
Modeling Rett Syndrome with Human Pluripotent Stem Cells: Mechanistic Outcomes and Future Clinical Perspectives

 ,
,  ,
,  and
and 
{kind=link}
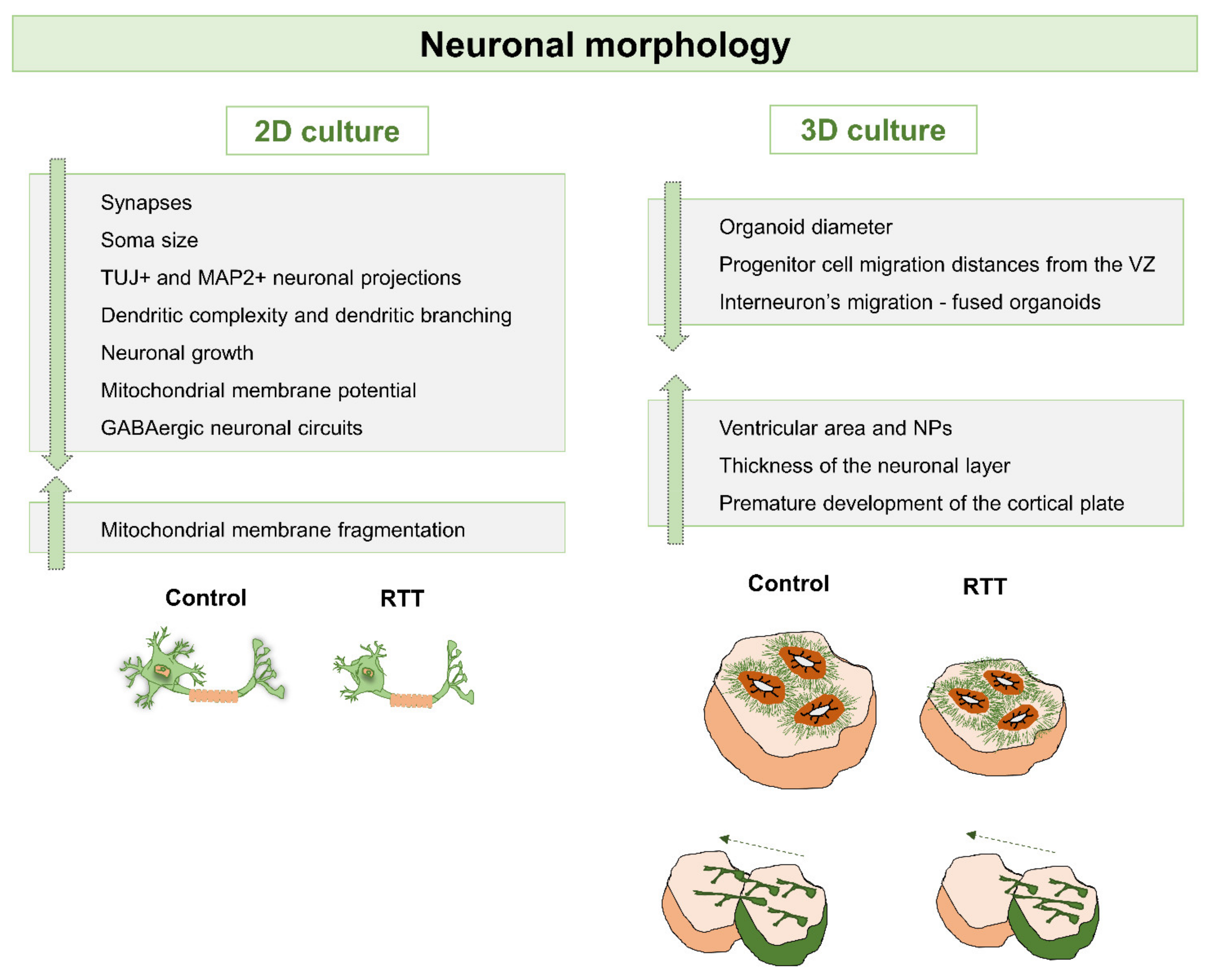{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Rett Syndrome—The Pathophysiology
3. An Overview of MECP2 Role as a Transcriptional Regulator
4. Modelling RTT with hiPSCs
4.1. 2D-Based Neuronal Differentiation Models
4.1.1. Evaluating Altered RTT Phenotype
4.1.2. Understanding MeCP2′s Molecular Functions
4.1.3. High-Content Molecular Analysis of RTT Cells
4.2. 3D Brain Models
4.2.1. Scaffold-Based 3D Models
4.2.2. 3D Brain Organoids
5. Future Clinical Translation of hPSC Technology in RTT
6. hPSC-Derived RTT Brain Models-Limitations and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ASD | autism spectrum disorder |
| AD | Alzheimer’s disease |
| AMPA | 2-Amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl) propanoic acid |
| AAV | adeno-associated virus |
| BBB | blood brain barrier |
| BDNF | brain-derived neurotrophic factor |
| BRD4 | bromodomain 4 |
| CDKL5 | cyclin dependent kinase like 5 |
| CNS | central nervous system |
| CTD | C-terminal domain |
| EB | embryoid bodies |
| E/I | excitatory/inhibitory imbalance |
| EVs | extracellular vesicles |
| FOXG1 | forkhead box G1 |
| FLT3 | fms-like tyrosine kinase 3 |
| GABA | gamma-aminobutyric acid |
| GFAP | glial fibrillary acidic protein |
| GluR2/3 | glutamate receptor subunit |
| HDAC | histone deacetylases |
| HDAC6 | histone deacetylases 6 |
| hESCs | human embryonic stem cells |
| hiPSCs | human induced pluripotent stem cells |
| Hsp | heat-shock proteins |
| HTS | high-throughput screening |
| HUVECs | human umbilical vein endothelial cells |
| IC | isogenic control |
| ID | intervening domain |
| IGF-1 | insulin-like growth factor-1 |
| IL | interleukin |
| ILVs | intraluminal vesicles |
| INs | interneurons |
| L1CAM L1 | cell adhesion molecule |
| LPS | lipopolysaccharides |
| MBD | methyl-CpG binding domain |
| MEA | multi electrode array |
| MeCP2 | methyl-CpG-binding protein 2 |
| MeCP2LOF | methyl-CpG-binding protein 2 loss of function |
| MeCP2-KO | methyl-CpG-binding protein 2 knocking out |
| MGE | medial ganglionic eminence |
| miRNAs | micro RNAs |
| mRNAs | messenger RNA |
| mtDNA | mitochondrial DNA |
| MVs | microvesicles |
| MVBs | multivesicular bodies |
| NCOR | nuclear co-repressor |
| NMDA | N-methyl-d-aspartate |
| NPs | neural progenitors |
| NPCs | neural progenitor cells |
| NTD | N-terminal domain |
| OL | oligodendrocytes |
| oRGCs | outer radial glia cells |
| oSVZ | outer subventricular zone |
| PSD-95 | postsynaptic density protein 95 |
| RG | radial glia |
| rhIGF1 | full-length recombinant human IGF1 |
| RNA | ribonucleic acid |
| RNA-seq | ribonucleic acid sequencing |
| ROS | reactive oxygen species |
| RTT | Rett Syndrome |
| siRNA | small interfering RNA |
| SMRT | silencing mediator of retinoic acid and thyroid hormone receptor |
| STT | somatostatin |
| TGF-β1 | transforming growth factor beta 1 |
| TRD | transcriptional repression domain |
| TrkB | tropomyosin-related kinase B |
| KCC2 | neuron-specific K+-Cl− cotransporter 2 |
| XCI | X-chromosome inactivation |
References
- Reichow, B.; Tara, A.G.; Isaac, L.; Volkmar, F.R.; Boy, M.Á. Brief Report: Systematic Review of Rett Syndrome in Males. J. Autism Dev. Disord. 2015, 45, 3377–3383. [Google Scholar] [CrossRef]
- Chahrour, M.; Zoghbi, H.Y. The Story of Rett Syndrome: From Clinic to Neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef]
- Wan, M.; Sung, S.; Lee, J.; Zhang, X.; Houwink-manville, I.; Song, H.; Amir, R.E.; Budden, S.; Naidu, S.; Pereira, J.L.P.; et al. Rett Syndrome and Beyond: Recurrent Spontaneous and Familial MECP2 Mutations at CpG Hotspots. Am. J. Hum. Genet. 1999, 65, 1520–1529. [Google Scholar] [CrossRef]
- Martínez De Paz, A.; Khajavi, L.; Martin, H.; Claveria-Gimeno, R.; Tom Dieck, S.; Cheema, M.S.; Sanchez-Mut, J.V.; Moksa, M.M.; Carles, A.; Brodie, N.I.; et al. MeCP2-E1 isoform is a dynamically expressed, weakly DNA-bound protein with different protein and DNA interactions compared to MeCP2-E2. Epigenetics Chromatin 2019, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cheadle, J.P.; Gill, H.; Fleming, N.; Maynard, J.; Kerr, A.; Leonard, H.; Krawczak, M.; Cooper, D.N.; Lynch, S.; Thomas, N.; et al. Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: Correlation of disease severity with mutation type and location. Hum. Mol. Genet. 2000, 9, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific mutations in Methyl-CpG-Binding Protein 2 confer different severity in Rett syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Caffarelli, C.; Gonnelli, S.; Pitinca, M.D.T.; Camarri, S.; Al Refaie, A.; Hayek, J.; Nuti, R. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with bone disease severity in Rett syndrome. BMC Med. Genet. 2020, 21, 21. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Pillai, R.B.; Shekar, K.V.; Lane, J.B.; Motil, K.J.; Skinner, S.A.; Tarquinio, D.C.; Glaze, D.G.; McGwin, G.; Kaufmann, W.E.; et al. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in rett syndrome. J. Med. Genet. 2014, 51, 152–158. [Google Scholar] [CrossRef]
