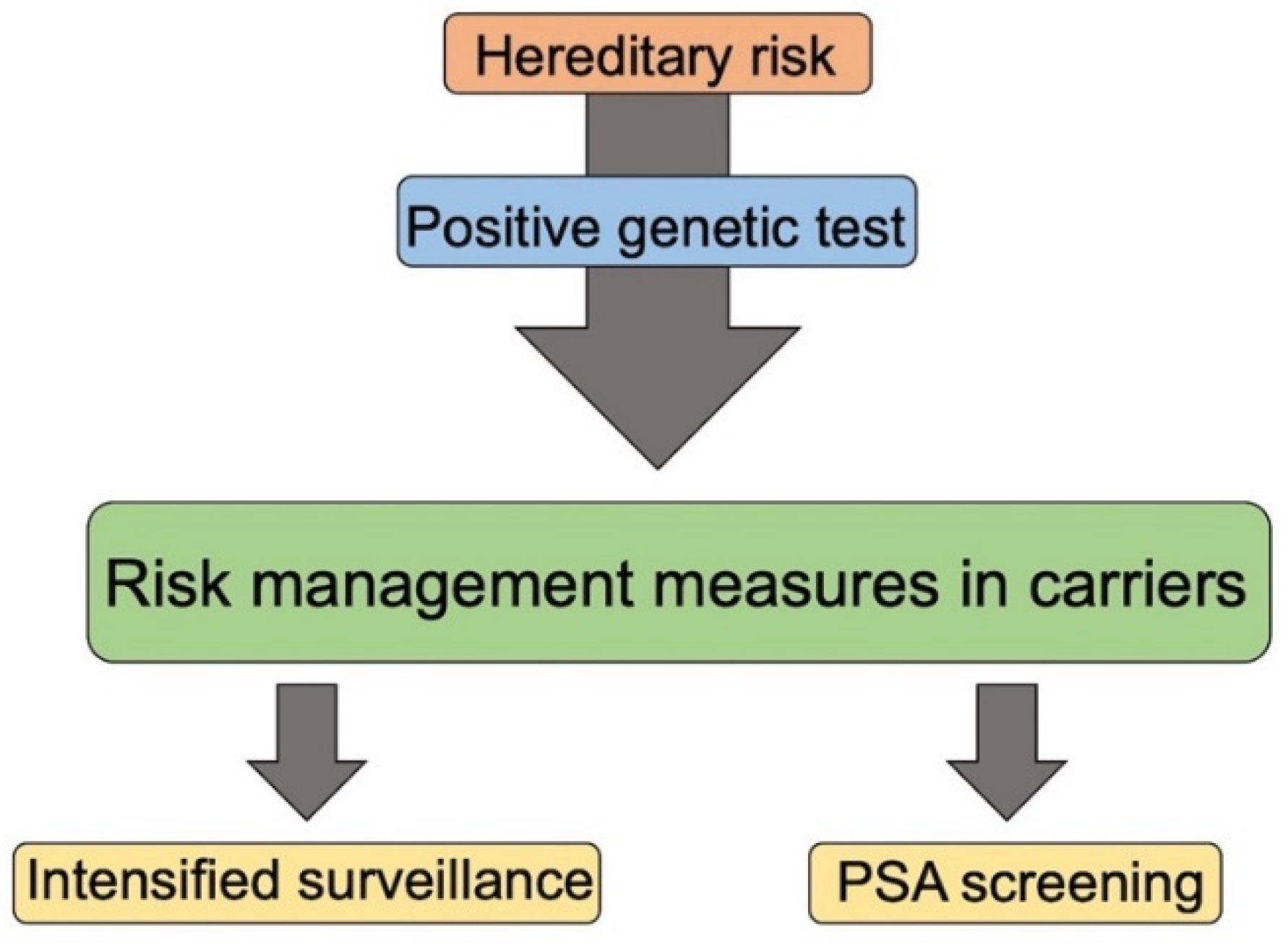
7. Genetic Counselling, Guidelines for Genetic Test and Surveillance
8. Liquid Biopsy
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Virtanen, V.; Paunu, K.; Ahlskog, J.K.; Varnai, R.; Sipeky, C.; Sundvall, M. PARP Inhibitors in Prostate Cancer—The Preclinical Rationale and Current Clinical Development. Genes 2019, 10, 565. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Castelli, G.; Pelosi, E. Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications. Medicines 2019, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Giri, V.N.; Beebe-Dimmer, J.L. Familial prostate cancer. Semin Oncol. 2016, 43, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Thalgott, M.; Kron, M.; Brath, J.M.; Ankerst, D.P.; Thompson, I.M.; Gschwend, J.E.; Herkommer, K. Men with family history of prostate cancer have a higher risk of disease recurrence after radical prostatectomy. World J. Urol. 2018, 36, 177–185. [Google Scholar] [CrossRef]
- Cooney, K.A. Inherited Predisposition to Prostate Cancer: From Gene Discovery to Clinical Impact. Trans. Am. Clin. Climatol. Assoc. 2017, 128, 14–23. [Google Scholar]
- Heidegger, I.; Tsaur, I.; Borgmann, H.; Surcel, C.; Kretschmer, A.; Mathieu, R.; Visschere, P.; Valerio, M.; van den Bergh, R.C.N.; Ost, P.; et al. EAU-YAU Prostate Cancer Working Party. Hereditary prostate cancer—Primetime for genetic testing? Cancer Treat. Rev. 2019, 81, 101927. [Google Scholar] [CrossRef]
- Leongamornlert, D.; Saunders, E.; Dadaev, T.; Tymrakiewicz, M.; Goh, C.; Jugurnauth-Little, S.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Barrowdale, D.; et al. Frequent germline deleterious mutations in DNA repair genes in familial prostate cancer cases are associated with advanced disease. Br. J. Cancer 2014, 110, 1663–1672. [Google Scholar] [CrossRef]
- Ren, Z.J.; Cao, D.H.; Zhang, Q.; Ren, P.W.; Liu, L.R.; Wei, Q.; Wei, W.R.; Dong, Q. First-degree family history of breast cancer is associated with prostate cancer risk: A systematic review and meta-analysis. BMC Cancer 2019, 19, 871. [Google Scholar] [CrossRef]
- Giri, V.N.; Knudsen, K.E.; Kelly, W.K.; Abida, W.; Andriole, G.L.; Bangma, C.H.; Bekelman, J.E.; Benson, M.C.; Blanco, A.; Burnett, A.; et al. Role of Genetic Testing for Inherited Prostate Cancer Risk: Philadelphia Prostate Cancer Consensus Conference 2017. J. Clin. Oncol. 2018, 36, 414–424. [Google Scholar] [CrossRef]
- Barber, L.; Gerke, T.; Markt, S.C.; Peisch, S.F.; Wilson, K.M.; Ahearn, T.; Giovannucci, E.; Parmigiani, G.; Mucci, L.A. Family History of Breast or Prostate Cancer and Prostate Cancer Risk. Clin. Cancer Res. 2018, 24, 5910–5917. [Google Scholar] [CrossRef]
- Rebbeck, T.R. Prostate Cancer Genetics: Variation by Race, Ethnicity, and Geography. Semin. Radiat. Oncol. 2017, 27, 3–10. [Google Scholar] [CrossRef]
