
Tau Is Truncated in Five Regions of the Normal Adult Human Brain

 ,
, 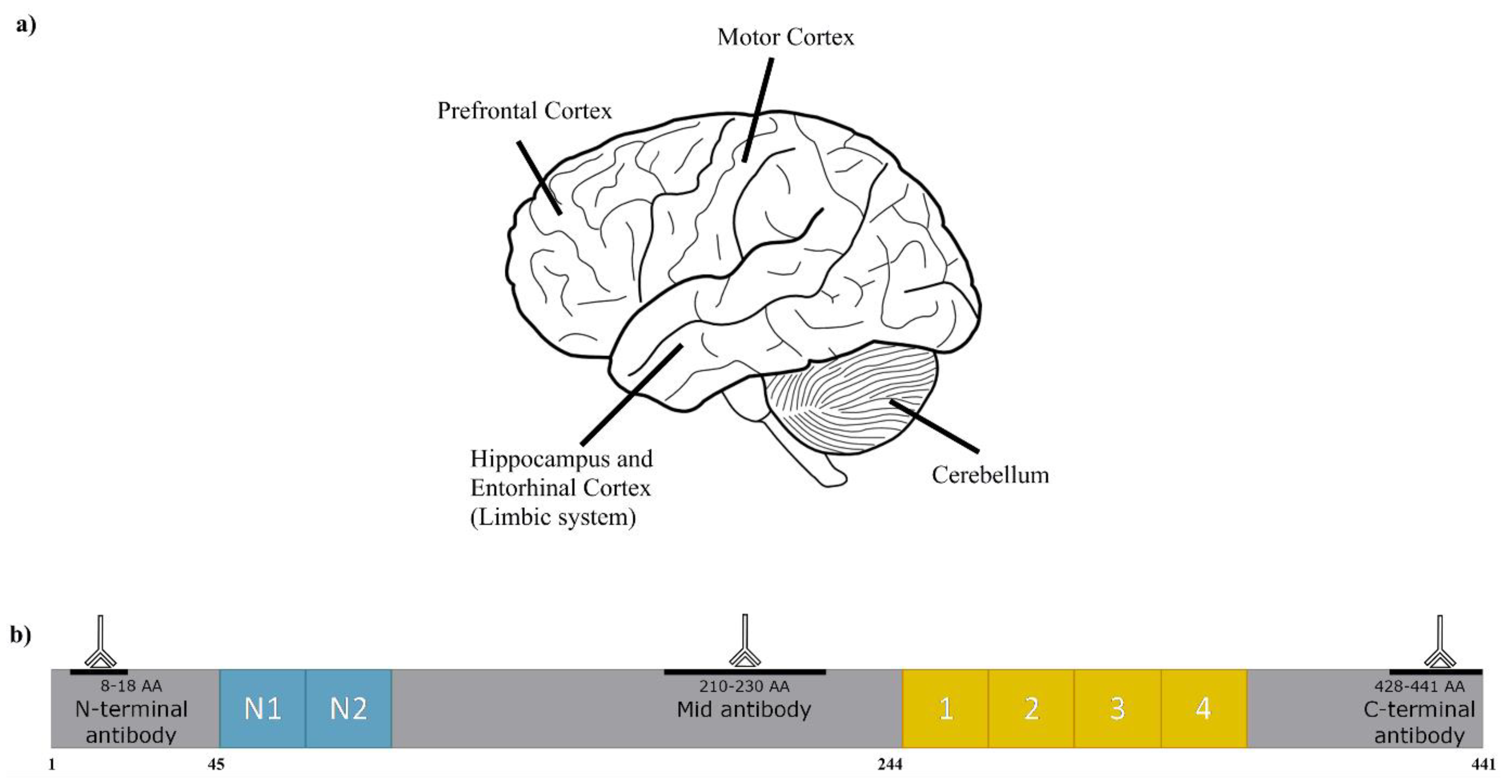{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
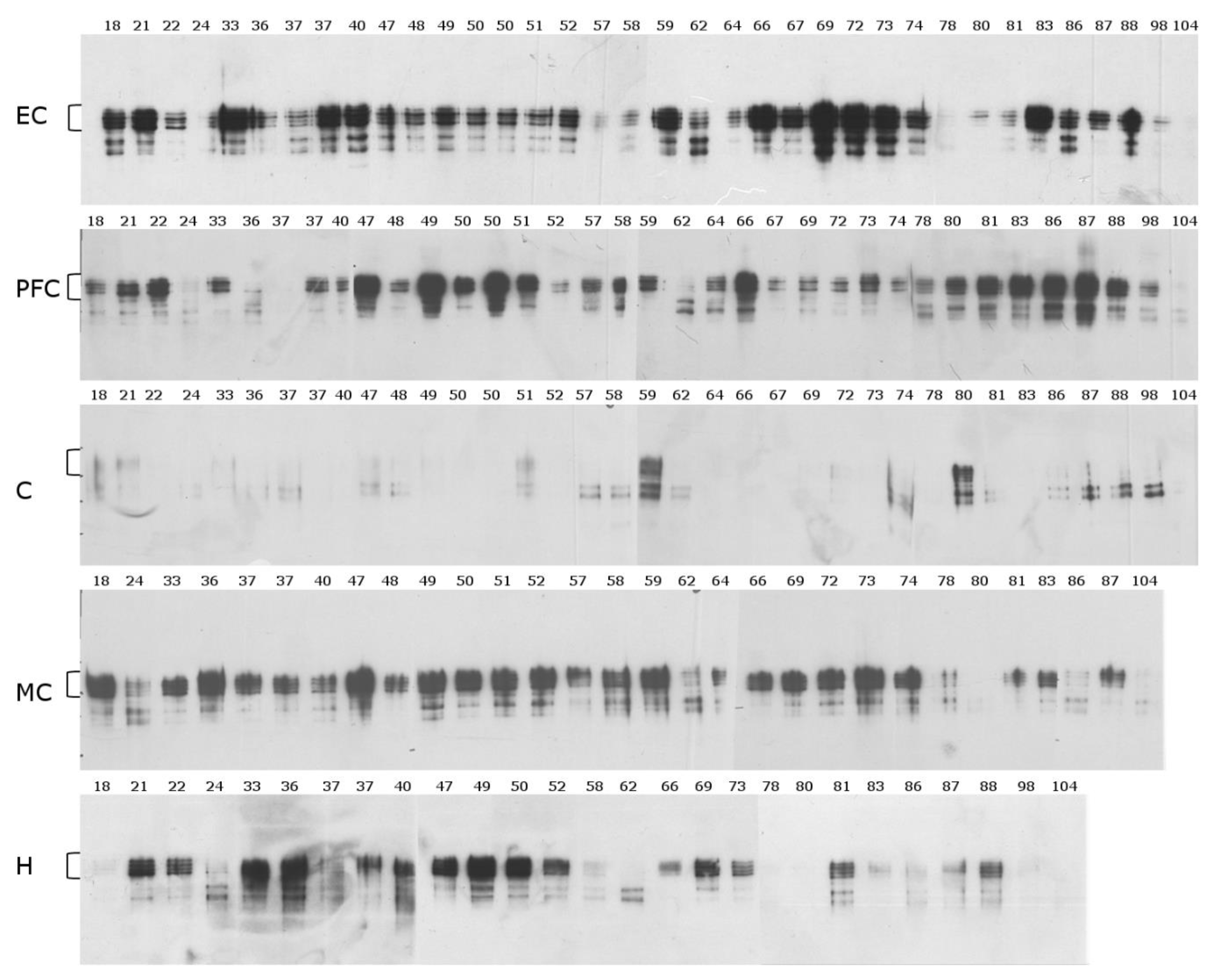
2.1. Mid-Sequence Antibody
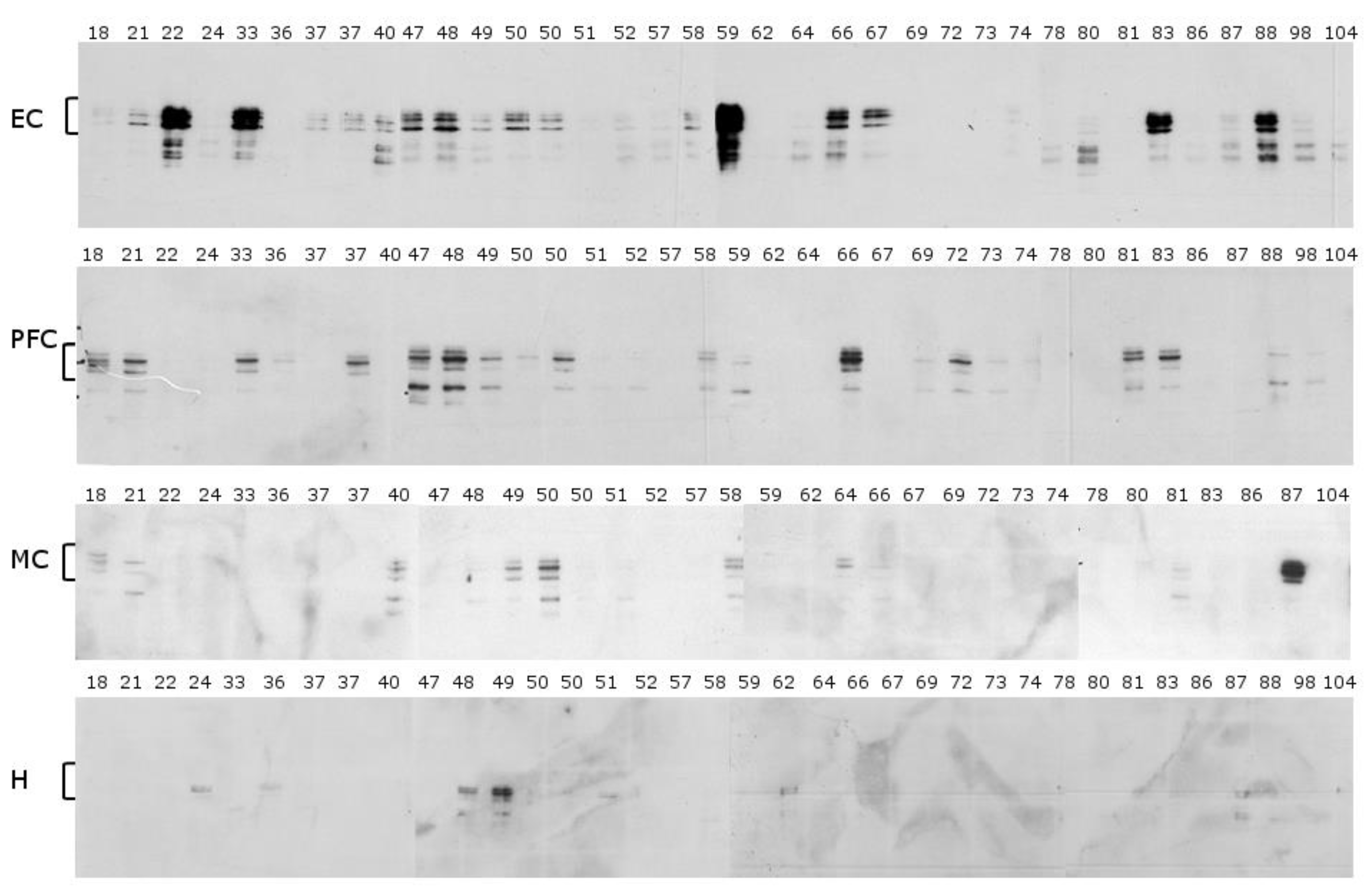
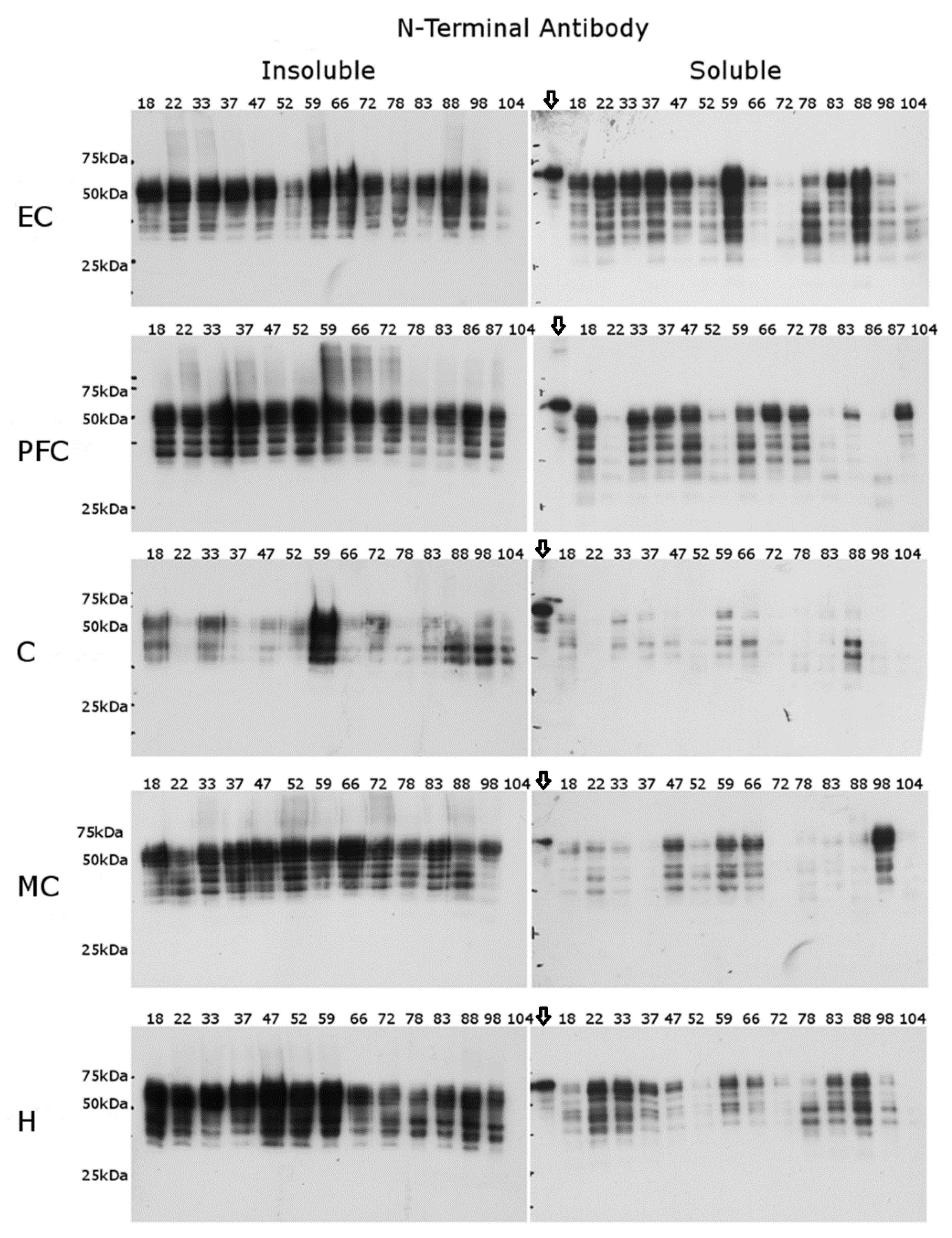
2.2. N-Terminal Antibody
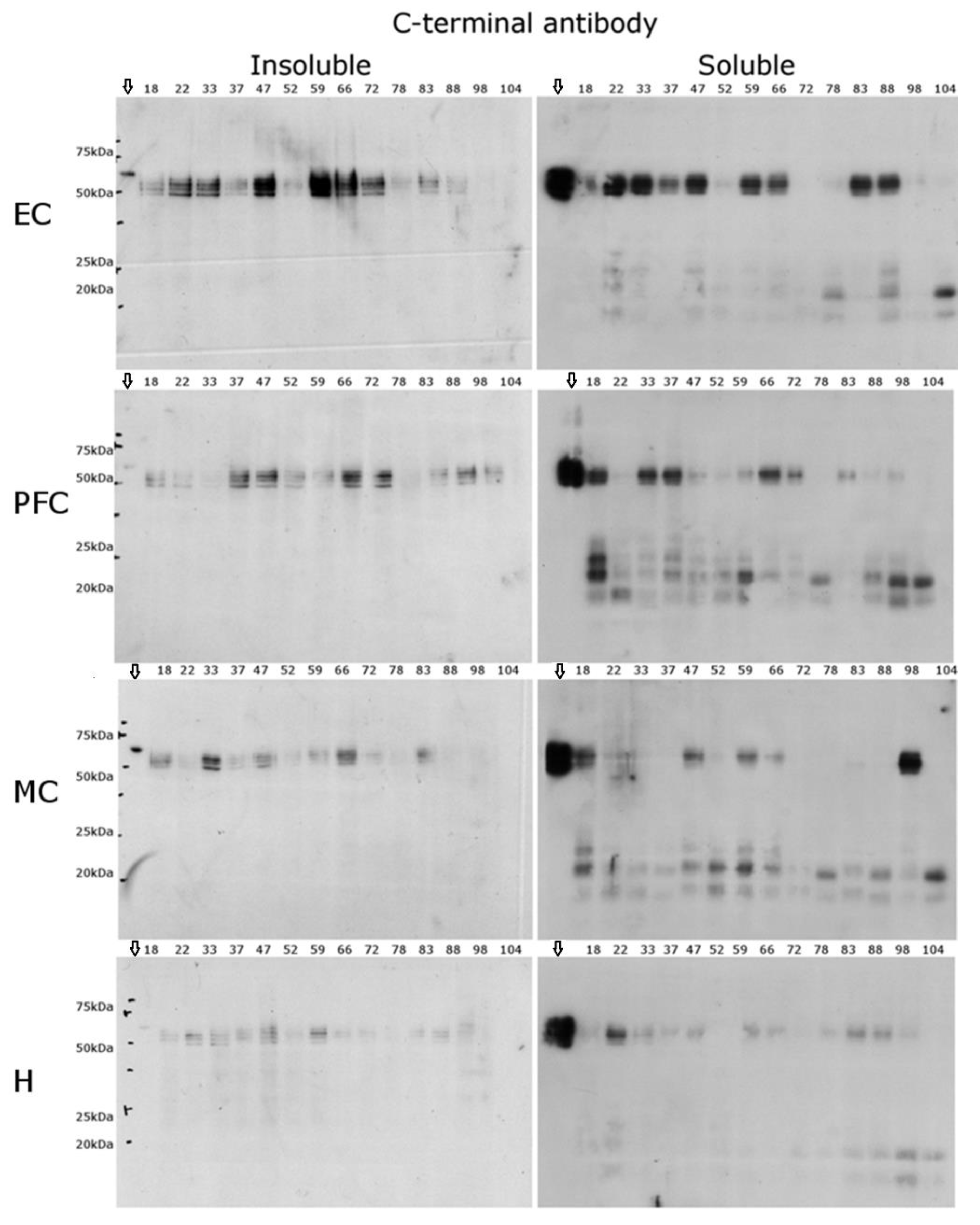
2.3. C-Terminal Antibody
2.4. Limitations of the Study
3. Discussion
4. Materials and Methods
4.1. Homogenisation of Human Brain Tissue
4.2. Western Blots
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| EC | entorhinal cortex |
| PFC | prefrontal cortex |
| H | hippocampus |
| MC | motor cortex |
| C | cerebellum |
References
- Josephs, K.A.; Whitwell, J.L.; Ahmed, Z.; Shiung, M.M.; Weigand, S.D.; Knopman, D.S.; Boeve, B.F.; Parisi, J.E.; Petersen, R.C.; Dickson, D.W.; et al. β-amyloid burden is not associated with rates of brain atrophy. Ann. Neurol. 2008, 63, 204–212. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 67–72. [Google Scholar] [CrossRef]
- Flament, S.; Delacourte, A.; Verny, M.; Hauw, J.J.; Javoy-Agid, F. Abnormal Tau proteins in progressive supranuclear palsy. Similarities and differences with the neurofibrillary degeneration of the Alzheimer type. Acta Neuropathol. 1991, 81, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Zhukareva, V.; Vogelsberg-Ragaglia, V.; Wszolek, Z.; Reed, L.; Miller, B.I.; Geschwind, D.H.; Bird, T.D.; McKeel, D.; Goate, A.; et al. Mutation-Specific Functional Impairments in Distinct Tau Isoforms of Hereditary FTDP-17. Science 1998, 282, 1914–1917. [Google Scholar] [CrossRef]
- Zhou, Y.; Shi, J.; Chu, D.; Hu, W.; Guan, Z.; Gong, C.-X.; Iqbal, K.; Liu, F. Relevance of Phosphorylation and Truncation of Tau to the Etiopathogenesis of Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 27. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Hayashi, A.; Takeuchi, M.; Dobashi, M.; Mori, Y.; Shinoda, H.; Aizawa, T.; Demura, M.; Kawano, K. Unfolding and aggregation of transthyretin by the truncation of 50 N-terminal amino acids. Proteins 2008, 72, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Mondragón-Rodríguez, S.; Basurto-Islas, G.; Santa-Maria, I.; Mena, R.; Binder, L.I.; Avila, J.; Smith, M.A.; Perry, G.; García-Sierra, F. Cleavage and conformational changes of tau protein follow phosphorylation during Alzheimer’s disease. Int. J. Exp. Pathol. 2008, 89, 81–90. [Google Scholar] [CrossRef]