- Hoffbuhr, K.C.; Moses, L.M.; Jerdonek, M.A.; Naidu, S.; Hoffman, E.P. Associations between MeCP2 mutations, X-chromosome inactivation, and phenotype. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 99–105. [Google Scholar] [CrossRef]
- Ishii, T.; Makita, Y.; Ogawa, A.; Amamiya, S.; Yamamoto, M. The role of different X-inactivation pattern on the variable clinical phenotype with Rett syndrome. Brain Dev. 2001, 23, 161–164. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Percy, A.K.; Schultz, R.J.; Fill, C. Patterns of X Chromosome Inactivation in the Rett Syndrome. Brain Dev. 1990, 12, 131–135. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Sun, Y.; Zoghbi, H.Y. Balanced X chromosome inactivation patterns in the Rett syndrome brain. Am. J. Med. Genet. 2002, 111, 164–168. [Google Scholar] [CrossRef]
- Ribeiro, M.C.; Macdonald, J.L. Sex differences in Mecp2 -mutant Rett syndrome model mice and the impact of cellular mosaicism in phenotype development. Brain Res. 2020, 1729, 146644. [Google Scholar] [CrossRef]
- Wochenschr, W.M. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochenschr. 1996, 116, 723–726. [Google Scholar]
- Amir, R.E.; Van Den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Evans, J.C.; Archer, H.L.; Colley, J.P.; Ravn, K.; Nielsen, J.B.; Kerr, A.; Williams, E.; Christodoulou, J.; Jardine, P.E.; Wright, M.J.; et al. Early onset seizures and Rett-like features associated with mutations in CDKL5. Eur. J. Hum. Genet. 2005, 13, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Philippe, C.; Amsallem, D.; Francannet, C.; Lambert, L.; Saunier, A.; Verneau, F.; Jonveaux, P. Phenotypic variability in Rett syndrome associated with FOXG1 mutations in females. Med. Genet. 2010, 47, 59–65. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cosentino, L.; Vigli, D.; Franchi, F.; Laviola, G.; Filippis, B. De Rett syndrome before regression: A time window of overlooked opportunities for diagnosis and intervention. Neurosci. Biobehav. Rev. 2019, 107, 115–135. [Google Scholar] [CrossRef] [PubMed]
- Halbach, N.; Smeets, E.; Steinbusch, C.; Maaskant, M.; van Waardenburg, D.; Curfs, L. Aging in Rett syndrome: A longitudinal study. Clin. Genet. 2013, 84, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Killian, J.T.; Lane, J.B.; Lee, H.-S.; Skinner, S.A.; Kaufmann, W.E.; Glaze, D.G.; Neul, J.L. Scoliosis in Rett Syndrome: Progression, Comorbidities, and Predictors. Pediatr. Neurol. 2017, 70, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Roze, E.; Sangla, S.; Bienvenu, T.; Leu-semenescu, S. Rett Syndrome: An Overlooked Diagnosis in Women with Stereotypic Hand Movements, Psychomotor Retardation, Parkinsonism, and Dystonia? Mov. Disord. 2007, 22, 387–433. [Google Scholar] [CrossRef] [PubMed]
- Tarquinio, D.C.; Hou, W.; Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Motil, K.J.; Skinner, S.A.; Lee, H.; Percy, A.K. The Changing Face of Survival in Rett Syndrome and MECP2 -Related Disorders. Pediatr. Neurol. 2015, 53, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Huppke, P.; Laccone, F.; Krämer, N.; Engel, W.; Hanefeld, F. Rett syndrome: Analysis of MECP2 and clinical characterization of 31 patients. Hum. Mol. Genet. 2000, 9, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Murakami, J.W.; Courchesne, E.; Haas, R.H.; Press, G.A.; Yeung-courchesne, R. Cerebellar and Cerebral Abnormalities in Rett Syndrome: A Quantitative MR Analysis. Ajr. Am. J. Roentgenol. 1992, 159, 177–183. [Google Scholar] [CrossRef]
- Cardoza, B.; Clarke, A.; Wilcox, J.; Gibbon, F.; Smith, P.E.M.; Archer, H.; Hryniewiecka-jaworska, A.; Kerr, M. Epilepsy in Rett syndrome: Association between phenotype and genotype, and implications for practice. Seizure 2011, 20, 646–649. [Google Scholar] [CrossRef]
- Glaze, D.G.; Percy, A.K.; Skinner, S.; Motil, K.J.; Neul, J.L.; Barrish, O.; Lane, J.B.; Geerts, S.P.; Annese, F.; Graham, J.; et al. Epilepsy and the natural history of Rett syndrome. Neurology 2010, 74, 909–913. [Google Scholar] [CrossRef]
