
Combined Transcriptomic and Proteomic Analysis of Perk Toxicity Pathways

 ,
,
Abstract
1. Introduction
2. Results
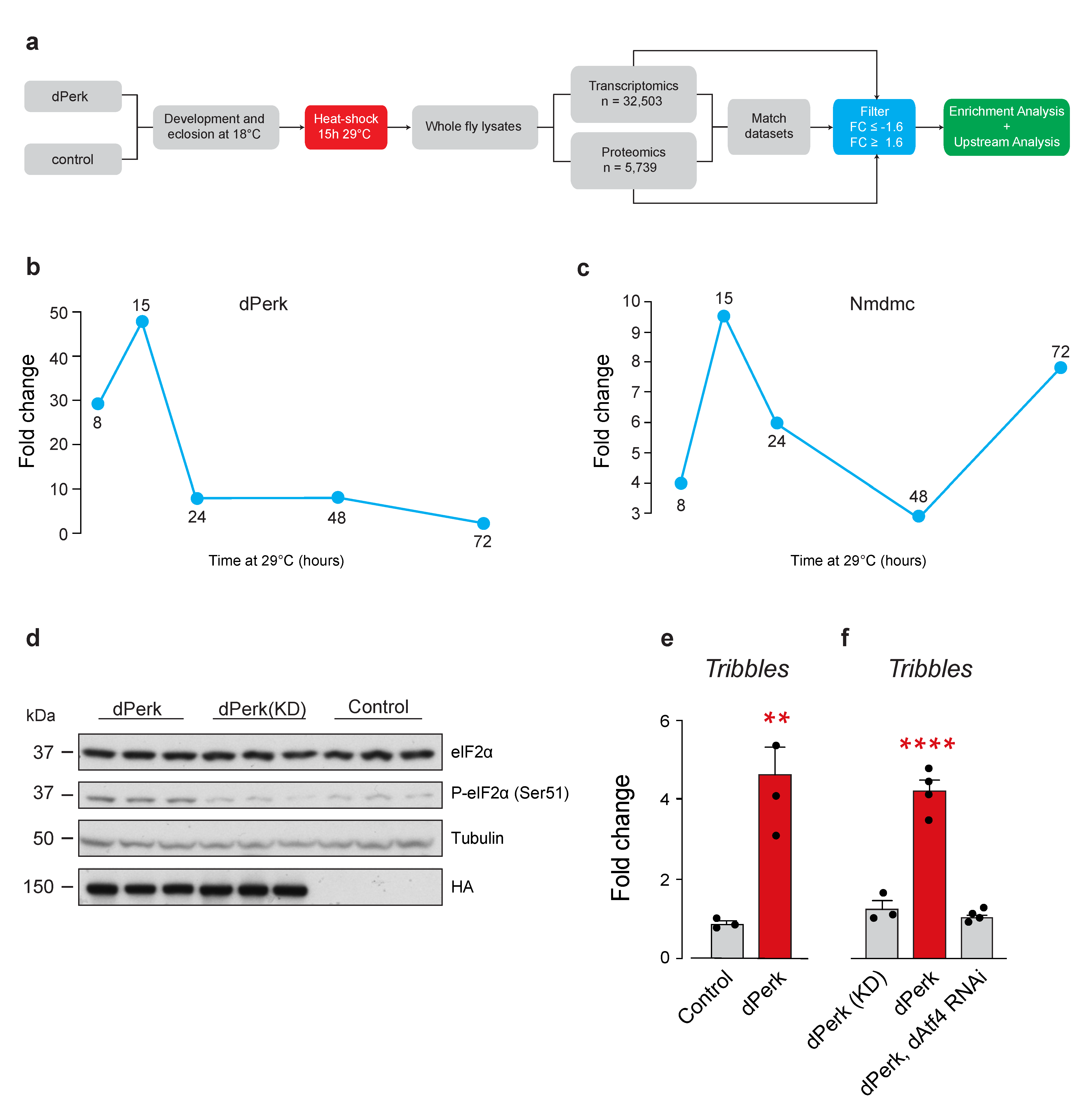
2.1. Drosophila tribbles Is Regulated by dPerk and dAtf4
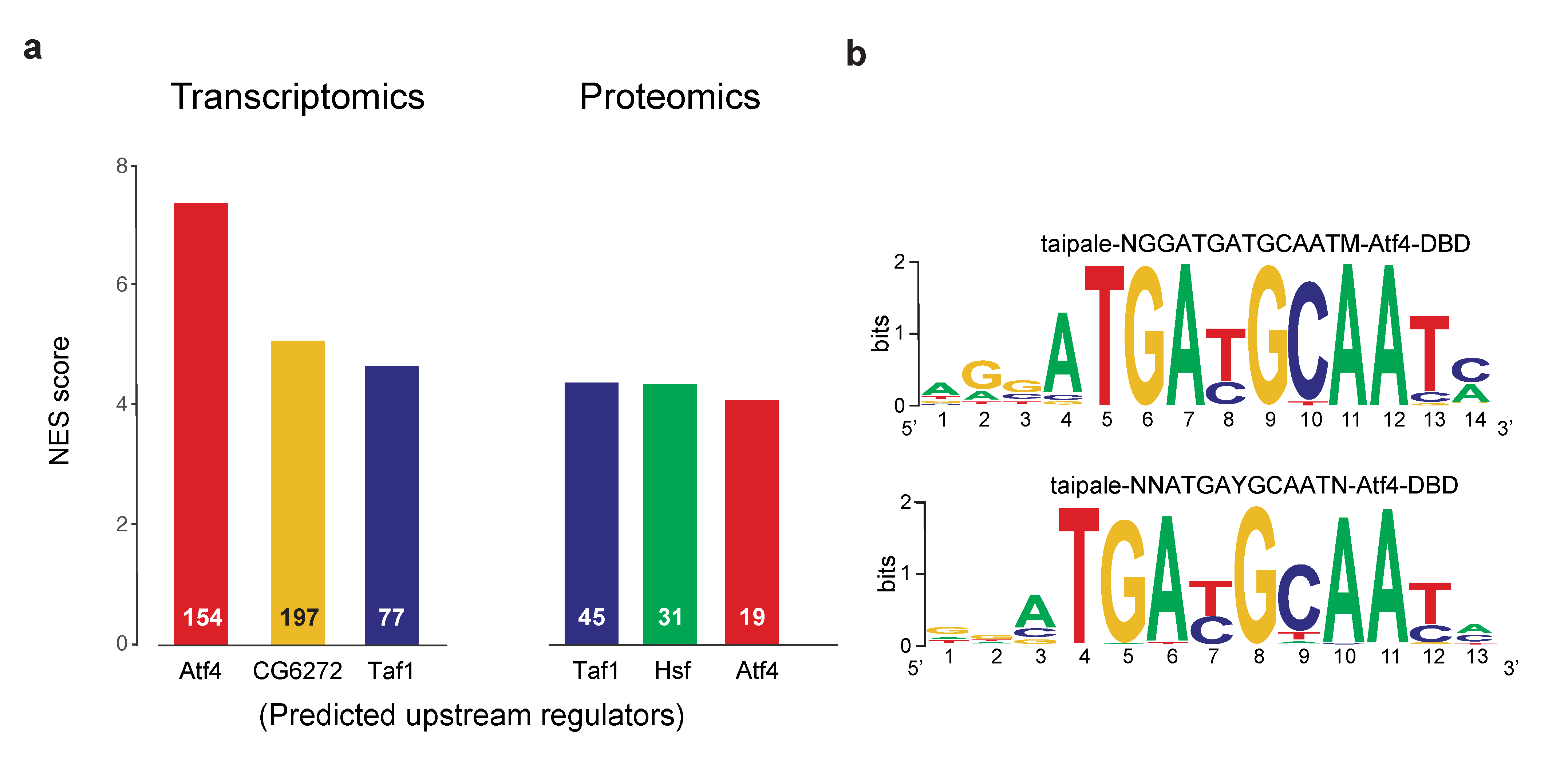
2.2. Divergence between the Upstream Transcriptional Regulators of dPerk-Induced Alterations in Transcripts and Proteins
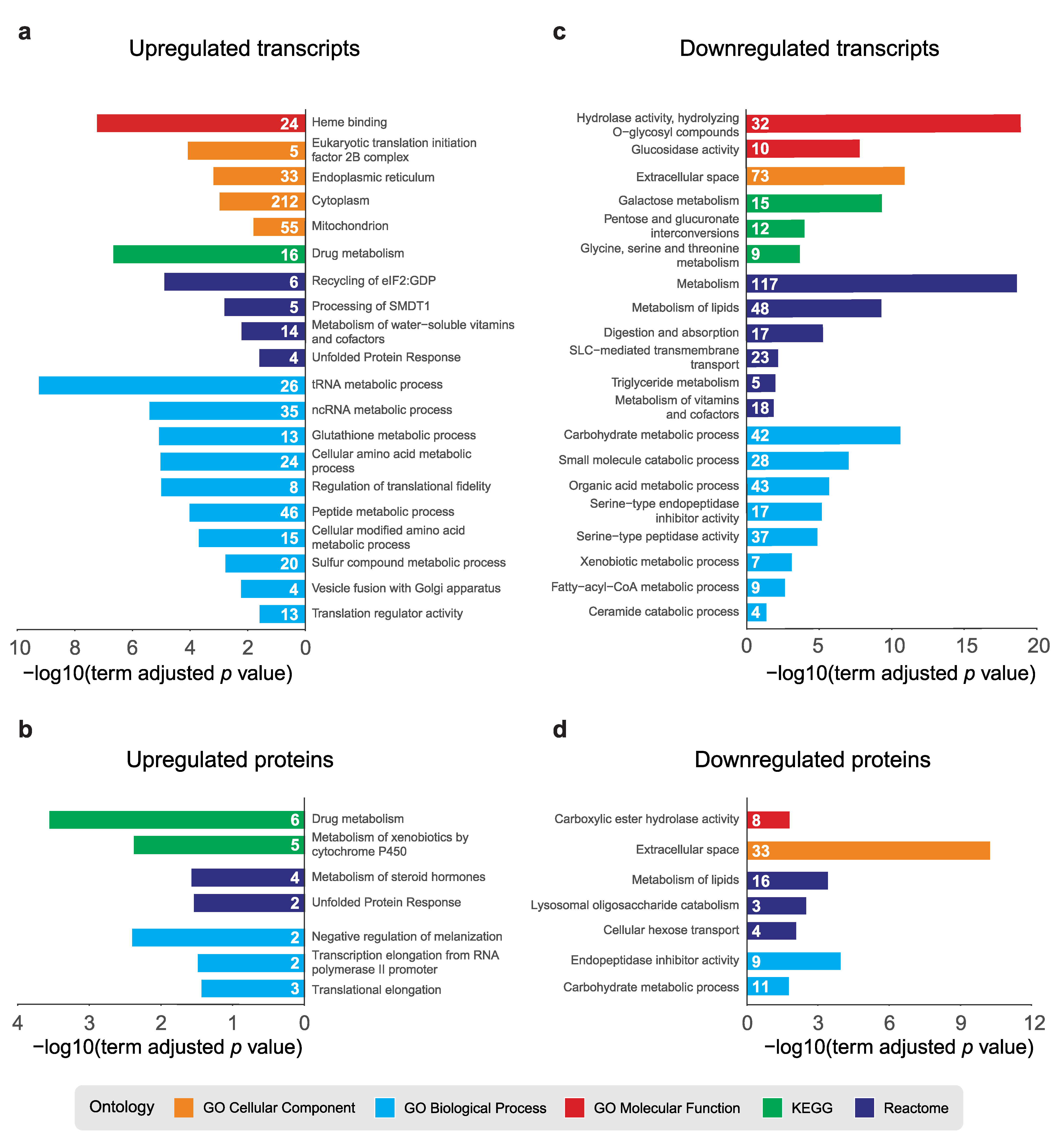
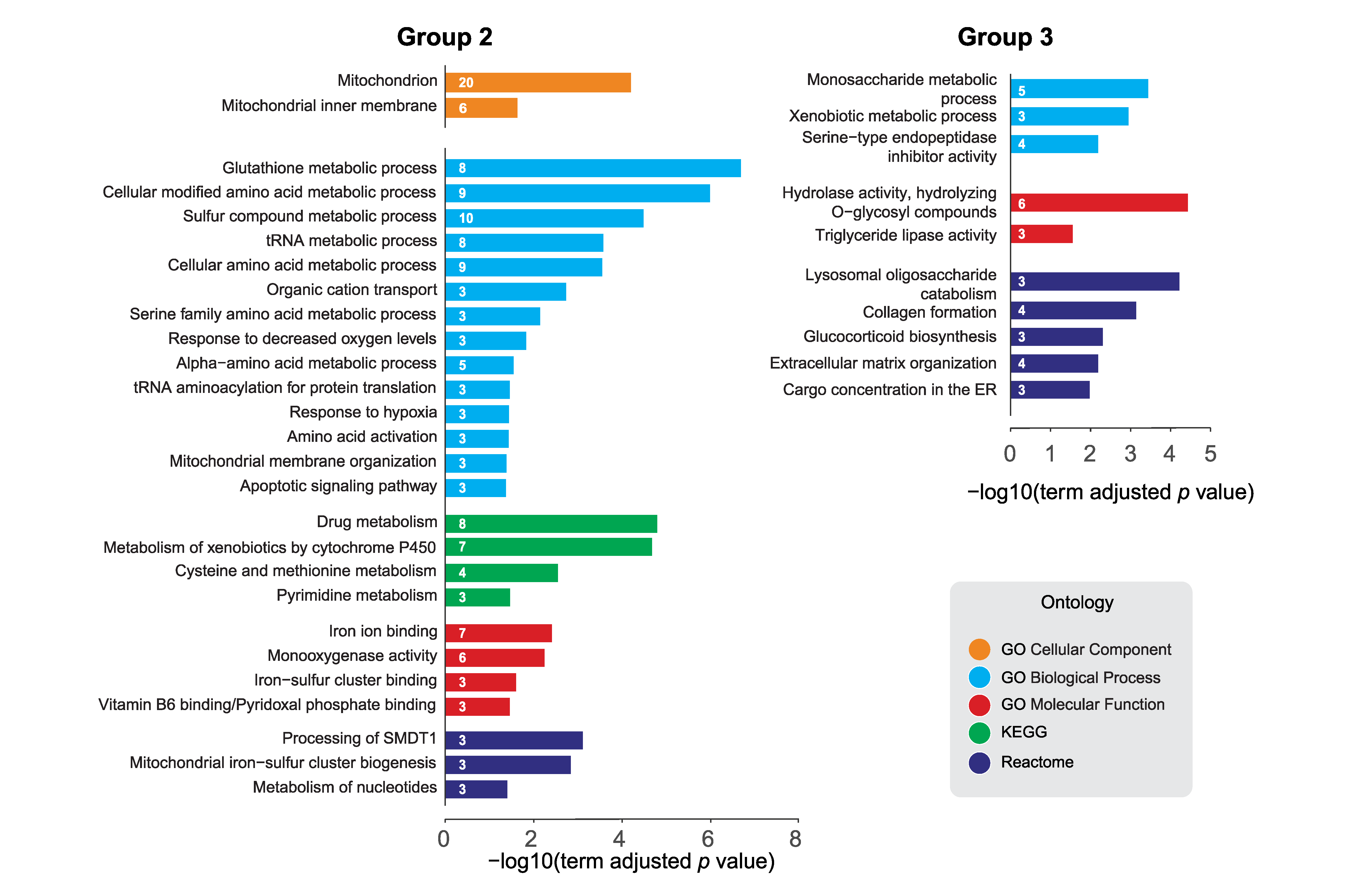
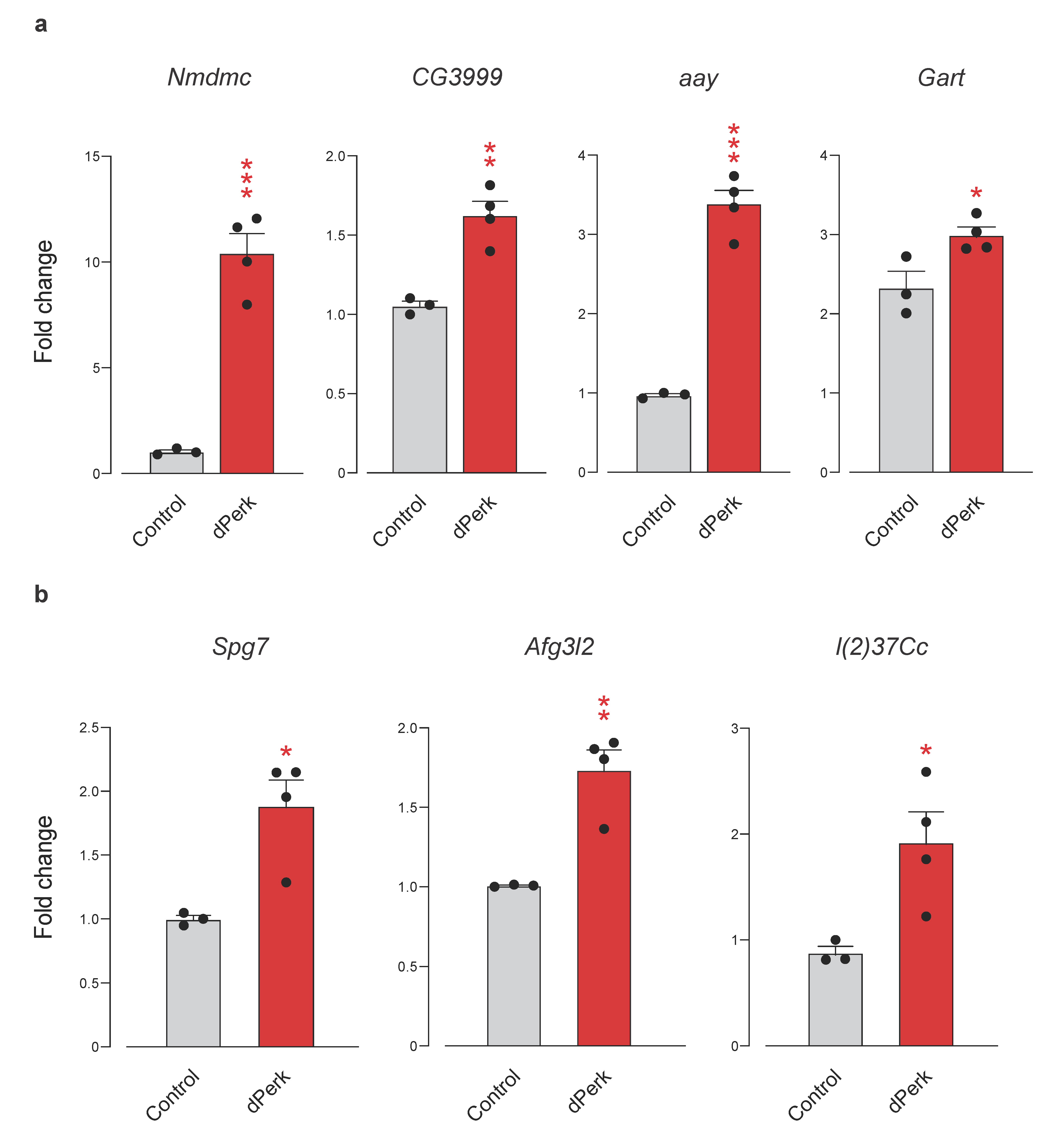
2.3. Pathway Analysis of dPerk-Dependent Alterations in Transcripts and Proteins
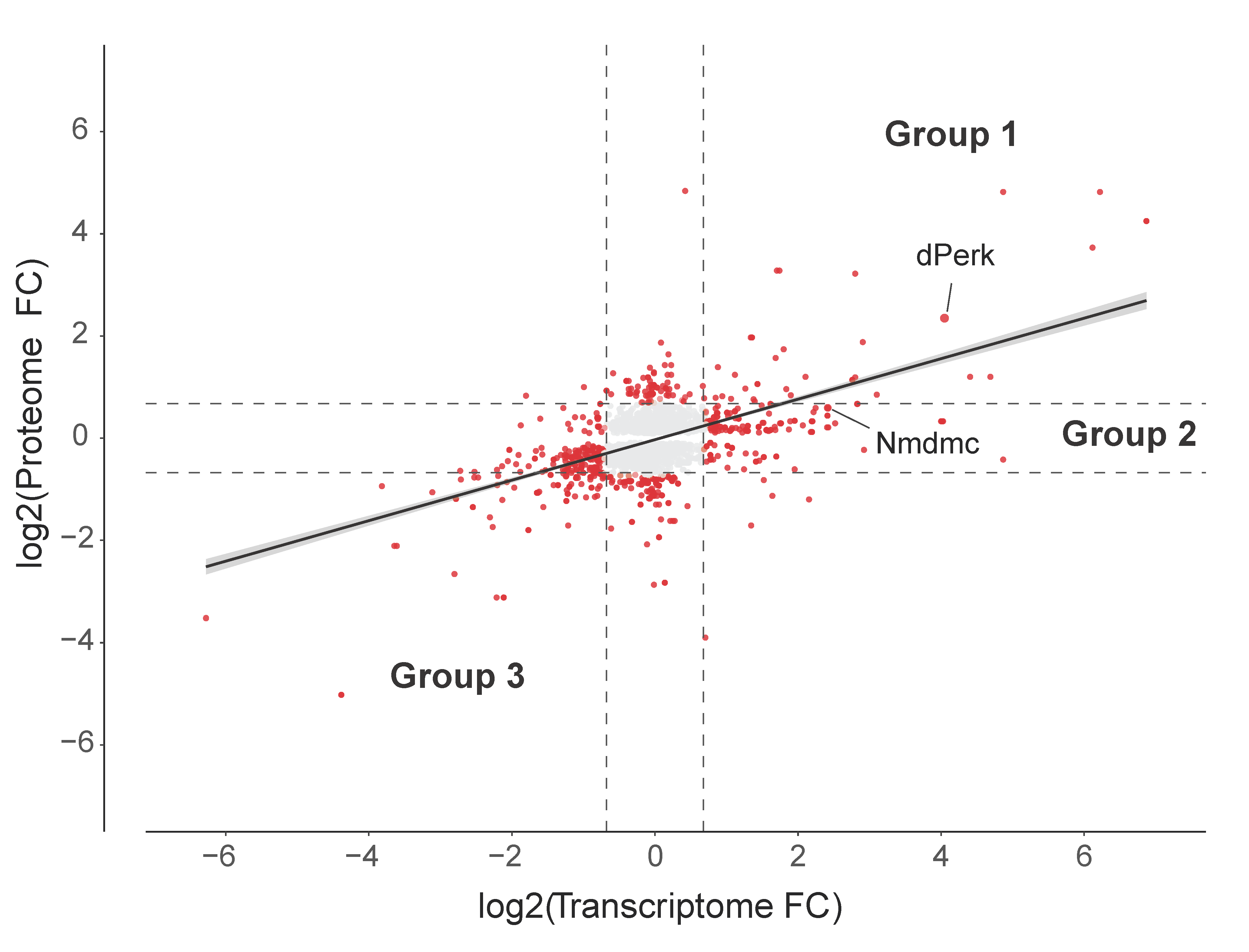
2.4. Analysis of the dPerk-Induced Correlations between Transcript and Protein Levels
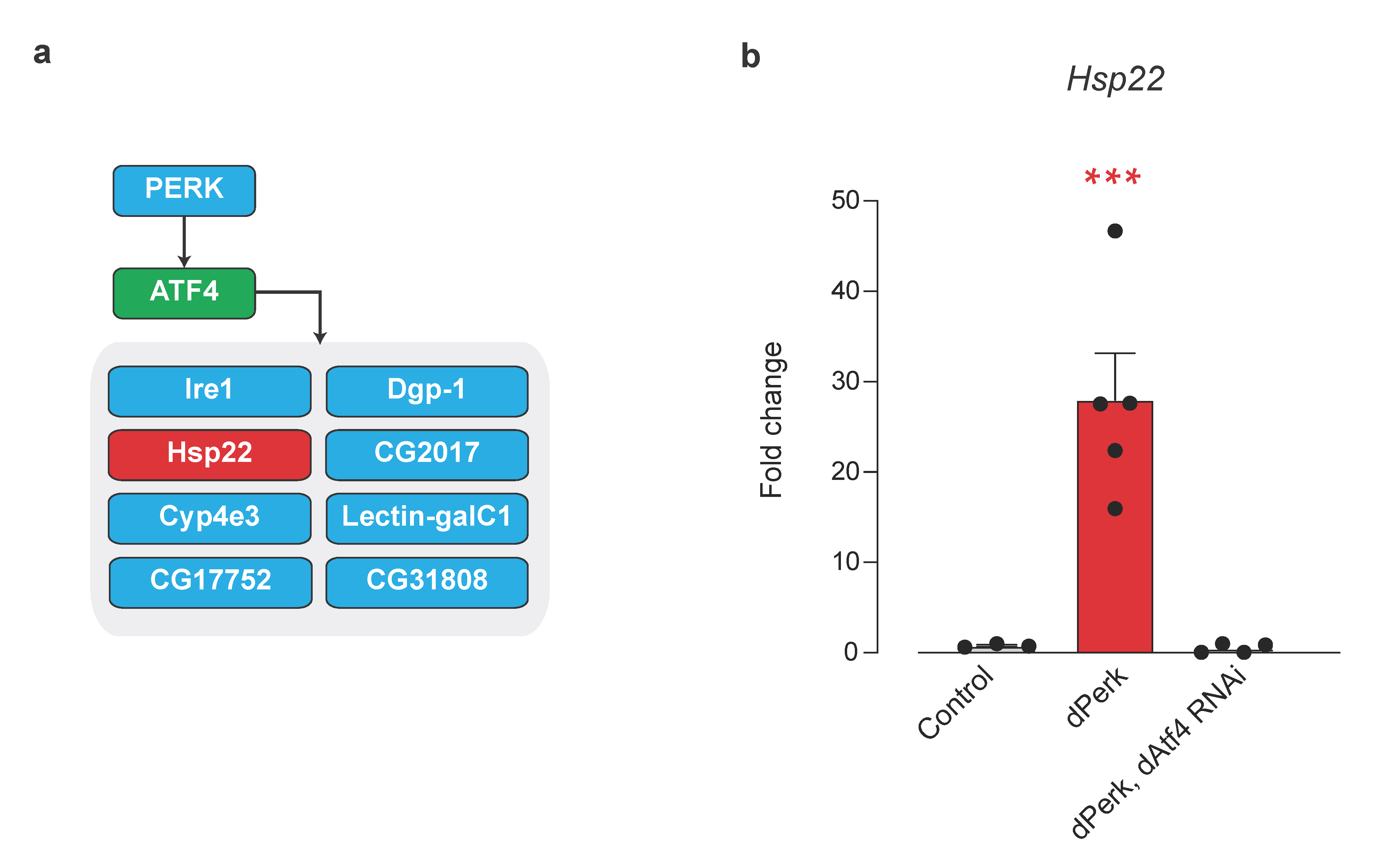
2.5. Small Mitochondrial Chaperone Hsp22 Is a Novel dPerk/dAtf4 Signalling Target
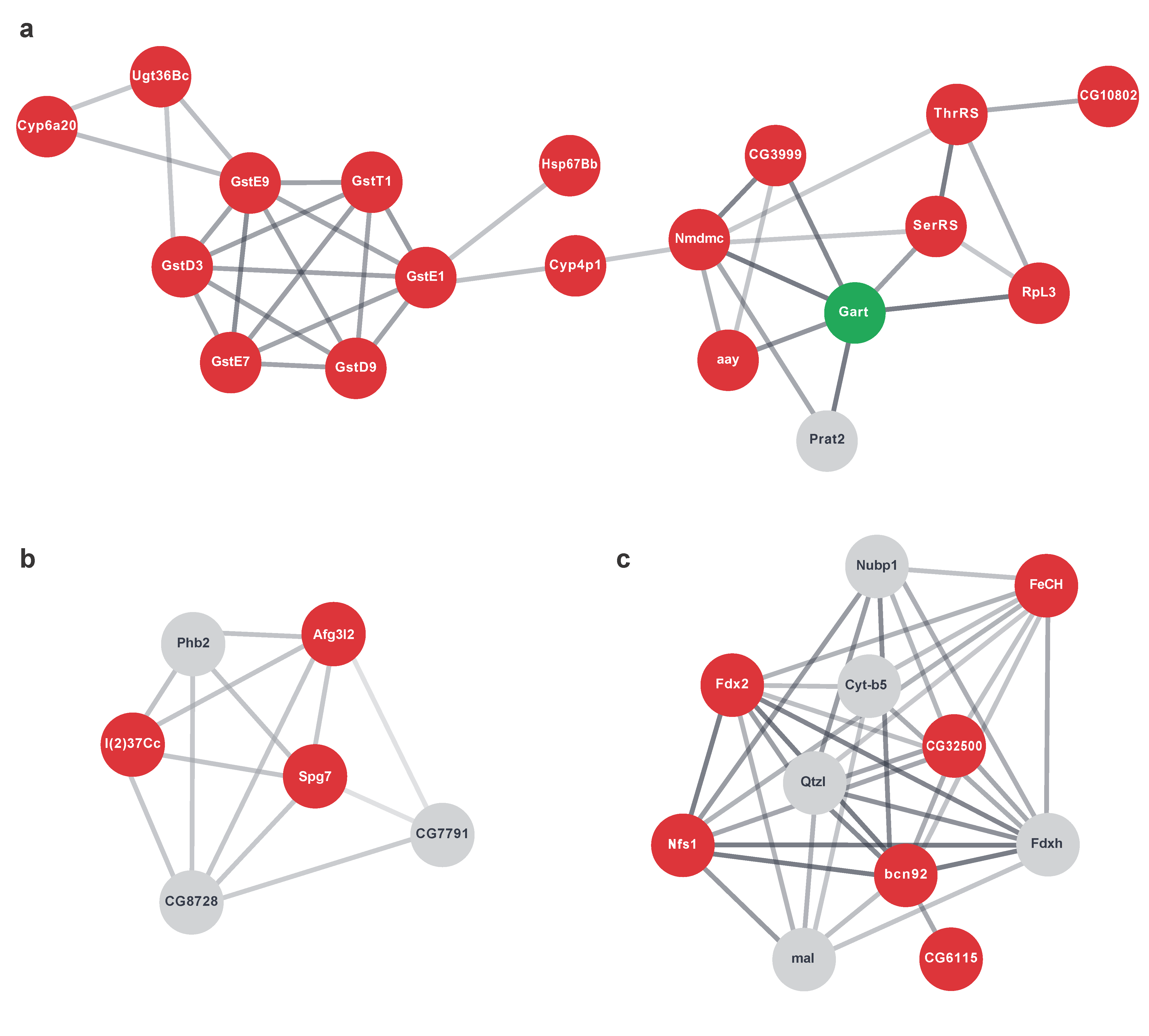
2.6. Networks of Translationally Repressed Signalling Pathways Caused by dPerk Expression
3. Discussion
4. Materials and Methods
4.1. Genetics and Drosophila Strains
4.2. Microarray Acquisition and Analysis
4.3. Proteomics Analysis
4.4. RNA Extraction and Quantitative Real-Time PCR
4.5. Protein Extraction and Western Blotting
4.6. Functional Pathway Enrichment and Upstream Analysis
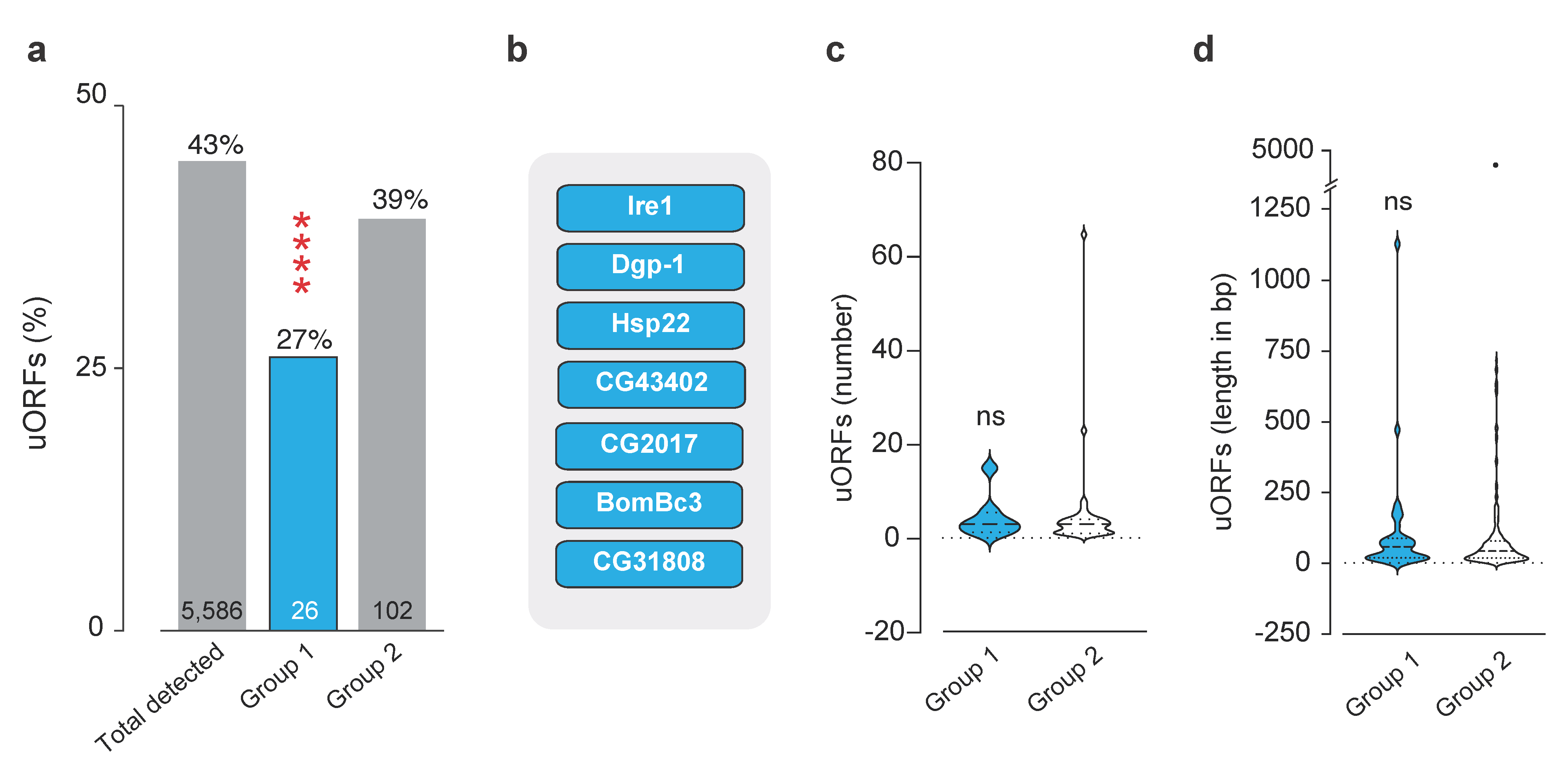
4.7. uORF Analysis
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hughes, D.; Mallucci, G.R. The unfolded protein response in neurodegenerative disorders—Therapeutic modulation of the PERK pathway. FEBS J. 2019, 286, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.; Mallucci, G.R. Modulating the unfolded protein response to prevent neurodegeneration and enhance memory. Neuropathol. Appl. Neurobiol. 2015, 41, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Novick, P.; Ferro-Novick, S. ER structure and function. Curr. Opin. Cell Biol. 2013, 25, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Kozutsumi, Y.; Segal, M.; Normington, K.; Gething, M.J.; Sambrook, J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1988, 332, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. Mammalian stress response: Induction of the glucose-regulated protein family. Curr. Opin. Cell Biol. 1992, 4, 267–273. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R., 3rd; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell. Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef]