- Kenessey, A.; Yen, S.-H.; Liu, W.-K.; Yang, X.-R.; Dunlop, D.S. Detection of d-aspartate in tau proteins associated with Alzheimer paired helical filaments. Brain Res. 1995, 675, 183–189. [Google Scholar] [CrossRef]
- Watanabe, A.; Takio, K.; Ihara, Y. Deamidation and Isoaspartate Formation in Smeared Tau in Paired Helical Filaments: Unusual Properties of the Microtubule-Binding Domain of Tau. J. Biol. Chem. 1999, 274, 7368–7378. [Google Scholar] [CrossRef]
- Reyes, J.F.; Fu, Y.; Vana, L.; Kanaan, N.M.; Binder, L.I. Tyrosine Nitration within the Proline-Rich Region of Tau in Alzheimer’s Disease. Am. J. Pathol. 2011, 178, 2275–2285. [Google Scholar] [CrossRef]
- Wang, J.-Z.; Grundke-Iqbal, I.; Iqbal, K. Glycosylation of microtubule−associated protein tau: An abnormal posttranslational modification in Alzheimer’s disease. Nat. Med. 1996, 2, 871–875. [Google Scholar] [CrossRef]
- Funk, K.E.; Thomas, S.N.; Schafer, K.N.; Cooper, G.L.; Liao, Z.; Clark, D.J.; Yang, A.J.; Kuret, J. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem. J. 2014, 462, 77–88. [Google Scholar] [CrossRef]
- Thomas, S.N.; Funk, K.E.; Wan, Y.; Liao, Z.; Davies, P.; Kuret, J.; Yang, A.J. Dual modification of Alzheimer’s disease PHF-tau protein by lysine methylation and ubiquitylation: A mass spectrometry approach. Acta Neuropathol. 2012, 123, 105–117. [Google Scholar] [CrossRef]
- Watanabe, A.; Hong, W.-K.; Dohmae, N.; Takio, K.; Morishima-Kawashima, M.; Ihara, Y. Molecular aging of tau: Disulfide-independent aggregation and non-enzymatic degradation in vitro and in vivo. J. Neurochem. 2004, 90, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Kovacech, B.; Novak, M. Tau Truncation is a Productive Posttranslation Modification of Neurofibrillary Degeneration in Alzheimer’s Disease. Curr. Alzheimer Res. 2010, 7, 708–716. [Google Scholar] [CrossRef]
- Derisbourg, M.; Leghay, C.; Chiappetta, G.; Fernandez-Gomez, F.-J.; Laurent, C.; Demeyer, D.; Carrier, S.; Buée-Scherrer, V.; Blum, D.; Vinh, J.; et al. Role of the Tau N-terminal region in microtubule stabilization revealed by new endogenous truncated forms. Sci. Rep. 2015, 5, 9659. [Google Scholar] [CrossRef]
- Quinn, J.P.; Corbett, N.J.; Kellett, K.A.B.; Hooper, N.M. Tau Proteolysis in the Pathogenesis of Tauopathies: Neurotoxic Fragments and Novel Biomarkers. J. Alzheimer’s Dis. JAD 2018, 63, 13–33. [Google Scholar] [CrossRef]
