
Excess TGF-β1 Drives Cardiac Mesenchymal Stromal Cells to a Pro-Fibrotic Commitment in Arrhythmogenic Cardiomyopathy

 ,
,  , , ,
, , ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
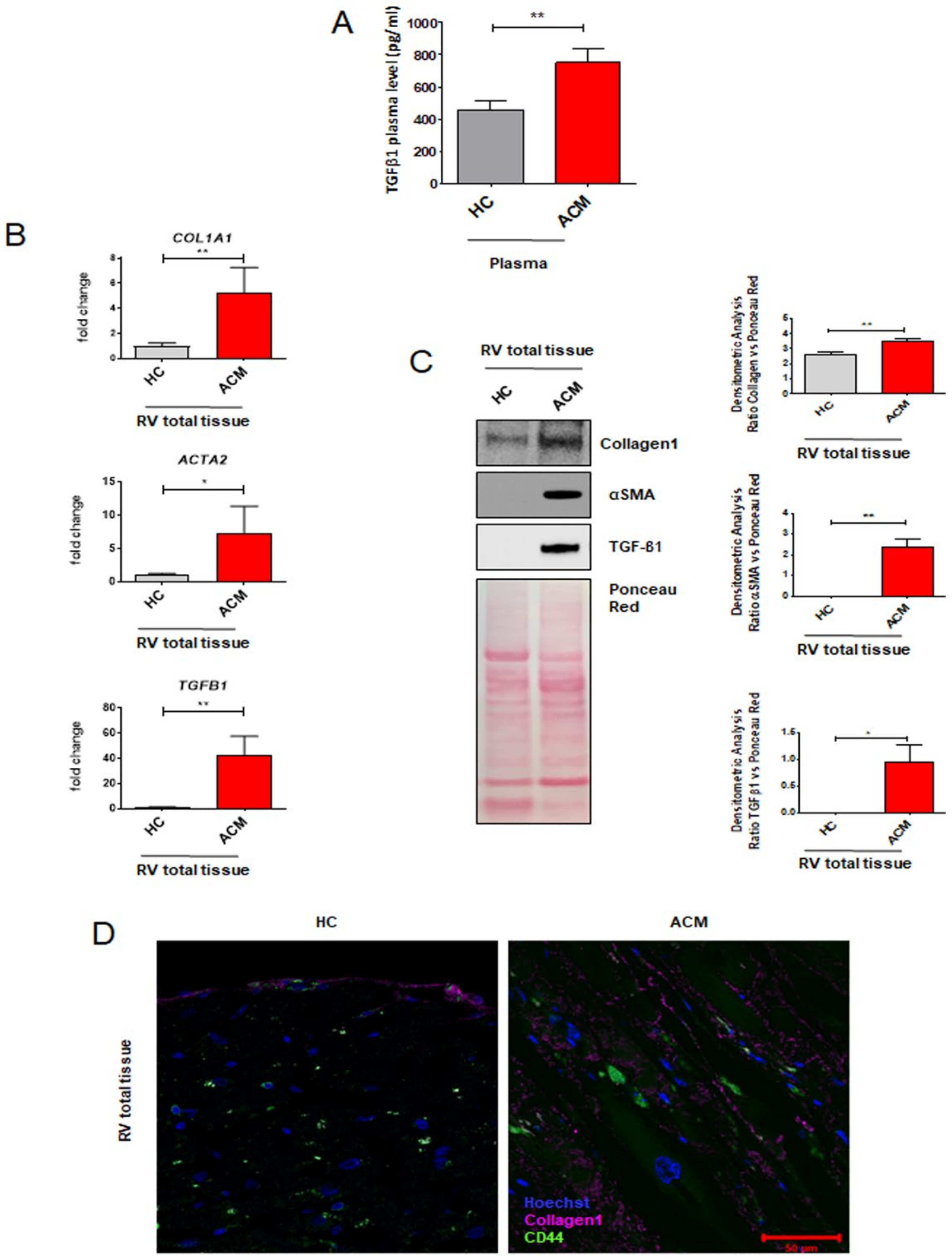
2.1. Arrhythmogenic Cardiomyopathy Patient-Derived Tissues Exhibit Higher Fibrosis and TGF-β1 Levels Than Healthy Controls
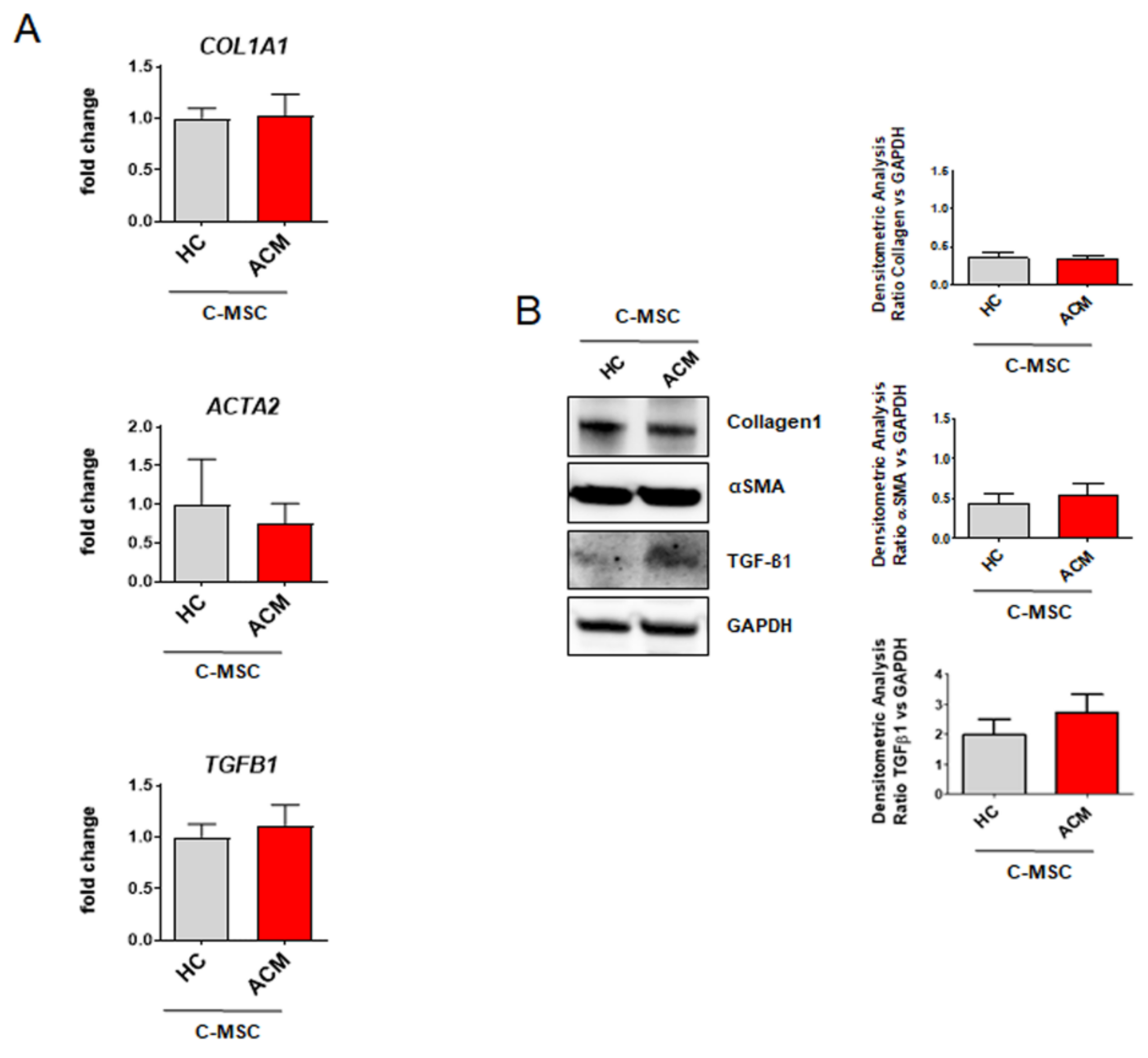
2.2. Cardiac Mesenchymal Stromal Cells Isolated from Arrhythmogenic Cardiomyopathy Patients and from Healthy Controls Exhibit Comparable Fibrotic Marker Levels
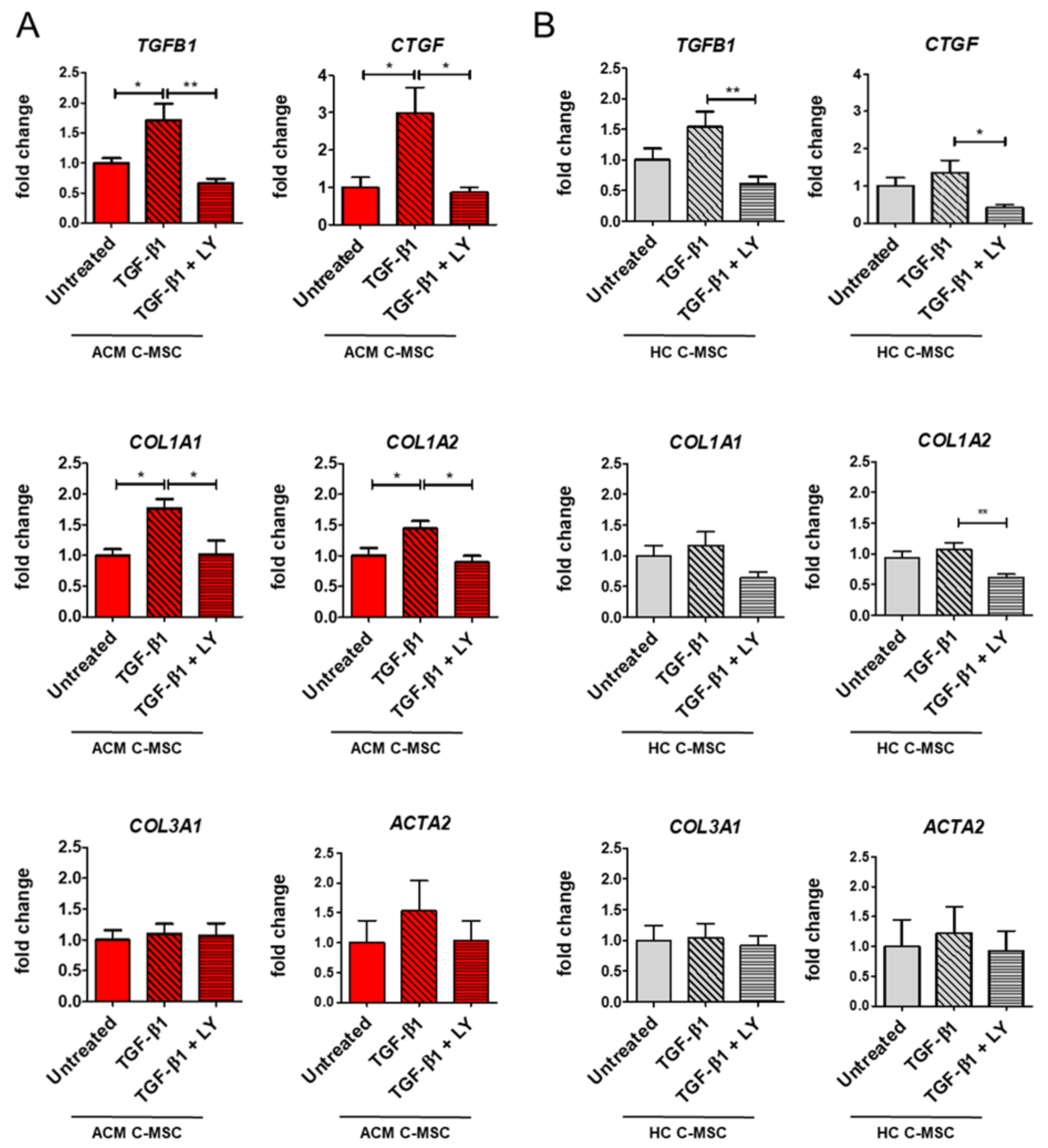
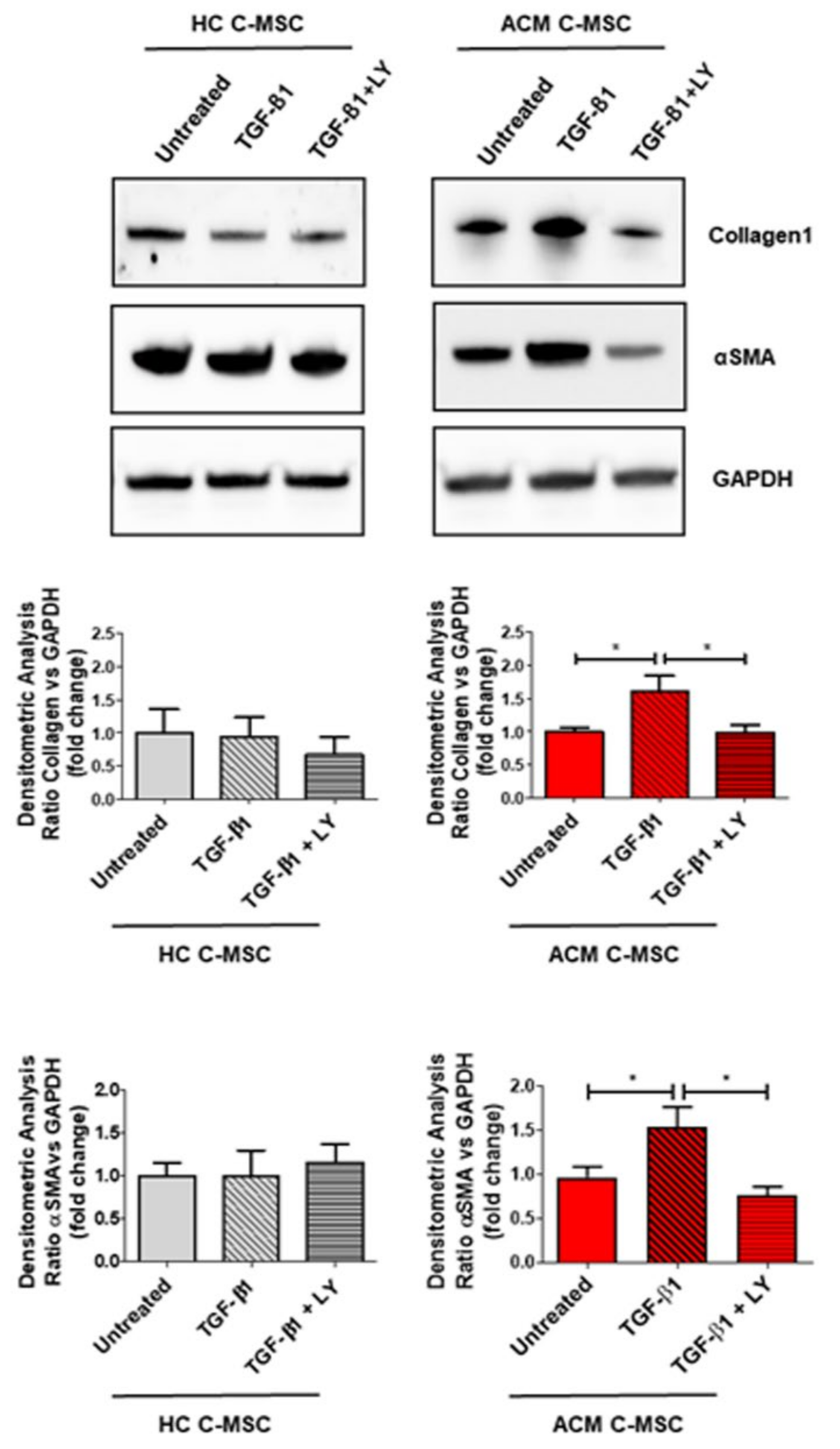
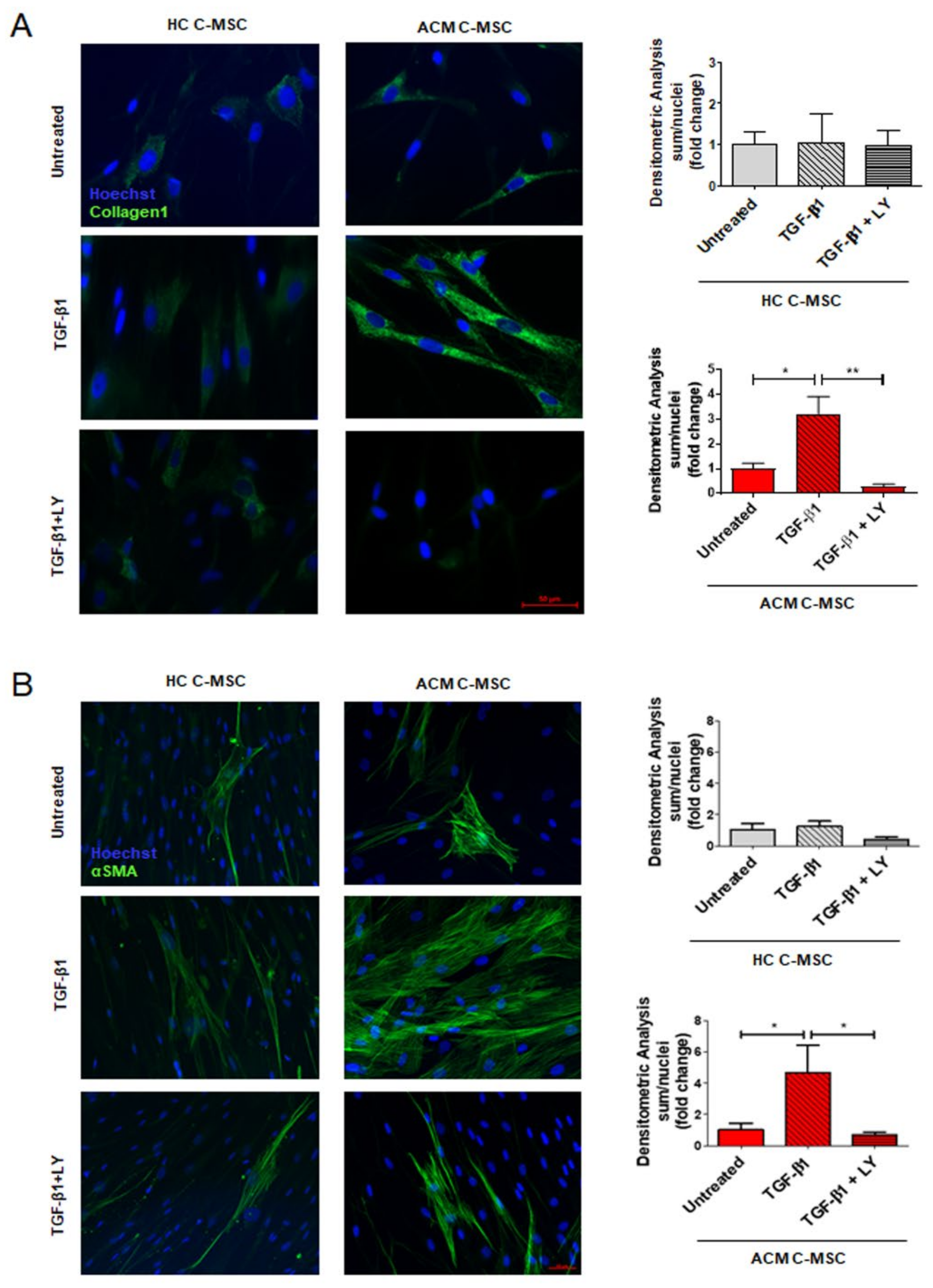
2.3. TGF-β1 Stimulation Drives Pro-Fibrotic Differentiation of Cardiac Mesenchymal Stromal Cell from Arrhythmogenic Cardiomyopathy Patients