- Spriggs, K.A.; Bushell, M.; Willis, A.E. Translational regulation of gene expression during conditions of cell stress. Mol. Cell 2010, 40, 228–237. [Google Scholar] [CrossRef]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef]
- Palam, L.R.; Baird, T.D.; Wek, R.C. Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J. Biol. Chem. 2011, 286, 10939–10949. [Google Scholar] [CrossRef]
- Starck, S.R.; Tsai, J.C.; Chen, K.; Shodiya, M.; Wang, L.; Yahiro, K.; Martins-Green, M.; Shastri, N.; Walter, P. Translation from the 5’ untranslated region shapes the integrated stress response. Science 2016, 351, aad3867. [Google Scholar] [CrossRef]
- Young, S.K.; Willy, J.A.; Wu, C.; Sachs, M.S.; Wek, R.C. Ribosome Reinitiation Directs Gene-specific Translation and Regulates the Integrated Stress Response. J. Biol. Chem. 2015, 290, 28257–28271. [Google Scholar] [CrossRef]
- Jaud, M.; Philippe, C.; Di Bella, D.; Tang, W.; Pyronnet, S.; Laurell, H.; Mazzolini, L.; Rouault-Pierre, K.; Touriol, C. Translational Regulations in Response to Endoplasmic Reticulum Stress in Cancers. Cells 2020, 9, 540. [Google Scholar] [CrossRef]
- Celardo, I.; Martins, L.M.; Gandhi, S. Unravelling mitochondrial pathways to Parkinson’s disease. Br. J. Pharmacol. 2014, 171, 1943–1957. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Mallucci, G.R.; Loh, S.H.; Martins, L.M. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016, 7, e2271. [Google Scholar] [CrossRef]
- Celardo, I.; Lehmann, S.; Costa, A.C.; Loh, S.H.; Miguel Martins, L. dATF4 regulation of mitochondrial folate-mediated one-carbon metabolism is neuroprotective. Cell Death Differ. 2017, 24, 638–648. [Google Scholar] [CrossRef]
- McGuire, S.E.; Le, P.T.; Osborn, A.J.; Matsumoto, K.; Davis, R.L. Spatiotemporal rescue of memory dysfunction in Drosophila. Science 2003, 302, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Jousse, C.; Deval, C.; Maurin, A.C.; Parry, L.; Cherasse, Y.; Chaveroux, C.; Lefloch, R.; Lenormand, P.; Bruhat, A.; Fafournoux, P. TRB3 inhibits the transcriptional activation of stress-regulated genes by a negative feedback on the ATF4 pathway. J. Biol. Chem. 2007, 282, 15851–15861. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Pomar, N.; Berlanga, J.J.; Campuzano, S.; Hernandez, G.; Elias, M.; de Haro, C. Functional characterization of Drosophila melanogaster PERK eukaryotic initiation factor 2alpha (eIF2alpha) kinase. Eur. J. Biochem. 2003, 270, 293–306. [Google Scholar] [CrossRef]