- Gamblin, T.C.; Chen, F.; Zambrano, A.; Abraha, A.; Lagalwar, S.; Guillozet, A.L.; Lu, M.; Fu, Y.; Garcia-Sierra, F.; LaPointe, N.; et al. Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 10032–10037. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.-M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Biernat, J.; Pickhardt, M.; Mandelkow, E.; Mandelkow, E.-M. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc. Natl. Acad. Sci. USA 2007, 104, 10252–10257. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Ferreira, A. The Generation of a 17 kDa Neurotoxic Fragment: An Alternative Mechanism by which Tau Mediates β-Amyloid-Induced Neurodegeneration. J. Neurosci. 2005, 25, 5365–5375. [Google Scholar] [CrossRef]
- Mena, R.; Edwards, P.C.; Harrington, C.R.; Mukaetova-Ladinska, E.B.; Wischik, C.M. Staging the pathological assembly of truncated tau protein into paired helical filaments in Alzheimer’s disease. Acta Neuropathol. 1996, 91, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, M.; Liu, X.; Kang, S.S.; Kwon, I.-S.; Duong, D.M.; Seyfried, N.T.; Hu, W.T.; Liu, Z.; Wang, J.-Z.; et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat. Med. 2014, 20, 1254. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Xu, W.; Jin, N.; Li, L.; Zhou, Y.; Chu, D.; Gong, C.X.; Iqbal, K.; Liu, F. Truncation of Tau selectively facilitates its pathological activities. J. Biol. Chem. 2020, 295, 13812–13828. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Bracamontes, V.J.; Escobar-Herrera, J.; Kristofikova, Z.; Rípova, D.; Florán-Garduño, B.; Garcia-Sierra, F. Early but not late conformational changes of tau in association with ubiquitination of neurofibrillary pathology in Alzheimer’s disease brains. Brain Res. 2020, 1744, 146953. [Google Scholar] [CrossRef]
- Hooi, M.; Truscott, R. Racemisation and human cataract. d-Ser, d-Asp/Asn and d-Thr are higher in the lifelong proteins of cataract lenses than in age-matched normal lenses. Age 2011, 33, 131–141. [Google Scholar] [CrossRef]
- Friedrich, M.G.; Wang, Z.; Schey, K.L.; Truscott, R.J.W. Mechanism of protein cleavage at asparagine leading to protein–protein cross-links. Biochem. J. 2019, 476, 3817–3834. [Google Scholar] [CrossRef]
- Friedrich, M.G.; Lam, J.; Truscott, R.J. Degradation of an old human protein: Age-dependent cleavage of gammaS-crystallin generates a peptide that binds to cell membranes. J. Biol. Chem. 2012, 287, 39012–39020. [Google Scholar] [CrossRef]