- Nectoux, J.; Bahi-Buisson, N.; Guellec, I.; Coste, J.; De Roux, N.; Rosas, H.; Tardieu, M.; Chelly, J.; Bienvenu, T. The p. Val66Met polymorphism in the BDNF gene protects against early seizures in Rett syndrome. Neurology 2008, 70, 2145–2152. [Google Scholar] [CrossRef]
- Ben Zeev, B.; Leonard, H.; De Klerk, N. The common BDNF polymorphism may be a modifier of disease severity in Rett syndrome. Neurology 2009, 72, 1242–1247. [Google Scholar] [CrossRef]
- Buoni, S.; Zannolli, R.; De Felice, C.; Saponari, S.; Strambi, M.; Teresa, M.; Castrucci, E.; Corbini, L.; Orsi, A.; Hayek, J. Drug-resistant epilepsy and epileptic phenotype-EEG association in MECP2 mutated Rett syndrome. Clin. Neurophysiol. 2008, 119, 2455–2458. [Google Scholar] [CrossRef] [PubMed]
- Krajnc, N. Management of epilepsy in patients with Rett syndrome: Perspectives and considerations. Clin. Risk Manag. 2015, 11, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Geerdink, N.; Rotteveel, J.J.; Lammens, M.; Sistermans, E.A.; Heikens, G.T.; Mullaart, R.A.; Hamel, B.C.J. MECP2 Mutation in a Boy with Severe Neonatal Encephalopathy: Clinical, Neuropathological and Molecular Findings. Neuropediatrics 2002, 33, 33–36. [Google Scholar] [CrossRef]
- Bourdon, V.; Philippe, C.; Martin, D.; Verlo, A.; Jonveaux, P. MECP2 Mutations or Polymorphisms in Mentally Retarded Boys. Mol. Diagn. 2003, 7, 3–7. [Google Scholar]
- Neul, J.L.; Benke, T.A.; Marsh, E.D.; Skinner, S.A.; Merritt, J.; Lieberman, D.N.; Standridge, S.; Feyma, T.; Heydemann, P.; Peters, S.; et al. The array of clinical phenotypes of males with mutations in Methyl-CpG binding protein 2. Am. J. Med. Genet. 2019, 180, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Schwartzman, J.S.; Bernardino, A.; Nishimura, A.; Gomes, R.R.; Zatz, M.; Mackenzie, U.; Paulo, S. Rett Syndrome in a Boy with a 47, XXY Karyotype Confirmed by a Rare Mutation in the MECP2 Gene. Neuropediatrics 2001, 32, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, X.; Wang, J.; Li, J.; Wu, Q. Genomic mosaicism in the pathogenesis and inheritance of a Rett syndrome cohort. Genet. Med. 2019, 21, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Pieras, J.I.; Mun, B.; Borrego, S.; Marcos, I.; Sanchez, J.; Madruga, M.; Antin, G. Somatic mosaicism for Y120X mutation in the MECP2 gene causes atypical Rett syndrome in a male. Brain Dev. 2011, 33, 608–611. [Google Scholar] [CrossRef]
- Van Esch, H.; Bauters, M.; Ignatius, J.; Jansen, M.; Raynaud, M.; Hollanders, K.; Lugtenberg, D.; Bienvenu, T.; Jensen, L.R.; Ge, J.; et al. Duplication of the MECP2 Region Is a Frequent Cause of Severe Mental Retardation and Progressive Neurological Symptoms in Males. Am. J. Hum. Genet. 2005, 77, 442–453. [Google Scholar] [CrossRef]
- Gaudio, D.; Fang, P.; Scaglia, F.; Ward, P.A.; Craigen, W.J. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet. Med. 2006, 8, 784–792. [Google Scholar] [CrossRef]
- Miguet, M.; Faivre, L.; Amiel, J.; Nizon, M.; Touraine, R.; Prieur, F.; Pasquier, L.; Lefebvre, M.; Thevenon, J.; Dubourg, C.; et al. Further delineation of the MECP2 duplication syndrome phenotype in 59 French male patients, with a particular focus on morphological and neurological features. J. Med. Genet. 2018, 55, 359–371. [Google Scholar] [CrossRef]
- Yu, B.; Yuan, B.; Dai, J.K.; Cheng, T.-l.; Xia, S.N.; He, L.J.; Yuan, Y.T.; Zhang, Y.F.; Xu, H.T.; Xu, F.Q.; et al. Reversal of Social Recognition Deficit in Adult Mice with MECP2 Duplication via Normalization of MeCP2 in the Medial Prefrontal Cortex. Neurosci. Bull. 2020, 36, 570–584. [Google Scholar] [CrossRef]
- Nageshappa, S.; Carromeu, C.; Trujillo, C.A.; Mesci, P.; Espuny-Camacho, I.; Pasciuto, E.; Vanderhaeghen, P.; Verfaillie, C.M.; Raitano, S.; Kumar, A.; et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol. Psychiatry 2016, 21, 178–188. [Google Scholar] [CrossRef]
- Liyanage, V.R.B.; Rastegar, M.; Mecp, M.Á.M.Á.; Rett, Á. Rett Syndrome and MeCP2. Neuromol. Med 2014, 16, 231–264. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, F.; Coort, S.L.M.; Cirillo, E.; Smeets, E.; Evelo, C.T.; Curfs, L.M.G. Rett syndrome—Biological pathways leading from MECP2 to disorder phenotypes. Orphanet J. Rare Dis. 2016, 11, 1–13. [Google Scholar] [CrossRef]
- Horvath, P.M.; Monteggia, L.M. MeCP2 as an activator of gene expression. Trends Neurosci. 2019, 41, 72–74. [Google Scholar] [CrossRef]
- Picard, N.; Fagiolini, M. MeCP2: An epigenetic regulator of critical periods. Curr. Opin. Neurobiol. 2019, 59, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Tillotson, R.; Bird, A. The Molecular Basis of MeCP2 Function in the Brain. J. Mol. Biol. 2020, 432, 1602–1623. [Google Scholar] [CrossRef] [PubMed]
- Pejhan, S.; Rastegar, M. Role of dna methyl-cpg-binding protein mecp2 in rett syndrome pathobiology and mechanism of disease. Biomolecules 2021, 11, 75. [Google Scholar] [CrossRef]