- Verfaillie, A.; Imrichova, H.; Van de Sande, B.; Standaert, L.; Christiaens, V.; Hulselmans, G.; Herten, K.; Naval Sanchez, M.; Potier, D.; Svetlichnyy, D.; et al. iRegulon: From a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 2014, 10, e1003731. [Google Scholar]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Levashina, E.A.; Langley, E.; Green, C.; Gubb, D.; Ashburner, M.; Hoffmann, J.A.; Reichhart, J.M. Constitutive activation of toll-mediated antifungal defense in serpin-deficient Drosophila. Science 1999, 285, 1917–1919. [Google Scholar] [CrossRef]
- Ligoxygakis, P.; Pelte, N.; Ji, C.; Leclerc, V.; Duvic, B.; Belvin, M.; Jiang, H.; Hoffmann, J.A.; Reichhart, J.M. A serpin mutant links Toll activation to melanization in the host defence of Drosophila. EMBO J. 2002, 21, 6330–6337. [Google Scholar] [CrossRef]
- Majumder, M.; Huang, C.; Snider, M.D.; Komar, A.A.; Tanaka, J.; Kaufman, R.J.; Krokowski, D.; Hatzoglou, M. A novel feedback loop regulates the response to endoplasmic reticulum stress via the cooperation of cytoplasmic splicing and mRNA translation. Mol. Cell Biol. 2012, 32, 992–1003. [Google Scholar] [CrossRef]
- Siu, F.; Bain, P.J.; LeBlanc-Chaffin, R.; Chen, H.; Kilberg, M.S. ATF4 is a mediator of the nutrient-sensing response pathway that activates the human asparagine synthetase gene. J. Biol. Chem. 2002, 277, 24120–24127. [Google Scholar] [CrossRef]
- Chung, K.T.; Shen, Y.; Hendershot, L.M. BAP, a mammalian BiP-associated protein, is a nucleotide exchange factor that regulates the ATPase activity of BiP. J. Biol. Chem. 2002, 277, 47557–47563. [Google Scholar] [CrossRef]
- Tyson, J.R.; Stirling, C.J. LHS1 and SIL1 provide a lumenal function that is essential for protein translocation into the endoplasmic reticulum. EMBO J. 2000, 19, 6440–6452. [Google Scholar] [CrossRef]
- Greene, J.C.; Whitworth, A.J.; Andrews, L.A.; Parker, T.J.; Pallanck, L.J. Genetic and genomic studies of Drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Hum. Mol. Genet. 2005, 14, 799–811. [Google Scholar] [CrossRef]