- Kohaar, I.; Petrovics, G.; Srivastava, S. A Rich Array of Prostate Cancer Molecular Biomarkers: Opportunities and Challenges. Int. J. Mol. Sci. 2019, 20, 1813. [Google Scholar] [CrossRef]
- Sokolova, A.O.; Cheng, H.H. Genetic Testing in Prostate Cancer. Curr. Oncol. Rep. 2020, 22, 5. [Google Scholar] [CrossRef] [PubMed]
- Rantapero, T.; Wahlfors, T.; Kähler, A.; Hultman, C.; Lindberg, J.; Tammela, T.L.; Nykter, M.; Schleutker, J.; Wiklund, F. Inherited DNA Repair Gene Mutations in Men with Lethal Prostate Cancer. Genes 2020, 11, 314. [Google Scholar] [CrossRef] [PubMed]
- Zhen, J.T.; Syed, J.; Nguyen, K.A.; Leapman, M.S.; Agarwal, N.; Brierley, K.; Llor, X.; Hofstatter, E.; Shuch, B. Genetic testing for hereditary prostate cancer: Current status and limitations. Cancer 2018, 124, 3105–3117. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Salami, S.S.; Spratt, D.E.; Kaffenberger, S.D.; Jacobs, M.F.; Morgan, T.M. Bringing Prostate Cancer Germline Genetics into Clinical Practice. J. Urol. 2019, 202, 223–230. [Google Scholar] [CrossRef]
- Nombela, P.; Lozano, R.; Aytes, A.; Mateo, J.; Olmos, D.; Castro, E. BRCA2 and Other DDR Genes in Prostate Cancer. Cancers 2019, 11, 352. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Cook, M.B.; Wang, Z.; Yeboah, E.D.; Tettey, Y.; Biritwum, R.B.; Adjei, A.A.; Tay, E.; Truelove, A.; Niwa, S.; Chung, C.C.; et al. A genome-wide association study of prostate cancer in West African men. Hum. Genet. 2014, 133, 509–521. [Google Scholar] [CrossRef]
- Tan, S.H.; Petrovics, G.; Srivastava, S. Prostate Cancer Genomics: Recent Advances and the Prevailing Underrepresentation from Racial and Ethnic Minorities. Int. J. Mol. Sci. 2018, 19, 1255. [Google Scholar] [CrossRef]
- Pritzlaff, M.; Tian, Y.; Reineke, P.; Stuenkel, A.J.; Allen, K.; Gutierrez, S.; Jackson, M.; Dolinsky, J.S.; LaDuca, H.; Xu, J.; et al. Diagnosing hereditary cancer predisposition in men with prostate cancer. Genet. Med. 2020, 22, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network (NCCN). Genetic/Familial High-Risk Assessment: Breast and Ovarian; Version 1.2018; National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2018. [Google Scholar]
- Pilarski, R. The Role of BRCA Testing in Hereditary Pancreatic and Prostate Cancer Families. Am. Soc. Clin. Oncol. Educ. Book. 2019, 39, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Ewing, C.M.; Ray, A.M.; Lange, E.M.; Zuhlke, K.A.; Robbins, C.M.; Tembe, W.D.; Wiley, K.E.; Isaacs, S.D.; Johng, D.; Wang, Y.; et al. Germline mutations in HOXB13 and prostate-cancer risk. N. Engl. J. Med. 2012, 12, 141–149. [Google Scholar] [CrossRef]