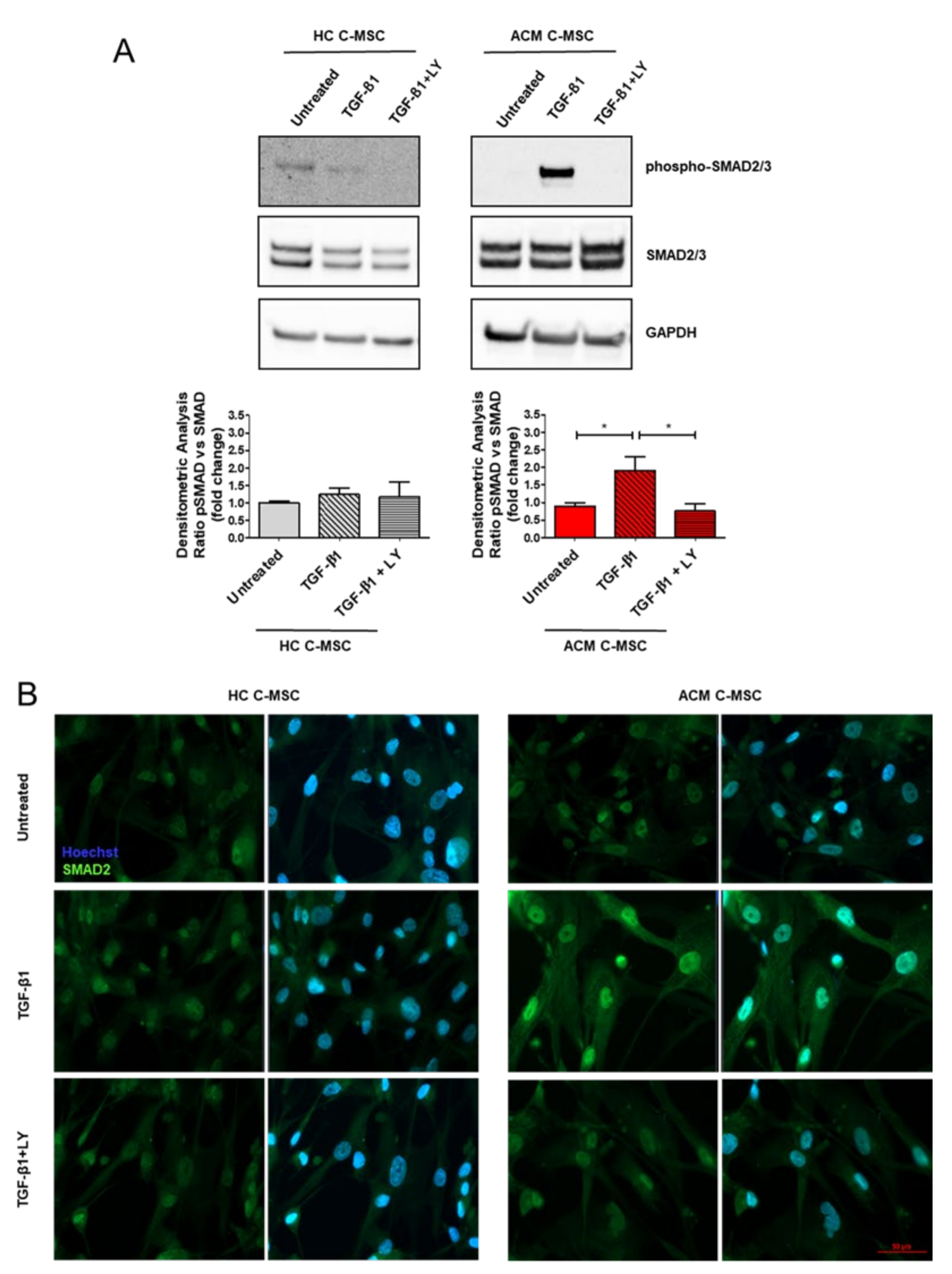
2.4. TGF-β1 Treatment Cause Activation of TGF-β1 Canonical Signaling Pathway in Arrhythmogenic Cardiomyopathy-Derived Cardiac Mesenchymal Stromal Cells
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Plasmatic TGF-β1 Concentration Assay
4.3. Immunofluorescence on Tissues and Cells
4.4. C-MSC Isolation, Culture, and Treatment
4.5. mRNA Extraction and qRT-PCR Assay
4.6. Protein Extraction and Western Blot Analysis
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72. [Google Scholar] [CrossRef]
- Stadiotti, I.; Pompilio, G.; Maione, A.S.; Pilato, C.A.; D’Alessandra, Y.; Sommariva, E. Arrhythmogenic cardiomyopathy: What blood can reveal? Heart Rhythm 2019, 16, 470–477. [Google Scholar] [CrossRef]