- Girardot, F.; Monnier, V.; Tricoire, H. Genome wide analysis of common and specific stress responses in adult drosophila melanogaster. BMC Genom. 2004, 5, 74. [Google Scholar] [CrossRef]
- Gruenewald, C.; Botella, J.A.; Bayersdorfer, F.; Navarro, J.A.; Schneuwly, S. Hyperoxia-induced neurodegeneration as a tool to identify neuroprotective genes in Drosophila melanogaster. Free Radic. Biol. Med. 2009, 46, 1668–1676. [Google Scholar] [CrossRef]
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.R.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep. 2018, 22, 2827–2836. [Google Scholar] [CrossRef]
- Iacono, M.; Mignone, F.; Pesole, G. uAUG and uORFs in human and rodent 5’untranslated mRNAs. Gene 2005, 349, 97–105. [Google Scholar] [CrossRef]
- Barbosa, C.; Peixeiro, I.; Romao, L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013, 9, e1003529. [Google Scholar] [CrossRef]
- Mokrejs, M.; Masek, T.; Vopalensky, V.; Hlubucek, P.; Delbos, P.; Pospisek, M. IRESite—A tool for the examination of viral and cellular internal ribosome entry sites. Nucleic Acids Res. 2010, 38, D131–D136. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Leal, N.S.; Martins, L.M. Mind the Gap: Mitochondria and the Endoplasmic Reticulum in Neurodegenerative Diseases. Biomedicines 2021, 9. [Google Scholar] [CrossRef]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Morrow, G.; Samson, M.; Michaud, S.; Tanguay, R.M. Overexpression of the small mitochondrial Hsp22 extends Drosophila life span and increases resistance to oxidative stress. FASEB J. 2004, 18, 598–599. [Google Scholar] [CrossRef]
- Morrow, G.; Battistini, S.; Zhang, P.; Tanguay, R.M. Decreased lifespan in the absence of expression of the mitochondrial small heat shock protein Hsp22 in Drosophila. J. Biol. Chem. 2004, 279, 43382–43385. [Google Scholar] [CrossRef]
- Kim, H.J.; Morrow, G.; Westwood, J.T.; Michaud, S.; Tanguay, R.M. Gene expression profiling implicates OXPHOS complexes in lifespan extension of flies over-expressing a small mitochondrial chaperone, Hsp22. Exp. Gerontol. 2010, 45, 611–620. [Google Scholar] [CrossRef]