- Nan, X.; Meehan, R.R.; Bird, A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993, 21, 4886–4892. [Google Scholar] [CrossRef]
- Nan, X.; Ng, H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Lett. Nat. 1998, 393, 386–389. [Google Scholar] [CrossRef]
- Lagger, S.; Connelly, J.C.; Schweikert, G.; Webb, S.; Selfridge, J.; Ramsahoye, B.H.; Yu, M.; He, C.; Sanguinetti, G.; Sowers, L.C.; et al. MeCP2 recognizes cytosine methylated tri- nucleotide and di-nucleotide sequences to tune transcription in the mammalian brain. PLoS Genet. 2017, 13, e1006793. [Google Scholar] [CrossRef] [PubMed]
- Kokura, K.; Kaul, S.C.; Wadhwa, R.; Nomura, T.; Khan, M.; Shinagawa, T.; Yasukawa, T.; Colmenares, C.; Ishii, S. The Ski Protein Family Is Required for MeCP2-mediated Transcriptional Repression. J. Biol. Chem. 2001, 276, 34115–34121. [Google Scholar] [CrossRef] [PubMed]
- Lyst, M.J.; Ekiert, R.; Ebert, D.H.; Merusi, C.; Nowak, J.; Selfridge, J.; Guy, J.; Kastan, N.R.; Robinson, N.D.; Alves, F.D.L.; et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013, 16, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Boxer, L.D.; Renthal, W.; Greben, A.W.; Griffith, E.C.; Bonev, B.; Greenberg, M.E.; Boxer, L.D.; Renthal, W.; Greben, A.W.; Whitwam, T.; et al. MeCP2 Represses the Rate of Transcriptional Initiation of Highly Methylated Long Genes. Mol. Cell 2020, 77, 294–309.e9. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, K.; Lavery, L.A.; Andrew, S.; Shaw, C.A.; Li, W. MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 5509–5514. [Google Scholar] [CrossRef] [PubMed]
- Kinde, B.; Gabel, H.W.; Gilbert, C.S.; Griffith, E.C.; Greenberg, M.E. Reading the unique DNA methylation landscape of the and MeCP2. Proc. Natl. Acad. Sci. USA 2015, 112, 6800–6806. [Google Scholar] [CrossRef]
- Gabel, H.W.; Kinde, B.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015, 522, 89–93. [Google Scholar] [CrossRef]
- Clemens, A.W.; Wu, D.Y.; Moore, J.R.; Christian, D.L.; Zhao, G.; Gabel, H.W.; Methylation, C.T.D.N.A.; Clemens, A.W.; Wu, D.Y.; Moore, J.R.; et al. MeCP2 Represses Enhancers through Chromosome Article MeCP2 Represses Enhancers through. Mol. Cell 2020, 77, 279–293.e8. [Google Scholar] [CrossRef]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.C.; Zoghbi, H.Y. MeCP2, a Key Contributor to Neurological Disease, Activates and Represses Transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef]
- Ito-ishida, A.; Baker, S.A.; Sillitoe, R.V.; Sun, Y.; Zhou, J.; Ono, Y.; Iwakiri, J.; Yuzaki, M.; Zoghbi, H.Y. MeCP2 Levels Regulate the 3D Structure of Heterochromatic Foci in Mouse Neurons. J. Neurosci. 2020, 40, 8746–8766. [Google Scholar] [CrossRef]
- Baker, S.A.; Chen, L.; Wilkins, A.D.; Yu, P.; Lichtarge, O.; Zoghbi, H.Y. An AT-Hook Domain in MeCP2 Determines the Clinical Course of Rett Syndrome and Related Disorders. Cell 2013, 152, 984–996. [Google Scholar] [CrossRef]
- Squillaro, T.; Hayek, G.; Farina, E.; Cipollaro, M.; Renieri, A.; Galderisi, U. A case report: Bone marrow mesenchymal stem cells from a rett syndrome patient are prone to senescence and show a lower degree of apoptosis. J. Cell. Biochem. 2008, 103, 1877–1885. [Google Scholar] [CrossRef]
- Squillaro, T.; Alessio, N.; Capasso, S.; Di Bernardo, G.; Melone, M.A.B.; Peluso, G.; Galderisi, U. Senescence phenomena and metabolic alteration in mesenchymal stromal cells from a mouse model of rett syndrome. Int. J. Mol. Sci. 2019, 20, 2508. [Google Scholar] [CrossRef]
- Ip, J.P.K.; Mellios, N.; Sur, M. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat. Rev. Neurosci. 2018, 19, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Colantuoni, C.; Jeon, O.H.; Hyder, K.; Chenchik, A.; Khimani, A.H.; Narayanan, V.; Hoffman, E.P.; Kaufmann, W.E.; Naidu, S.B.; Pevsner, J. Gene expression profiling in postmortem Rett Syndrome brain: Differential gene expression and patient classification. Neurobiol. Dis. 2001, 8, 847–865. [Google Scholar] [CrossRef]
- Zeng, R.; Sidik, H.; Robinson, K.S.; Zhong, F.L.; Reversade, B. Generation of four H1 hESC sublines carrying a hemizygous knock-out/mutant MECP2. Stem Cell Res. 2019, 40, 101533. [Google Scholar] [CrossRef] [PubMed]
- Thanh, T.; Le, H.; Tran, N.T.; Mai, T.; Dao, L.; Nguyen, D.D.; Do, H.D.; Ha, T.L.; Kühn, R.; Nguyen, T.L.; et al. Efficient and Precise CRISPR/Cas9- Mediated MECP2 Modifications in Human-Induced Pluripotent Stem Cells. Front. Genet. 2019, 10, 1–10. [Google Scholar]