- Beebe-Dimmer, J.L.; Hathcock, M.; Yee, C.; Okoth, L.A.; Ewing, C.M.; Isaacs, W.B.; Cooney, K.A.; Thibodeau, S.N. The HOXB13 G84E mutation is associated with an increased risk for prostate cancer and other malignancies. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Shi, Z.; Na, R.; Wang, C.H.; Resurreccion, W.K.; Zheng, S.L.; Hulick, P.J.; Cooney, K.A.; Helfand, B.T.; Isaacs, W.B.; et al. Germline HOXB13 G84E mutation carriers and risk to twenty common types of cancer: Results from the UK Biobank. Br. J. Cancer 2020, 123, 1356–1359. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; Caliendo, G.; D’Elia, G.; Resse, M.; Casamassimi, A.; Minucci, P.B.; Dello Ioio, C.; Cioffi, M.; Molinari, A.M. Five Italian Families with Two Mutations in BRCA Genes. Genes 2020, 11, 1451. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.M.; Evans, D.G.; Hope, Q.; Norman, A.R.; Barbachano, Y.; Bullock, S.; Kote-Jarai, Z.; Meitz, J.; Falconer, A.; Osin, P.; et al. Prostate cancer in BRCA2 germline mutation carriers is associated with poorer prognosis. Br. J. Cancer 2010, 7, 918–924. [Google Scholar] [CrossRef]
- Powell, S.N.; Kachnic, L.A. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene 2003, 22, 5784–5789. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.; Goh, C.L.; Eeles, R.A. Prostate cancer screening in BRCA and Lynch syndrome mutation carriers. Am. Soc. Clin. Oncol. Educ. Book 2013. [Google Scholar] [CrossRef]
- Mayrhofer, M.; De Laere, B.; Whitington, T.; Van Oyen, P.; Ghysel, C.; Ampe, J.; Ost, P.; Demey, W.; Hoekx, L.; Schrijvers, D.; et al. Cell-free DNA profiling of metastatic prostate cancer reveals microsatellite instability, structural rearrangements and clonal hematopoiesis. Genome Med. 2018, 10, 85. [Google Scholar] [CrossRef]
- Nanda, N.; Roberts, N.J. ATM serine/threonine kinase and its role in pancreatic risk. Genes. 2020, 11, 108. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.H.; Sokolova, A.O.; Schaeffer, E.M.; Small, E.J.; Higano, C.S. Germline and Somatic Mutations in Prostate Cancer for the Clinician. J. Natl. Compr. Cancer Network 2019, 17, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Athie, A.; Arce-Gallego, S.; Gonzalez, M.; Morales-Barrera, R.; Suarez, C.; Casals Galobart, T.; Hernandez Viedma, G.; Carles, J.; Mateo, J. Targeting DNA Repair Defects for Precision Medicine in Prostate Cancer. Curr. Oncol. Rep. 2019, 21, 42. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; D’Elia, G.; Caliendo, G.; Casamassimi, A.; Federico, A.; Passariello, L.; Cioffi, M.; Molinari, A.M. Prevalence of mutations in BRCA and MMR genes in patients affected with hereditary endometrial cancer. Med. Oncol. 2021, 38, 13. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.A.; Leininger, A. The genetic basis of Lynch syndrome and its implications for clinical practice and risk management. Appl. Clin. Genet. 2014, 7, 147–158. [Google Scholar] [CrossRef]