- Nemcovicova, I.; Nemcovic, M.; Sestak, S.; Plskova, M.; Wilson, I.B.; Mucha, J. Expression, purification and preliminary crystallographic analysis of Drosophila melanogaster lysosomal alpha-mannosidase. Acta Cryst. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 965–970. [Google Scholar] [CrossRef]
- Chiba, S. Molecular mechanism in alpha-glucosidase and glucoamylase. Biosci. Biotechnol. Biochem. 1997, 61, 1233–1239. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef]
- Tsai, M.F.; Phillips, C.B.; Ranaghan, M.; Tsai, C.W.; Wu, Y.; Willliams, C.; Miller, C. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. Elife 2016, 5, e15545. [Google Scholar] [CrossRef]
- Atorino, L.; Silvestri, L.; Koppen, M.; Cassina, L.; Ballabio, A.; Marconi, R.; Langer, T.; Casari, G. Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J. Cell Biol. 2003, 163, 777–787. [Google Scholar] [CrossRef]
- Koppen, M.; Metodiev, M.D.; Casari, G.; Rugarli, E.I.; Langer, T. Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol. Cell Biol. 2007, 27, 758–767. [Google Scholar] [CrossRef]
- Lill, R.; Muhlenhoff, U. Maturation of iron-sulfur proteins in eukaryotes: Mechanisms, connected processes, and diseases. Annu. Rev. Biochem. 2008, 77, 669–700. [Google Scholar] [CrossRef]
- Marelja, Z.; Leimkuhler, S.; Missirlis, F. Iron Sulfur and Molybdenum Cofactor Enzymes Regulate the Drosophila Life Cycle by Controlling Cell Metabolism. Front. Physiol. 2018, 9, 50. [Google Scholar] [CrossRef]
- Sellers, V.M.; Wang, K.F.; Johnson, M.K.; Dailey, H.A. Evidence that the fourth ligand to the [2Fe-2S] cluster in animal ferrochelatase is a cysteine. Characterization of the enzyme from Drosophila melanogaster. J. Biol. Chem. 1998, 273, 22311–22316. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Gonen, N.; Sabath, N.; Burge, C.B.; Shalgi, R. Widespread PERK-dependent repression of ER targets in response to ER stress. Sci. Rep. 2019, 9, 4330. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Morrow, G.; Inaguma, Y.; Kato, K.; Tanguay, R.M. The small heat shock protein Hsp22 of Drosophila melanogaster is a mitochondrial protein displaying oligomeric organization. J. Biol. Chem. 2000, 275, 31204–31210. [Google Scholar] [CrossRef] [PubMed]