- Tchieu, J.; Kuoy, E.; Chin, M.H.; Trinh, H.; Patterson, M.; Sean, P.; Aimiuwu, O.; Lindgren, A.; Hakimian, S.; Zack, J.A.; et al. Female human iPS cells retain inactive X-chromosome. Cell Stem Cell 2010, 7, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.Y.L.; Horvath, L.M.; Carrel, L.; Ellis, J.; Colman, A. X-chromosome inactivation in Rett syndrome human induced pluripotent stem cells. Front. Psychiatry 2012, 3, 1–16. [Google Scholar] [CrossRef]
- Bar, S.; Seaton, L.R.; Weissbein, U.; Eldar-geva, T.; Bar, S.; Seaton, L.R.; Weissbein, U.; Eldar-geva, T.; Benvenisty, N. Global Characterization of X Chromosome Report Global Characterization of X Chromosome Inactivation in Human Pluripotent Stem Cells. Cell Rep. 2019, 27, 20–29.e3. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A Model for Neural Development and Treatment of Rett Syndrome Using Human Induced Pluripotent Stem Cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Ananiev, G.; Williams, E.C.; Li, H.; Chang, Q. Isogenic Pairs of Wild Type and Mutant Induced Pluripotent Stem Cell ( iPSC ) Lines from Rett Syndrome Patients as In Vitro Disease Model. PLoS ONE 2011, 6, e25255. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Hysolli, E.; Park, I.H. Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 14169–14174. [Google Scholar] [CrossRef] [PubMed]
- Djuric, U.; Cheung, A.Y.L.; Zhang, W.; Mok, R.S.; Piekna, A.; Hendry, J.A.; Ross, P.J.; Pasceri, P.; Kim, D.; Salter, M.W.; et al. MECP2e1 isoform mutation affects the form and function of neurons from Rett syndrome patient iPS cells. Neurobiol. Dis. 2015, 76, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef]
- Fernandes, T.G.; Duarte, S.T.; Ghazvini, M.; Gaspar, C.; Santos, D.C. Neural commitment of human pluripotent stem cells under defined conditions recapitulates neural development and generates patient-specific neural cells. Biotechnol. J. 2015, 10, 1578–1588. [Google Scholar] [CrossRef]
- Chin, E.W.M.; Marcy, G.; Yoon, S.I.; Ma, D.; Rosales, F.J.; Augustine, G.J.; Goh, E.L.K. Choline Ameliorates Disease Phenotypes in Human iPSC Models of Rett Syndrome. Neuromol. Med. 2016, 18, 364–377. [Google Scholar] [CrossRef]
- Bu, Q.; Wang, A.; Hamzah, H.; Waldman, A.; Jiang, K.; Dong, Q.; Li, R.; Kim, J.; Turner, D.; Chang, Q. CREB signaling is involved in rett syndrome pathogenesis. J. Neurosci. 2017, 37, 3671–3685. [Google Scholar] [CrossRef]
- Yoo, M.; Carromeu, C.; Kwon, O.; Muotri, A.; Schachner, M. The L1 adhesion molecule normalizes neuritogenesis in Rett syndrome-derived neural precursor cells. Biochem. Biophys. Res. Commun. 2017, 494, 504–510. [Google Scholar] [CrossRef]
- Tang, X.; Kim, J.; Zhou, L.; Wengert, E.; Zhang, L.; Wu, Z.; Carromeu, C.; Muotri, A.R. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA 2015, 113, 751–756. [Google Scholar] [CrossRef]
- Tang, X.; Drotar, J.; Li, K.; Clairmont, C.D.; Brumm, A.S.; Sullins, A.J.; Wu, H.; Liu, X.S.; Wang, J.; Gray, N.S.; et al. Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci. Transl. Med. 2019, 11, 1–14. [Google Scholar] [CrossRef]
- Ohashi, M.; Korsakova, E.; Allen, D.; Lee, P.; Fu, K.; Vargas, B.S.; Cinkornpumin, J.; Salas, C.; Park, J.C.; Germanguz, I.; et al. Loss of MECP2 Leads to Activation of P53 and Neuronal Senescence. Stem Cell Rep. 2018, 10, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Landucci, E.; Brindisi, M.; Bianciardi, L.; Catania, L.M.; Daga, S.; Croci, S.; Frullanti, E.; Fallerini, C.; Butini, S.; Brogi, S.; et al. iPSC-derived neurons profi ling reveals GABAergic circuit disruption and acetylated α -tubulin defect which improves after iHDAC6 treatment in Rett syndrome. Exp. Cell Res. 2018, 368, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Sullivan, G.J.; Weissman, S.M.; Park, I.; Xiang, Y.; Tanaka, Y.; Patterson, B.; Hwang, S.; et al. Dysregulation of BRD4 Function Underlies the Functional Abnormalities of MeCP2 Mutant Neurons. Mol. Cell 2020, 79, 84–98.e9. [Google Scholar] [CrossRef] [PubMed]
- Varderidou-minasian, S.; Hinz, L.; Hagemans, D.; Posthuma, D.; Altelaar, M.; Heine, V.M. Quantitative proteomic analysis of Rett iPSC-derived neuronal progenitors. Mol. Autism 2020, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Mesci, P.; Carromeu, C.; McClatchy, D.R.; Schiapparelli, L.; Yates, J.R.; Muotri, A.R.; Cline, H.T. Exosomes regulate neurogenesis and circuit assembly. Proc. Natl. Acad. Sci. USA 2019, 116, 16086–16094. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Han, X.; Blanchi, B.; Guan, W.; Ge, W.; Yu, Y.; Sun, Y.E. Graded and pan-neural disease phenotypes of Rett Syndrome linked with dosage of functional MeCP2. Protein Cell 2020, 1–14. [Google Scholar] [CrossRef]
- Benito-kwiecinski, S.; Lancaster, M.A. Brain Organoids: Human Neurodevelopment in a Dish. Cold Spring Harb. Perspect. Biol. 2020, 12, 1–18. [Google Scholar] [CrossRef]