- Haraldsdottir, S.; Hampel, H.; Wei, L.; Wu, C.; Frankel, W.; Bekaii-Saab, T.; De La Chapelle, A.; Goldberg, R.M. Prostate cancer incidence in males with Lynch syndrome. Genet. Med. 2014, 16, 553–557. [Google Scholar] [CrossRef]
- Brandão, A.; Paulo, P.; Teixeira, M.R. Hereditary Predisposition to Prostate Cancer: From Genetics to Clinical Implications. Int. J. Mol. Sci. 2020, 21, 5036. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; Caliendo, G.; Schiano, C.; Casamassimi, A.; Molinari, A.M.; Napoli, C.; Cioffi, M. Analysis of PALB2 in a cohort of Italian breast cancer patients: Identification of a novel PALB2 truncating mutation. Fam. Cancer 2015, 14, 341–348. [Google Scholar] [CrossRef]
- Nicolosi, P.; Ledet, E.; Yang, S.; Michalski, S.; Freschi, B.; O’Leary, E.; Esplin, E.D.; Nussbaum, R.L.; Sartor, O. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 2019, 5, 523–528. [Google Scholar] [CrossRef]
- De Bono, J.S.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes inWomen With Ovarian Cancer. J. Natl. Cancer Inst. 2015, 107, 1. [Google Scholar] [CrossRef]
- Kote-Jarai, Z.; Jugurnauth, S.; Mulholland, S.; Leongamornlert, D.A.; Guy, M.; Edwards, S.; Tymrakiewitcz, M.; O’brien, L.; Hall, A.; Wilkinson, R.; et al. A recurrent truncating germline mutation in the BRIP1/FANCJ gene and susceptibility to prostate cancer. Br. J. Cancer 2009, 100, 426–430. [Google Scholar] [CrossRef]
- Rupnik, A.; Grenon, M.; Lowndes, N. The MRN complex. Curr. Biol. 2008, 18, R455–R457. [Google Scholar] [CrossRef]
- Cybulski, C.; Górski, B.; Debniak, T.; Gliniewicz, B.; Mierzejewski, M.; Masojć, B.; Jakubowska, A.; Matyjasik, J.; Złowocka, E.; Sikorski, A.; et al. NBS1 is a prostate cancer susceptibility gene. Cancer Res. 2004, 64, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Paulo, P.; Pinto, P.; Peixoto, A.; Santos, C.; Pinto, C.; Rocha, P.; Veiga, I.; Soares, G.; Machado, C.; Ramos, F.; et al. Validation of a Next-Generation Sequencing Pipeline for the Molecular Diagnosis of Multiple Inherited Cancer Predisposing Syndromes. J. Mol. Diagn. 2017, 19, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, Z.; Chou, J.; Feng, F.Y.; Ryan, C.J. Prostate cancer in the era of “Omic” medicine: Recognizing the importance of DNA damage repair pathways. Ann. Transl. Med. 2018, 6, 161. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1- dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef]
- Dodson, G.E.; Limbo, O.; Nieto, D.; Russell, P. Phosphorylation-regulated binding of Ctp1 to Nbs1 is critical for repair of DNA double-strand breaks. Cell Cycle 2010, 9, 1516–1522. [Google Scholar] [CrossRef]