- Grey, A.C.; Schey, K.L. Age-Related Changes in the Spatial Distribution of Human Lens α-Crystallin Products by MALDI Imaging Mass Spectrometry. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4319–4329. [Google Scholar] [CrossRef] [PubMed]
- Oz, S.; Ivashko-Pachima, Y.; Gozes, I. The ADNP Derived Peptide, NAP Modulates the Tubulin Pool: Implication for Neurotrophic and Neuroprotective Activities. PLoS ONE 2012, 7, e51458. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Yin, Y.; Yin, Y.; Zhang, H.; Wan, H.; Wang, L.; Zuo, Y.; Gao, D.; Li, M.; Li, J.; et al. Tau-induced upregulation of C/EBPβ-TRPC1-SOCE signaling aggravates tauopathies: A vicious cycle in Alzheimer neurodegeneration. Aging Cell 2020, 19, e13209. [Google Scholar] [CrossRef] [PubMed]
- Labisso, W.L.; Raulin, A.C.; Nwidu, L.L.; Kocon, A.; Wayne, D.; Erdozain, A.M.; Morentin, B.; Schwendener, D.; Allen, G.; Enticott, J.; et al. The Loss of α- and β-Tubulin Proteins Are a Pathological Hallmark of Chronic Alcohol Consumption and Natural Brain Ageing. Brain Sci. 2018, 8, 175. [Google Scholar] [CrossRef] [PubMed]
- Maphis, N.M.; Jiang, S.; Binder, J.; Wright, C.; Gopalan, B.; Lamb, B.T.; Bhaskar, K. Whole Genome Expression Analysis in a Mouse Model of Tauopathy Identifies MECP2 as a Possible Regulator of Tau Pathology. Front. Mol. Neurosci. 2017, 10, 69. [Google Scholar] [CrossRef]
- Kubo, A.; Misonou, H.; Matsuyama, M.; Nomori, A.; Wada-Kakuda, S.; Takashima, A.; Kawata, M.; Murayama, S.; Ihara, Y.; Miyasaka, T. Distribution of endogenous normal tau in the mouse brain. J. Comp. Neurol. 2019, 527, 985–998. [Google Scholar] [CrossRef]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The Many Faces of Tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [PubMed]
- Mukaetova-Ladinska, E.B.; Harrington, C.R.; Hills, R.; O’Sullivan, A.; Roth, M.; Wischik, C.M. Regional Distribution of Paired Helical Filaments and Normal Tau Proteins in Aging and in Alzheimer’s Disease with and without Occipital Lobe Involvement. Dement. Geriatic Cogn. Disord. 1992, 3, 61–69. [Google Scholar] [CrossRef]
- Čaušević, M.; Farooq, U.; Lovestone, S.; Killick, R. β-Amyloid precursor protein and tau protein levels are differently regulated in human cerebellum compared to brain regions vulnerable to Alzheimer’s type neurodegeneration. Neurosci. Lett. 2010, 485, 162–166. [Google Scholar] [CrossRef]