- Casella, M.; Gasperetti, A.; Sicuso, R.; Conte, E.; Catto, V.; Sommariva, E.; Bergonti, M.; Vettor, G.; Rizzo, S.; Pompilio, G.; et al. Characteristics of Patients With Arrhythmogenic Left Ventricular Cardiomyopathy: Combining Genetic and Histopathologic Findings. Circ. Arrhythm. Electrophysiol. 2020, 13, e009005. [Google Scholar] [CrossRef]
- Hoorntje, E.T.; Te Rijdt, W.P.; James, C.A.; Pilichou, K.; Basso, C.; Judge, D.P.; Bezzina, C.R.; van Tintelen, J.P. Arrhythmogenic cardiomyopathy: Pathology, genetics, and concepts in pathogenesis. Cardiovasc. Res. 2017, 113, 1521–1531. [Google Scholar] [CrossRef]
- Lin, C.Y.; Lin, Y.J.; Li, C.H.; Chung, F.P.; Lo, M.T.; Lin, C.; Chang, H.C.; Chang, S.L.; Lo, L.W.; Hu, Y.F.; et al. Heterogeneous distribution of substrates between the endocardium and epicardium promotes ventricular fibrillation in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Eurospace 2018, 20, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Maione, A.S.; Pilato, C.A.; Casella, M.; Gasperetti, A.; Stadiotti, I.; Pompilio, G.; Sommariva, E. Fibrosis in Arrhythmogenic Cardiomyopathy: The Phantom Thread in the Fibro-Adipose Tissue. Front. Physiol. 2020, 11, 279. [Google Scholar] [CrossRef] [PubMed]
- Furtado, M.B.; Costa, M.W.; Rosenthal, N.A. The cardiac fibroblast: Origin, identity and role in homeostasis and disease. Differ. Res. Biol. Divers. 2016, 92, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; SC, J.L.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef]
- Moore-Morris, T.; Guimaraes-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef]