- Trujillo, C.A.; Adams, J.W.; Negraes, P.D.; Carromeu, C.; Tejwani, L.; Acab, A.; Tsuda, B.; Thomas, C.A.; Sodhi, N.; Fichter, K.M.; et al. Pharmacological reversal of synaptic and network pathology in human MECP 2 -KO neurons and cortical organoids. EMBO Mol. Med. 2020, 13, e12523. [Google Scholar]
- Lancaster, M.A.; Renner, M.; Martin, C.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–391. [Google Scholar] [CrossRef]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini- bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef]
- Bagley, J.A.; Reumann, D.; Bian, S.; Lévi-strauss, J.; Knoblich, J.A. Fused cerebral organoids model interactions between brain regions. Nat. Methods 2017, 12, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Zhang, X.; Renier, N.; Wu, Z.; Atkin, T.; Sun, Z.; Ozair, M.Z.; Tchieu, J.; Zimmer, B.; Fattahi, F.; et al. Combined small-molecule inhibition accelerates the derivation of functional cortical neurons from human pluripotent stem cells. Nat. Biotechnol. 2017, 35, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.N.; Freitas, B.C.; Qian, H.; Lux, J.; Acab, A.; Trujillo, C.A.; Herai, R.H.; Huu, V.A.N.; Wen, J.H.; Joshi-Barr, S.; et al. Layered hydrogels accelerate iPSC-derived neuronal maturation and reveal migration defects caused by MeCP2 dysfunction. Proc. Natl. Acad. Sci. USA 2016, 113, 3185–3190. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Mattiassi, S.; Loeblein, M.; Chin, E.; Ma, D.; Coquet, P.; Viasnoff, V.; Teo, E.H.T.; Goh, E.L.; Yim, E.K.F. Human Rett-derived neuronal progenitor cells in 3D graphene scaffold as an in vitro platform to study the effect of electrical stimulation on neuronal differentiation. Biomed. Mater. 2018, 13, 1–17. [Google Scholar] [CrossRef]
- Mellios, N.; Feldman, D.A.; Sheridan, S.D.; Ip, J.P.K.; Kwok, S.; Amoah, S.K.; Rosen, B.; Rodriguez, B.A.; Crawford, B.; Swaminathan, R.; et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol. Psychiatry 2018, 23, 1051–1065. [Google Scholar] [CrossRef]
- Gomes, A.R.; Fernandes, T.G.; Vaz, S.H.; Silva, T.P.; Bekman, E.P.; Xapelli, S.; Duarte, S.; Ghazvini, M.; Gribnau7, J.; Muotri, A.R.; et al. Modeling Rett Syndrome With Human Patient-Specific Forebrain Organoids. Front. Cell Dev. Biol. 2020, 8, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kwok, S.; Li, Y.; Wang, H.; Muffat, J.; Cheng, A.W.; Orlando, D.A.; Love, J.; Feldman, D.A.; Bateup, H.S.; Gao, Q.; et al. Global Transcriptional and Translational Repression in Human-Embryonic-Stem-Cell-Derived Rett Syndrome Neurons. Cell Stem Cell. 2013, 13, 446–458. [Google Scholar]
- Tropea, D.; Giacometti, E.; Wilson, N.R.; Beard, C.; Mccurry, C.; Dong, D.; Flannery, R.; Jaenisch, R.; Sur, M. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. USA 2009, 106, 2029–2034. [Google Scholar] [CrossRef]
- Castro, J.; Garcia, R.I.; Kwok, S.; Banerjee, A.; Petravicz, J.; Woodson, J. Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 9941–9946. [Google Scholar] [CrossRef]
- Khwaja, O.S.; Ho, E.; Barnes, K.V.; Leary, H.M.O.; Pereira, L.M.; Finkelstein, Y. Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 4596–4601. [Google Scholar] [CrossRef]
- Leary, H.M.O.; Kaufmann, W.E.; Barnes, K.V.; Rakesh, K.; Kapur, K.; Tarquinio, D.C.; Cantwell, N.G.; Roche, K.J.; Rose, S.A.; Alexandra, C.; et al. Placebo-controlled crossover assessment of mecasermin for the treatment of Rett syndrome. Neurology 2018, 5, 323–332. [Google Scholar]
- Glaze, D.G.; Neul, J.L.; Kaufmann, W.E.; Berry-kravis, E. Double-blind, randomized, placebo-controlled study of trofinetide in pediatric Rett syndrome. Neurology 2019, 92, e1912–e1925. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Gluckman, P.; Yang, P.; Krissansen, G.; Sun, X.; Zhou, Y.; Wen, J.; Phillips, G.; Shorten, P.R.; McMahon, C.D.; et al. Cyclic glycine-proline regulates IGF-1 homeostasis by altering the binding of IGFBP-3 to IGF-1. Sci. Rep. 2014, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, K.; Wang, A.; Harris, P.W.R.; Vickers, M.H.; Guan, J. Cyclic glycine-proline administration normalizes high-fat diet-induced synaptophysin expression in obese rats. Neuropeptides 2019, 76, 101935. [Google Scholar] [CrossRef] [PubMed]
- Devesa, J.; Devesa, O.; Carrillo, M.; Casteleiro, N.; Devesa, A.; Llorente, D.; González, C. Rett Syndrome: Treatment with IGF-I, Melatonin, Blackcurrant Extracts, and Rehabilitation. Reports 2018, 1, 14. [Google Scholar] [CrossRef]
- Sánchez, A.; Calpena, A.C.; Clares, B. Evaluating the oxidative stress in inflammation: Role of melatonin. Int. J. Mol. Sci. 2015, 16, 16981–17004. [Google Scholar] [CrossRef]