- Morrow, G.; Kim, H.J.; Pellerito, O.; Bourrelle-Langlois, M.; Le Pecheur, M.; Groebe, K.; Tanguay, R.M. Changes in Drosophila mitochondrial proteins following chaperone-mediated lifespan extension confirm a role of Hsp22 in mitochondrial UPR and reveal a mitochondrial localization for cathepsin D. Mech. Ageing Dev. 2016, 155, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Schober, F.A.; Moore, D.; Atanassov, I.; Moedas, M.F.; Clemente, P.; Vegvari, A.; Fissi, N.E.; Filograna, R.; Bucher, A.L.; Hinze, Y.; et al. The one-carbon pool controls mitochondrial energy metabolism via complex I and iron-sulfur clusters. Sci. Adv. 2021, 7, eabf0717. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Konig, T.; Troder, S.E.; Bakka, K.; Korwitz, A.; Richter-Dennerlein, R.; Lampe, P.A.; Patron, M.; Muhlmeister, M.; Guerrero-Castillo, S.; Brandt, U.; et al. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol. Cell 2016, 64, 148–162. [Google Scholar] [CrossRef]
- Lindquist, S. Translational efficiency of heat-induced messages in Drosophila melanogaster cells. J. Mol. Biol. 1980, 137, 151–158. [Google Scholar] [CrossRef]
- Lindquist, S.; Petersen, R. Selective translation and degradation of heat-shock messenger RNAs in Drosophila. Enzyme 1990, 44, 147–166. [Google Scholar] [CrossRef]
- Duncan, R.F. Rapamycin conditionally inhibits Hsp90 but not Hsp70 mRNA translation in Drosophila: Implications for the mechanisms of Hsp mRNA translation. Cell Stress Chaperones 2008, 13, 143–155. [Google Scholar] [CrossRef]
- Zhang, H.; Dou, S.; He, F.; Luo, J.; Wei, L.; Lu, J. Genome-wide maps of ribosomal occupancy provide insights into adaptive evolution and regulatory roles of uORFs during Drosophila development. PLoS Biol. 2018, 16, e2003903. [Google Scholar] [CrossRef]
- Andreev, D.E.; O’Connor, P.B.; Fahey, C.; Kenny, E.M.; Terenin, I.M.; Dmitriev, S.E.; Cormican, P.; Morris, D.W.; Shatsky, I.N.; Baranov, P.V. Translation of 5’ leaders is pervasive in genes resistant to eIF2 repression. Elife 2015, 4, e03971. [Google Scholar] [CrossRef]
- Young, S.K.; Wek, R.C. Upstream Open Reading Frames Differentially Regulate Gene-specific Translation in the Integrated Stress Response. J. Biol. Chem. 2016, 291, 16927–16935. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef]