- Huen, M.S.; Sy, S.M.; Chen, J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 2010, 11, 138–148. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Bailis, J.M. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010, 20, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Sy, S.M.; Huen, M.S.; Zhu, Y.; Chen, J. PALB2 regulates recombinational repair through chromatin association and oligomerization. J. Biol. Chem. 2009, 284, 18302–18310. [Google Scholar] [CrossRef]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef]
- Reilly, N.M.; Novara, L.; Di Nicolantonio, F.; Bardelli, A. Exploiting DNA repair defects in colorectal cancer. Mol. Oncol. 2019, 13, 681–700. [Google Scholar] [CrossRef]
- Sundararajan, S.; Ahmed, A.; Goodman, O.B., Jr. The relevance of BRCA genetics to prostate cancer pathogenesis and treatment. Clin. Adv. Hematol. Oncol. 2011, 9, 748–755. [Google Scholar] [PubMed]
- Rubin, M.A.; Demichelis, F. The Genomics of Prostate Cancer: A Historic Perspective. Cold Spring Harb. Perspect Med. 2019, 9, a034942. [Google Scholar] [CrossRef]
- Castro, E.; Jugurnauth-Little, S.; Karlsson, Q.; Al-Shahrour, F.; Piñeiro-Yañez, E.; Van de Poll, F.; Leongamornlert, D.; Dadaev, T.; Govindasami, K.; Guy, M.; et al. UKGPCS, EMBRACE and IMPACT studies. High burden of copy number alterations and c-MYC amplification in prostate cancer from BRCA2 germline mutation carriers. Ann. Oncol. 2015, 26, 2293–2300. [Google Scholar] [CrossRef]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 175, 889. [Google Scholar] [CrossRef]
- Chen, Z.; Greenwood, C.; Isaacs, W.B.; Foulkes, W.D.; Sun, J.; Zheng, S.L.; Condreay, L.D.; Xu, J. The G84E mutation of HOXB13 is associated with increased risk for prostate cancer: Results from the REDUCE trial. Carcinogenesis 2013, 34, 1260–1264. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Fisher, C.; Foster, C.S.; Jameson, C.; Barbachanno, Y.; Bartlett, J.; Bancroft, E.; Doherty, R.; Kote-Jarai, Z.; Peock, S.; et al. Prostate cancer in male BRCA1 and BRCA2 mutation carriers has a more aggressive phenotype. J. Cancer 2008, 98, 502–507. [Google Scholar] [CrossRef]
- Hatano, Y.; Tamada, M.; Matsuo, M.; Hara, A. Molecular Trajectory of BRCA1 and BRCA2 Mutations. Front Oncol. 2020. [Google Scholar] [CrossRef]
- Gallagher, D.J.; Gaudet, M.M.; Pal, P.; Kirchhoff, T.; Balistreri, L.; Vora, K.; Bhatia, J.; Stadler, Z.; Fine, S.W.; Reuter, V.; et al. Germline BRCA mutations denote a clinicopathologic subset of prostate cancer. Clin. Cancer Res. 2010, 16, 2115–2121. [Google Scholar] [CrossRef]
- Agalliu, I.; Gern, R.; Leanza, S.; Burk, R.D. Associations of High-Grade Prostate Cancer with Brca1 and Brca2 Founder Mutations. Clin. Cancer Res. 2009, 15, 1112–1120. [Google Scholar] [CrossRef]