- De Felice, C.; Della Ragione, F.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Ciccoli, L.; Scalabrì, F.; Marracino, F.; Madonna, M.; Belmonte, G.; et al. Oxidative brain damage in Mecp2-mutant murine models of Rett syndrome. Neurobiol. Dis. 2014, 68, 66–77. [Google Scholar] [CrossRef]
- Shulyakova, N.; Andreazza, A.C.; Mills, L.R.; Eubanks, J.H. Mitochondrial dysfunction in the pathogenesis of rett syndrome: Implications for mitochondria-targeted therapies. Front. Cell. Neurosci. 2017, 11, 1–9. [Google Scholar] [CrossRef]
- De Leersnyder, H.; Zisapel, N.; Laudon, M. Prolonged-release melatonin for children with neurodevelopmental disorders. Pediatr. Neurol. 2011, 45, 23–26. [Google Scholar] [CrossRef]
- Lykken, E.A.; Shyng, C.; Edwards, R.J.; Rozenberg, A.; Gray, S.J. Recent progress and considerations for AAV gene therapies targeting the central nervous system. J. Neurodev. Disord. 2018, 10, 1–10. [Google Scholar] [CrossRef]
- Katz, D.M.; Bird, A.; Coenraads, M.; Gray, S.J.; Menon, D.U.; Philpot, B.D.; Tarquinio, D.C. Rett Syndrome: Crossing the Threshold to Clinical Translation. Trends Neurosci. 2016, 39, 100–113. [Google Scholar] [CrossRef]
- Croci, S.; Lucia, M.; Katia, C.; Daga, S.; Donati, F.; Tiziana, F.; Elisa, P.; Diego, F.; Lamacchia, V.; Tita, R.; et al. AAV-mediated FOXG1 gene editing in human Rett primary cells. Eur. J. Hum. Genet. 2020, 28, 1446–1458. [Google Scholar] [CrossRef]
- Garg, S.K.; Lioy, D.T.; Mcgann, J.C.; Bissonnette, J.M.; Murtha, M.J.; Foust, K.D.; Kaspar, B.K.; Bird, A.; Mandel, G. Systemic Delivery of MeCP2 Rescues Behavioral and Cellular Deficits in Female Mouse Models of Rett Syndrome. Neurobiol. Dis. Syst. 2013, 33, 13612–13620. [Google Scholar] [CrossRef] [PubMed]
- Gadalla, K.K.E.; Vudhironarit, T.; Hector, R.D.; Sinnett, S.; Bahey, N.G.; Bailey, M.E.S.; Gray, S.J.; Cobb, S.R. Development of a Novel AAV Gene Therapy Cassette with Improved Safety Features and Efficacy in a Mouse Model of Rett Syndrome. Mol. Methods Clin. Dev. 2017, 5, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Sinnett, S.E.; Hector, R.D.; Gadalla, K.K.E.; Heindel, C.; Chen, D.; Zaric, V.; Bailey, M.E.S.; Cobb, S.R.; Gray, S.J. Improved MECP2 Gene Therapy Extends the Survival of MeCP2-Null Mice without Apparent Toxicity after Intracisternal Delivery. Mol. Methods Clin. Dev. 2017, 5, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Lioy, D.T.; Ma, L.; Impey, S.; Mandel, G.; Goodman, R.H. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat. Neurosci. 2007, 10, 1513–1514. [Google Scholar] [CrossRef]
- Ribeiro, A.O.; Campos, V.F.; Lemke, N.; Pinhal, D. Understanding the Modus Operandi of MicroRNA Regulatory Clusters. Cells 2019, 8, 1–18. [Google Scholar]
- Miller, D.J.; Bhaduri, A.; Sestan, N.; Kriegstein, A. Shared and derived features of cellular diversity in the human cerebral cortex. Curr. Opin. Neurobiol. 2019, 56, 117–124. [Google Scholar] [CrossRef]
- Hodge, R.; Bakken, T.; Miller, J.; Smith, K.; Barkan, E.; Graybuck, L.; Close, J.; Long, B.; Johansen, N.; Penn, O.; et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 2019, 573, 61–68. [Google Scholar] [CrossRef]
- Anand, R.; Graham, S.M.; Hartman, R.D.; Forrest, E.C. Sarizotan for the treatment of severe apnea in patients with rett syndrome (RTT): Rationale and design of international 6-month, randomized, placebo-controlled phase III trial (STARs). J. Neurol. Sci. 2017, 381, 963. [Google Scholar] [CrossRef]
- Abdala, A.P.; Lioy, D.T.; Garg, S.K.; Knopp, S.J.; Paton, J.F.R.; Bissonnette, J.M. Effect of Sarizotan, a 5-HT 1a and D2-Like Receptor Agonist, on Respiration in Three Mouse Models of Rett Syndrome. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1031–1039. [Google Scholar] [CrossRef]
- Luo, C.; Lancaster, M.A.; Castanon, R.; Nery, J.R.; Knoblich, J.A.; Ecker, J.R.; Luo, C.; Lancaster, M.A.; Castanon, R.; Nery, J.R.; et al. Cerebral Organoids Recapitulate Epigenomic Signatures of the Human Fetal Brain Resource Cerebral Organoids Recapitulate Epigenomic Signatures of the Human Fetal Brain. Cell Rep. 2016, 17, 3369–3384. [Google Scholar] [CrossRef]
- Chiaradia, I.; Lancaster, M.A. Brain organoids for the study of human neurobiology at the interface of in vitro and in vivo. Nat. Neurosci. 2020, 23, 1496–1508. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.D.; Pas, S.P. Building Models of Brain Disorders with Three-Dimensional Organoids. Neuron 2018, 100, 389–405. [Google Scholar] [CrossRef]