- Ercan, E.; Eid, S.; Weber, C.; Kowalski, A.; Bichmann, M.; Behrendt, A.; Matthes, F.; Krauss, S.; Reinhardt, P.; Fulle, S.; et al. A validated antibody panel for the characterization of tau post-translational modifications. Mol. Neurodegener. 2017, 12, 87. [Google Scholar] [CrossRef]
- Chen, H.-H.; Liu, P.; Auger, P.; Lee, S.-H.; Adolfsson, O.; Rey-Bellet, L.; Lafrance-Vanasse, J.; Friedman, B.A.; Pihlgren, M.; Muhs, A.; et al. Calpain-mediated tau fragmentation is altered in Alzheimer’s disease progression. Sci. Rep. 2018, 8, 16725. [Google Scholar] [CrossRef]
- Sato, C.; Barthélemy, N.R.; Mawuenyega, K.G.; Patterson, B.W.; Gordon, B.A.; Jockel-Balsarotti, J.; Sullivan, M.; Crisp, M.J.; Kasten, T.; Kirmess, K.M.; et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 2018, 97, 1284–1298.e1287. [Google Scholar] [CrossRef]
- Fornasiero, E.F.; Mandad, S.; Wildhagen, H.; Alevra, M.; Rammner, B.; Keihani, S.; Opazo, F.; Urban, I.; Ischebeck, T.; Sakib, M.S.; et al. Precisely measured protein lifetimes in the mouse brain reveal differences across tissues and subcellular fractions. Nat. Commun. 2018, 9, 4230. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Friedrich, M.G.; Truscott, R.J.W.; Schey, K.L. Cleavage C-terminal to Asp leads to covalent crosslinking of long-lived human proteins. Biochim. Et Biophys. Acta (BBA) Proteins Proteom. 2019, 1867, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Lyons, B.; Kwan, A.H.; Truscott, R.J. Spontaneous cleavage of proteins at serine and threonine is facilitated by zinc. Aging Cell 2016, 15, 237–244. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Johnson, G.V.W.; Jope, R.S.; Binder, L.I. Proteolysis of tau by calpain. Biochem. Biophys. Res. Commun. 1989, 163, 1505–1511. [Google Scholar] [CrossRef]
- Lyons, B.; Jamie, J.; Truscott, R.W. Separate mechanisms for age-related truncation and racemisation of peptide-bound serine. Amino Acids 2014, 46, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Morales-Corraliza, J.; Mazzella, M.J.; Berger, J.D.; Diaz, N.S.; Choi, J.H.K.; Levy, E.; Matsuoka, Y.; Planel, E.; Mathews, P.M. In Vivo Turnover of Tau and APP Metabolites in the Brains of Wild-Type and Tg2576 Mice: Greater Stability of sAPP in the β-Amyloid Depositing Mice. PLoS ONE 2009, 4, e7134. [Google Scholar] [CrossRef]