- Na, R.; Zheng, S.L.; Han, M.; Yu, H.; Jiang, D.; Shah, S.; Ewing, C.M.; Zhang, L.; Novakovic, K.; Petkewicz, J.; et al. Germline Mutations in ATM and BRCA1/2 Distinguish Risk for Lethal and Indolent Prostate Cancer and are Associated with Early Age at Death. Eur. Urol. 2017, 71, 740–747. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Liang, C.; Wang, H.; Chen, Y.; Silberstein, J.L.; Piana, D.; Lai, Z.; Chen, Y.; et al. Germline DNA-repair Gene Mutations and Outcomes in Men with Metastatic Castration-resistant Prostate Cancer Receiving First-line Abiraterone and Enzalutamide. Eur. Urol. 2018, 74, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Horak, P.; Weischenfeldt, J.; Von Amsberg, G.; Beyer, B.; Schutte, A.; Uhrig, S.; Gieldon, L.; Klink, B.; Feuerbach, L.; Hübschmann, D.; et al. Response to olaparib in a PALB2 germline mutated prostate cancer and genetic events associated with resistance. Cold Spring Harb Mol. Case Stud. 2019, 5, a003657. [Google Scholar] [CrossRef] [PubMed]
- Lonergan, P.E.; Tindall, D.J. Androgen receptor signaling in prostate cancer development and progression. J. Carcinog. 2011, 10, 20. [Google Scholar] [CrossRef]
- Ku, S.Y.; Gleave, M.E.; Beltran, H. Towards precision oncology in advanced prostate cancer. Nat. Rev. Urol. 2019, 16, 645–654. [Google Scholar] [CrossRef]
- Teo, M.Y.; Rathkopf, D.E.; Kantoff, P. Treatment of Advanced Prostate Cancer. Annu. Rev. Med. 2019, 70, 479–499. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.; Kote-Jarai, Z.; Mikropoulos, C.; Eeles, R. Prostate Cancer Germline Variations and Implications for Screening and Treatment. Cold Spring Harb Perspect Med. 2018, 8, a030379. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.K.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Rodrigues, D.N.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed]
- Zafeiriou, Z.; Bianchini, D.; Chandler, R.; Rescigno, P.; Yuan, W.; Carreira, S.; Barrero, M.; Petremolo, A.; Miranda, S.; Riisnaes, R.; et al. Genomic Analysis of Three Metastatic Prostate Cancer Patients with Exceptional Responses to Carboplatin Indicating Di_erent Types of DNA Repair Deficiency. Eur. Urol. 2018, 75, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Shaukat, F.; Isaacsson Velho, P.; Kaur, H.; Shenderov, E.; Pardoll, D.M.; Lotan, T.L. Clinical Features and Therapeutic Outcomes in Men with Advanced Prostate Cancer and DNA Mismatch Repair Gene Mutations. Eur. Urol. 2019, 75, 378–382. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Velho, P.I.; Antonarakis, e.S. PD-1/PD-L1 pathway inhibitors in advanced prostate cancer. Expert Rev. Clin. Pharmacol. 2018, 11, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Network NCC. Prostate Cancer (Version 2.2018) 2018. Available online: https://www.nccn.org/professionals/physician_gls/default.aspx (accessed on 27 January 2021).
- U.S. Food and Drug Administration, FoundationOne® Liquid CDx 2020. Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf19/P190032C.pdf (accessed on 27 January 2021).