- Gao, X.; Wan, J.; Liu, B.; Ma, M.; Shen, B.; Qian, S.B. Quantitative profiling of initiating ribosomes in vivo. Nat. Methods 2015, 12, 147–153. [Google Scholar] [CrossRef]
- Fritsch, C.; Herrmann, A.; Nothnagel, M.; Szafranski, K.; Huse, K.; Schumann, F.; Schreiber, S.; Platzer, M.; Krawczak, M.; Hampe, J.; et al. Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Res. 2012, 22, 2208–2218. [Google Scholar] [CrossRef]
- Reich, S.; Nguyen, C.D.L.; Has, C.; Steltgens, S.; Soni, H.; Coman, C.; Freyberg, M.; Bichler, A.; Seifert, N.; Conrad, D.; et al. A multi-omics analysis reveals the unfolded protein response regulon and stress-induced resistance to folate-based antimetabolites. Nat. Commun. 2020, 11, 2936. [Google Scholar] [CrossRef]
- Gatto, L.; Lilley, K.S. MSnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 2012, 28, 288–289. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; D’Agostino McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Larkin, A.; Marygold, S.J.; Antonazzo, G.; Attrill, H.; Dos Santos, G.; Garapati, P.V.; Goodman, J.L.; Gramates, L.S.; Millburn, G.; Strelets, V.B.; et al. FlyBase: Updates to the Drosophila melanogaster knowledge base. Nucleic Acids Res. 2021, 49, D899–D907. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Brief Summary | Transcript FC | Protein FC |
|---|---|---|---|
| ER proteins | |||
| PERK | UPR kinase | 16.6 | 4.99 |
| Sil1 | Nucleotide exchange factor required for protein translocation and folding in the endoplasmic reticulum | 8.58 | 1.8 |
| AsnS | Asparagine synthase | 4.3 | 2.3 |
| Ire1 | UPR kinase and endoribonuclease | 2.09 | 1.71 |
| Translation | |||
| Dgp-1 | Translational GTPase predicted to be involved in translational elongation | 21.2 | 2.3 |
| CG2017 | Translational GTPase predicted to be involved in translational elongation | 2.6 | 1.69 |
| Detoxification | |||
| Cyp6a17 | Cytochrome P450 enzyme | 117 | 19.03 |
| Cyp9b2 | Cytochrome P450 enzyme | 6.96 | 2.28 |
| Cyp4e3 | Cytochrome P450 enzyme | 3.22 | 2.97 |
| Cyp4ad1 | Cytochrome P450 enzyme | 1.61 | 1.72 |
| Ugt37A3 | UDP-glycosyltransferase | 69.4 | 13.27 |
| Ugt86Dd | UDP-glycosyltransferase | 3.48 | 3.34 |
| GstD2 | Glutathione S transferase | 7.49 | 3.68 |
| GstE8 | Glutathione S transferase | 2.7 | 2.08 |
| Mitochondrial proteins | |||
| Hsp22 | Mitochondrial molecular chaperone | 29.2 | 28.25 |