- Shimizu, T.; Watanabe, A.; Ogawara, M.; Mori, H.; Shirasawa, T. Isoaspartate Formation and Neurodegeneration in Alzheimer’s Disease. Arch. Biochem. Biophys. 2000, 381, 225–234. [Google Scholar] [CrossRef]
- Truscott, R.J.W.; Schey, K.L.; Friedrich, M.G. Old Proteins in Man: A Field in its Infancy. Trends Biochem. Sci. 2016, 41, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S. Propensity for spontaneous succinimide formation from aspartyl and asparaginyl residues in cellular proteins. Int. J. Pept. Protein Res. 1987, 30, 808–821. [Google Scholar] [CrossRef] [PubMed]
- Hains, P.; Truscott, R.J. Age-dependent deamidation of life-long proteins in the human lens. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3107–3114. [Google Scholar] [CrossRef]
- Su, S.-P.; Lyons, B.; Friedrich, M.; McArthur, J.D.; Song, X.; Xavier, D.; Truscott, R.J.W.; Aquilina, J.A. Molecular signatures of long-lived proteins: Autolytic cleavage adjacent to serine residues. Aging Cell 2012, 11, 1125–1127. [Google Scholar] [CrossRef]
- Hooi, M.Y.S.; Raftery, M.J.; Truscott, R.J.W. Age-dependent racemization of serine residues in a human chaperone protein. Protein Sci. 2013, 22, 93–100. [Google Scholar] [CrossRef]
- Klein, T.; Eckhard, U.; Dufour, A.; Solis, N.; Overall, C.M. Proteolytic Cleavage—Mechanisms, Function, and “Omic” Approaches for a Near-Ubiquitous Posttranslational Modification. Chem. Rev. 2018, 118, 1137–1168. [Google Scholar] [CrossRef]
- Lambeth, T.R.; Riggs, D.L.; Talbert, L.E.; Tang, J.; Coburn, E.; Kang, A.S.; Noll, J.; Augello, C.; Ford, B.D.; Julian, R.R. Spontaneous Isomerization of Long-Lived Proteins Provides a Molecular Mechanism for the Lysosomal Failure Observed in Alzheimer’s Disease. ACS Cent. Sci. 2019, 5, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Korlimbinis, A.; Berry, Y.; Thibault, D.; Schey, K.L.; Truscott, R.J. Protein aging: Truncation of aquaporin 0 in human lens regions is a continuous age-dependent process. Exp. Eye Res. 2009, 88, 966–973. [Google Scholar] [CrossRef]
- Basurto-Islas, G.; Luna-Munoz, J.; Guillozet-Bongaarts, A.L.; Binder, L.I.; Mena, R.; Garcia-Sierra, F. Accumulation of Aspartic Acid421- and Glutamic Acid391-Cleaved Tau in Neurofibrillary Tangles Correlates With Progression in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2008, 67, 470–483. [Google Scholar] [CrossRef]