- Telang, J.M.; Lane, B.R.; Cher, M.L.; Miller, D.C.; Dupree, J.M. Prostate cancer family history and eligibility for active surveillance: A systematic review of the literature. BJU Int. 2017, 120, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Akizhanova, M.; Iskakova, E.E.; Kim, V.; Wang, X.; Kogay, R.; Turebayeva, A.; Sun, Q.; Zheng, T.; Wu, S.; Miao, L.; et al. PSA and Prostate Health Index based prostate cancer screening in a hereditary migration complicated population: Implications in precision diagnosis. J. Cancer 2017, 8, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Bova, G.S.; Partin, A.W.; Isaacs, S.D.; Carter, B.S.; Beaty, T.L.; Isaacs, W.B. and Walsh, P.C. Biological aggressiveness of hereditary prostate cancer: Long-term evaluation following radical prostatectomy. J. Urol. 1998, 160, 660–663. [Google Scholar] [CrossRef]
- Hampel, H.; Bennett, R.L.; Buchanan, A.; Pearlman, R.; Wiesner, G.L. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet. Med. 2015, 17, 70–87. [Google Scholar] [CrossRef] [PubMed]
- US Preventive Services Task Force; Grossman, D.C.; Curry, S.J.; Owens, D.K.; Bibbins-Domingo, K.; Caughey, A.B.; Davidson, K.W.; Doubeni, C.A.; Ebell, M.; Epling, J.W., Jr.; et al. Screening for Prostate Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2018, 319, 1901–1913. [Google Scholar] [CrossRef] [PubMed]
- Brunese, L.; Mercaldo, F.; Reginelli, A.; Santone, A. Formal methods for prostate cancer Gleason score and treatment prediction using radiomic biomarkers. Magn. Reason. Imaging. 2020, 66, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Page, E.C.; Bancroft, E.K.; Brook, M.N.; Assel, M.; Al Battat, M.H.; Thomas, S.; Taylor, N.; Chamberlain, A.; Pope, J.; Ni Raghallaigh, H.; et al. Interim Results from the IMPACT Study: Evidence for Prostate-specific Antigen Screening in BRCA2 Mutation Carriers. Eur. Urol. 2019, 76, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Mikropoulos, C.; Selkirk, C.G.H.; Saya, S.; Bancroft, E.; Vertosick, E.; Dadaev, T.; Brendler, C.; Page, E.; Dias, A.; Evans, D.G.; et al. IMPACT study collaborators. Prostate-specific antigen velocity in a prospective prostate cancer screening study of men with genetic predisposition. Br. J. Cancer 2018, 118, 266–276. [Google Scholar] [CrossRef]
- Storebjerg, T.M.; Hoyer, S.; Kirkegaard, P.; Bro, F.; LuCamp Study Group; Orntoft, T.F.; Borre, M.; Sorensen, K.D. Prevalence of the HOXB13 G84E mutation in Danish men undergoing radical prostatectomy and its correlations with prostate cancer risk and aggressiveness. BJU Int. 2016, 118, 646–653. [Google Scholar] [CrossRef]
- Casanova-Salas, I.; Athie, A.; Boutros, P.C.; Del Re, M.; Miyamoto, D.T.; Pienta, K.J.; Posadas, E.M.; Sowalsky, A.G.; Stenzl, A.; Wyatt, A.W.; et al. Quantitative and Qualitative Analysis of Blood-based Liquid Biopsies to Inform Clinical Decision-making in Prostate Cancer. Eur. Urol. 2021, 7, S0302–S2838. [Google Scholar] [CrossRef]
- Campos-Fernández, E.; Barcelos, L.S.; de Souza, A.G.; Goulart, L.R.; Alonso-Goulart, V. Research landscape of liquid biopsies in prostate cancer. Am. J. Cancer Res. 2019, 9, 1309–1328. [Google Scholar]
- Hennigan, S.T.; Trostel, S.Y.; Terrigino, N.T.; Voznesensky, O.S.; Schaefer, R.J.; Whitlock, N.C.; Wilkinson, S.; Carrabba, N.V.; Atway, R.; Shema, S.; et al. Low Abundance of Circulating Tumor DNA in Localized Prostate Cancer. JCO Precis. Oncol. 2019, 3. [Google Scholar] [CrossRef]
- Kamps, R.; Brandão, R.D.; Bosch, B.J.; Paulussen, A.D.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. Int. J. Mol. Sci. 2017, 18, 308. [Google Scholar] [CrossRef]
- Paulo, P.; Maia, S.; Pinto, C.; Pinto, P.; Monteiro, A.; Peixoto, A.; Teixeira, M.R. Targeted next generation sequencing identifies functionally deleterious germline mutations in novel genes in early-onset/familial prostate cancer. PLoS Genet. 2018, 14, e1007355. [Google Scholar] [CrossRef] [PubMed]