| Pepck2 | Mitochondrial and cytoplasmic phosphoenolpyruvate carboxykinase, plays a role in gluconeogenesis | 2.63 | 1.65 |
| CG34423 | Mitochondrial ATPase inhibitor | 2.4 | 1.96 |
| Immunity-related proteins | |||
| Lectin-galC1 | Galactose binding protein involved in the induction of bacterial agglutination and cell-cell adhesion | 3.26 | 9.71 |
| BomBc3 | Member of the Bombamin family of small, secreted immune-induced peptides that are induced by Toll signalling and may function to confer resistance to bacterial infection | 2.56 | 3.92 |
| Other | |||
| CG11911 | Predicted to have serine-type endopeptidase activity and be involved in proteolysis | 6.96 | 9.32 |
| Uro | Urate oxidase, involved in allantoin biosynthetic process | 3.73 | 1.79 |
| Ccp84Ab | Predicted to be a structural constituent of chitin-based larval cuticle | 1.92 | 1.77 |
| CG17752 | A member of the SLC22 family, predicted to have transmembrane transporter activity | 1.8 | 1.73 |
| CG7632 | Predicted to have hydrolase activity | 3.57 | 1.95 |
| CG43402 | Uncharacterized protein | 6.77 | 2.2 |
| CG12868 | Uncharacterized protein | 2.17 | 2.36 |
| CG31808 | Uncharacterized protein | 1.84 | 2.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popovic, R.; Celardo, I.; Yu, Y.; Costa, A.C.; Loh, S.H.Y.; Martins, L.M. Combined Transcriptomic and Proteomic Analysis of Perk Toxicity Pathways. Int. J. Mol. Sci. 2021, 22, 4598. https://doi.org/10.3390/ijms22094598
Popovic R, Celardo I, Yu Y, Costa AC, Loh SHY, Martins LM. Combined Transcriptomic and Proteomic Analysis of Perk Toxicity Pathways. International Journal of Molecular Sciences. 2021; 22(9):4598. https://doi.org/10.3390/ijms22094598
Chicago/Turabian StylePopovic, Rebeka, Ivana Celardo, Yizhou Yu, Ana C. Costa, Samantha H. Y. Loh, and L. Miguel Martins. 2021. "Combined Transcriptomic and Proteomic Analysis of Perk Toxicity Pathways" International Journal of Molecular Sciences 22, no. 9: 4598. https://doi.org/10.3390/ijms22094598
APA StylePopovic, R., Celardo, I., Yu, Y., Costa, A. C., Loh, S. H. Y., & Martins, L. M. (2021). Combined Transcriptomic and Proteomic Analysis of Perk Toxicity Pathways. International Journal of Molecular Sciences, 22(9), 4598. https://doi.org/10.3390/ijms22094598