- García-Sierra, F.; Wischik, C.M.; Harrington, C.R.; Luna-Muñoz, J.; Mena, R. Accumulation of C-terminally truncated tau protein associated with vulnerability of the perforant pathway in early stages of neurofibrillary pathology in Alzheimer’s disease. J. Chem. Neuroanat. 2001, 22, 65–77. [Google Scholar] [CrossRef]
- Novak, M.; Kabat, J.; Wischik, C.M. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. Embo J. 1993, 12, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Novak, M.; Edwards, P.C.; Klug, A.; Tichelaar, W.; Crowther, R.A. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4884–4888. [Google Scholar] [CrossRef]
- Liu, P.; Smith, B.R.; Montonye, M.L.; Kemper, L.J.; Leinonen-Wright, K.; Nelson, K.M.; Higgins, L.; Guerrero, C.R.; Markowski, T.W.; Zhao, X.; et al. A soluble truncated tau species related to cognitive dysfunction is elevated in the brain of cognitively impaired human individuals. Sci. Rep. 2020, 10, 3869. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.; Jakes, R.; Edwards, P.C.; Milstein, C.; Wischik, C.M. Difference between the tau protein of Alzheimer paired helical filament core and normal tau revealed by epitope analysis of monoclonal antibodies 423 and 7.51. Proc. Natl. Acad. Sci. USA 1991, 88, 5837–5841. [Google Scholar] [CrossRef]
- Harrington, C.R.; Mukaetova-Ladinska, E.B.; Hills, R.; Edwards, P.C.; Montejo de Garcini, E.; Novak, M.; Wischik, C.M. Measurement of distinct immunochemical presentations of tau protein in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 5842–5846. [Google Scholar] [CrossRef] [PubMed]
- Rissman, R.A.; Poon, W.W.; Blurton-Jones, M.; Oddo, S.; Torp, R.; Vitek, M.P.; LaFerla, F.M.; Rohn, T.T.; Cotman, C.W. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J. Clin. Investig. 2004, 114, 121–130. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Dolan, P.J.; Jin, Y.N.; Johnson, G.V.W. Truncated tau and Aβ cooperatively impair mitochondria in primary neurons. Neurobiol. Aging 2012, 33, 619.e625–619.e635. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Stringaro, A.; Colone, M.; D’Aguanno, S.; Meli, G.; Ciotti, M.; Sancesario, G.; Cattaneo, A.; Bussani, R.; et al. A NH2 tau fragment targets neuronal mitochondria at AD synapses: Possible implications for neurodegeneration. J. Alzheimer’s Dis. JAD 2010, 21, 445–470. [Google Scholar] [CrossRef]