- Susswein, L.R.; Marshall, M.L.; Vogel Nusbaum, R.; Postula, K.J.; Weissman, S.M.; Yackowski, L.; Vaccari, E.M.; Bissonnette, J.; Booker, J.K.; Cremona, M.L.; et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet. Med. 2016, 18, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Ni Raghallaigh, H.; Eeles, R. Genetic predisposition to prostate cancer: An update. Fam. Cancer 2021, 24. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Johns Hopkins group (1998) [83] | One of the following criteria must be included for the consideration of familial prostate cancer (PCa): 1. Three or more first-degree relatives with PCa; 2. Three successive generations of PCa; 3. Two relatives with PCa diagnosed at age ≤55 years |
| American College of Medical Genetics (2015) [84] | Genetic testing should be considered if one of the following criteria is met: 1. Three or more first-degree relatives with PCa; 2. Two or more first-degree relatives diagnosed with PCa at age ≤55 years; 3. Gleason grade > 7 PCa and a family history of ≥2 individuals with breast, ovarian, or pancreatic cancer |
| American Society of Urology (AUA 2017) European Association of Urology (EAU 2019) [6] | Recommend offering germline genetic testing for BRCA1, BRCA2, ATM, PALB2, and FANCA to all patients with high risk or metastatic disease regardless of family history. For those patients with a lower-risk disease, germline genetic testing should be considered when: 1. There is a strong family history (brother or father or multiple family members diagnosed with PCa <60 years), 2. Known germline abnormalities and/or more than one family member with breast, ovarian, or pancreatic cancer (suggestive of BRCA2 mutations), 3. More than one family member with Lynch syndrome (LS) |
| National Comprehensive Cancer Network (2018) [22] | Recommend genetic testing of BRCA in PCa patients with one of the conditions listed below: 1. A history of Gleason grade ≥7 PCa regardless of age and ≥1 close relative with breast cancer (age ≤ 50 years) and/or invasive ovarian cancer; 2. Patients with prostate cancer (Gleason ≥ 7) who have two relatives with breast, pancreatic, or PCa (Gleason ≥ 7) diagnosed at any age; 3. A personal history of metastatic PCa (radiographic evidence of or biopsy-proven disease) |
| National Comprehensive Cancer Network (updated version 2019) [6] | States that. if next generation sequencing (NGS) is used, the panel must include BRCA1, BRCA2, ATM, CHEK2, PALB2, MLH1, MSH2, MSH6, and PMS2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vietri, M.T.; D’Elia, G.; Caliendo, G.; Resse, M.; Casamassimi, A.; Passariello, L.; Albanese, L.; Cioffi, M.; Molinari, A.M. Hereditary Prostate Cancer: Genes Related, Target Therapy and Prevention. Int. J. Mol. Sci. 2021, 22, 3753. https://doi.org/10.3390/ijms22073753
Vietri MT, D’Elia G, Caliendo G, Resse M, Casamassimi A, Passariello L, Albanese L, Cioffi M, Molinari AM. Hereditary Prostate Cancer: Genes Related, Target Therapy and Prevention. International Journal of Molecular Sciences. 2021; 22(7):3753. https://doi.org/10.3390/ijms22073753
Chicago/Turabian StyleVietri, Maria Teresa, Giovanna D’Elia, Gemma Caliendo, Marianna Resse, Amelia Casamassimi, Luana Passariello, Luisa Albanese, Michele Cioffi, and Anna Maria Molinari. 2021. "Hereditary Prostate Cancer: Genes Related, Target Therapy and Prevention" International Journal of Molecular Sciences 22, no. 7: 3753. https://doi.org/10.3390/ijms22073753
APA StyleVietri, M. T., D’Elia, G., Caliendo, G., Resse, M., Casamassimi, A., Passariello, L., Albanese, L., Cioffi, M., & Molinari, A. M. (2021). Hereditary Prostate Cancer: Genes Related, Target Therapy and Prevention. International Journal of Molecular Sciences, 22(7), 3753. https://doi.org/10.3390/ijms22073753