- Fedorchak, N.J.; Iyer, N.; Ashton, R.S. Bioengineering tissue morphogenesis and function in human neural organoids. Semin. Cell Dev. Biol. 2020, 111, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Song, H.; Ming, G. Brain organoids: Advances, applications and challenges. Co. Biol. 2019, 146, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Corsini, N.S.; Wolfinger, S.; Gustafson, E.H.; Burkard, T.R.; Otani, T.; Livesey, F.J.; Juergen, A. Guided self-organization and cortical plate formation in human brain organoids. Nat. Biotechnol. 2017, 35, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Tanaka, Y.; Cakir, B.; Patterson, B.; Kim, K.; Sun, P.; Kang, Y.; Zhong, M.; Liu, X.; Patra, P.; et al. hESC-derived thalamic organoids form reciprocal projections when fused with cortical organoids. Cell Stem Cell. 2019, 24, 487–497. [Google Scholar] [CrossRef]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Lee, S.; Weissman, S.M.; Park, I.; Xiang, Y.; Tanaka, Y.; Patterson, B.; Kang, Y.; et al. Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and Interneuron Migration Resource Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development. Stem Cell 2017, 21, 383–398.e7. [Google Scholar]
- Muguruma, K.; Nishiyama, A.; Kawakami, H.; Hashimoto, K.; Sasai, Y. Self-Organization of Polarized Cerebellar Tissue in 3D Culture of Human Pluripotent Stem Cells. Cell Rep. 2015, 10, 537–550. [Google Scholar] [CrossRef]
- Silva, T.P.; Bekman, E.; Fernandes, T.G.; Vaz, S.H.; Carlos, A.; Diogo, M.M.; Cabral, J.M.; Carmo-fonseca, M. Maturation of human pluripotent stem cell-derived cerebellar neurons in the absence of co-culture. Front. Bioeng. Biotechnol. 2020, 8, 2296–4185. [Google Scholar] [CrossRef]
- Nayler, S.; Agarwal, D.; Curion, F.; Bowden, R.; Becker, E.B.E. Single-cell sequencing of human iPSC-derived cerebellar organoids shows recapitulation of cerebellar development. bioRxiv 2020, 1–30. [Google Scholar] [CrossRef]
- Louise, S.; Modamio, J.; Mendes-pinheiro, B.; Sophia, A.; Betsou, F.; Christian, J. Reproducible generation of human midbrain organoids for in vitro modeling of Parkinson ’ s disease. Stem Cell Res. 2020, 46, 101870. [Google Scholar]
- Smits, L.M.; Magni, S.; Kinugawa, K.; Grzyb, K.; Luginbühl, J.; Sabate-soler, S.; Bolognin, S.; Shin, J.W.; Mori, E.; Skupin, A.; et al. Single-cell transcriptomics reveals multiple neuronal cell types in human midbrain-specific organoids. Cell Tissue Res. 2020, 382, 463–476. [Google Scholar] [CrossRef]
- Ogura, T.; Sakaguchi, H.; Miyamoto, S.; Takahashi, J. Three-dimensional induction of dorsal, intermediate and ventral spinal cord tissues from human pluripotent stem cells. Co. Biol. 2018, 145, 1–12. [Google Scholar] [CrossRef]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Jacob, F.; Song, M.M.; Nguyen, H.N.; Song, H.; Ming, G. Generation of human brain region—Specific organoids using a miniaturized spinning bioreactor. Nature 2018, 13, 565–580. [Google Scholar] [CrossRef]
- Silva, T.P.; Fernandes, T.G.; Nogueira, D.E.S.; Rodrigues, C.A.V.; Bekman, E.P.; Jung, S.; Lee, B.; Carmo-fonseca, M.; Cabral, J.M.S. Scalable Generation of Mature Cerebellar Organoids from Human Pluripotent Stem Cells and Characterization by Immunostaining. JOVE 2020, 160, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Sun, L.; Id, M.W.; Id, J.L.; Id, S.Z.; Li, R.; Li, P.; Guo, L.; Fang, A.; Id, R.C.; et al. Vascularized human cortical organoids (vOrganoids) model cortical development in vivo. PLoS Biol. 2020, 18, e3000705. [Google Scholar] [CrossRef] [PubMed]
- Salmon, I.; Grebenyuk, S.; Rahman, A.; Fattah, A.; Rustandi, G.; Verfaillie, C.; Ranga, A. Engineering neurovascular organoids with 3D printed microfluidic chips. bioRxiv 2021, 1–22. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, A.R.; Fernandes, T.G.; Cabral, J.M.S.; Diogo, M.M. Modeling Rett Syndrome with Human Pluripotent Stem Cells: Mechanistic Outcomes and Future Clinical Perspectives. Int. J. Mol. Sci. 2021, 22, 3751. https://doi.org/10.3390/ijms22073751
Gomes AR, Fernandes TG, Cabral JMS, Diogo MM. Modeling Rett Syndrome with Human Pluripotent Stem Cells: Mechanistic Outcomes and Future Clinical Perspectives. International Journal of Molecular Sciences. 2021; 22(7):3751. https://doi.org/10.3390/ijms22073751
Chicago/Turabian StyleGomes, Ana Rita, Tiago G. Fernandes, Joaquim M.S. Cabral, and Maria Margarida Diogo. 2021. "Modeling Rett Syndrome with Human Pluripotent Stem Cells: Mechanistic Outcomes and Future Clinical Perspectives" International Journal of Molecular Sciences 22, no. 7: 3751. https://doi.org/10.3390/ijms22073751
APA StyleGomes, A. R., Fernandes, T. G., Cabral, J. M. S., & Diogo, M. M. (2021). Modeling Rett Syndrome with Human Pluripotent Stem Cells: Mechanistic Outcomes and Future Clinical Perspectives. International Journal of Molecular Sciences, 22(7), 3751. https://doi.org/10.3390/ijms22073751