- Al-Hilaly, Y.K.; Foster, B.E.; Biasetti, L.; Lutter, L.; Pollack, S.J.; Rickard, J.E.; Storey, J.M.D.; Harrington, C.R.; Xue, W.F.; Wischik, C.M.; et al. Tau (297–391) forms filaments that structurally mimic the core of paired helical filaments in Alzheimer’s disease brain. FEBS Lett. 2020, 594, 944–950. [Google Scholar] [CrossRef]
- Pollack, S.J.; Trigg, J.; Khanom, T.; Biasetti, L.; Marshall, K.E.; Al-Hilaly, Y.K.; Rickard, J.E.; Harrington, C.R.; Wischik, C.M.; Serpell, L.C. Paired Helical Filament-Forming Region of Tau (297–391) Influences Endogenous Tau Protein and Accumulates in Acidic Compartments in Human Neuronal Cells. J. Mol. Biol. 2020, 432, 4891–4907. [Google Scholar] [CrossRef]
- Kovac, A.; Zilkova, M.; Deli, M.A.; Zilka, N.; Novak, M. Human truncated tau is using a different mechanism from amyloid-beta to damage the blood-brain barrier. J. Alzheimer’s Dis. JAD 2009, 18, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Norris, S.E.; Friedrich, M.G.; Mitchell, T.W.; Truscott, R.J.W.; Else, P.L. Human prefrontal cortex phospholipids containing docosahexaenoic acid increase during normal adult aging, whereas those containing arachidonic acid decrease. Neurobiol. Aging 2015, 36, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Elobeid, A.; Libard, S.; Leino, M.; Popova, S.N.; Alafuzoff, I. Altered Proteins in the Aging Brain. J. Neuropathol. Exp. Neurol. 2016, 75, 316–325. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friedrich, M.G.; Skora, A.; Hancock, S.E.; Mitchell, T.W.; Else, P.L.; Truscott, R.J.W. Tau Is Truncated in Five Regions of the Normal Adult Human Brain. Int. J. Mol. Sci. 2021, 22, 3521. https://doi.org/10.3390/ijms22073521
Friedrich MG, Skora A, Hancock SE, Mitchell TW, Else PL, Truscott RJW. Tau Is Truncated in Five Regions of the Normal Adult Human Brain. International Journal of Molecular Sciences. 2021; 22(7):3521. https://doi.org/10.3390/ijms22073521
Chicago/Turabian StyleFriedrich, Michael G., Amanda Skora, Sarah E. Hancock, Todd W. Mitchell, Paul L. Else, and Roger J. W. Truscott. 2021. "Tau Is Truncated in Five Regions of the Normal Adult Human Brain" International Journal of Molecular Sciences 22, no. 7: 3521. https://doi.org/10.3390/ijms22073521
APA StyleFriedrich, M. G., Skora, A., Hancock, S. E., Mitchell, T. W., Else, P. L., & Truscott, R. J. W. (2021). Tau Is Truncated in Five Regions of the Normal Adult Human Brain. International Journal of Molecular Sciences, 22(7), 3521. https://doi.org/10.3390/ijms22073521