- Carlson, S.; Trial, J.; Soeller, C.; Entman, M.L. Cardiac mesenchymal stem cells contribute to scar formation after myocardial infarction. Cardiovasc. Res. 2011, 91, 99–107. [Google Scholar] [CrossRef]
- Stadiotti, I.; Piacentini, L.; Vavassori, C.; Chiesa, M.; Scopece, A.; Guarino, A.; Micheli, B.; Polvani, G.; Colombo, G.I.; Pompilio, G.; et al. Human Cardiac Mesenchymal Stromal Cells from Right and Left Ventricles Display Differences in Number, Function, and Transcriptomic Profile. Front. Physiol. 2020, 11, 604. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J. Mol. Cell Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ. Res. 2010, 106, 1675–1680. [Google Scholar] [CrossRef]
- Sommariva, E.; Brambilla, S.; Carbucicchio, C.; Gambini, E.; Meraviglia, V.; Dello Russo, A.; Farina, F.M.; Casella, M.; Catto, V.; Pontone, G.; et al. Cardiac mesenchymal stromal cells are a source of adipocytes in arrhythmogenic cardiomyopathy. Eur. Heart J. 2016, 37, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Vallee, A.; Lecarpentier, Y.; Guillevin, R.; Vallee, J.N. Interactions between TGF-beta1, canonical WNT/beta-catenin pathway and PPAR gamma in radiation-induced fibrosis. Oncotarget 2017, 8, 90579–90604. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Kim, S.Y.; Kim, M.J.; Lim, J.H.; Cho, M.S.; Kang, J.L. PPARgamma activation following apoptotic cell instillation promotes resolution of lung inflammation and fibrosis via regulation of efferocytosis and proresolving cytokines. Mucosal Immunol. 2015, 8, 1031–1046. [Google Scholar] [CrossRef]
- Lakshmi, S.P.; Reddy, A.T.; Reddy, R.C. Transforming growth factor beta suppresses peroxisome proliferator-activated receptor gamma expression via both SMAD binding and novel TGF-beta inhibitory elements. Biochem. J. 2017, 474, 1531–1546. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Chen, S.N.; Ruggiero, A.; Gurha, P.; Czernuszewicz, G.Z.; Willerson, J.T.; Marian, A.J. Cardiac Fibro-Adipocyte Progenitors Express Desmosome Proteins and Preferentially Differentiate to Adipocytes Upon Deletion of the Desmoplakin Gene. Circ. Res. 2016, 119, 41–54. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Ho, J.E.; Liu, C.; Lyass, A.; Courchesne, P.; Pencina, M.J.; Vasan, R.S.; Larson, M.G.; Levy, D. Galectin-3, a marker of cardiac fibrosis, predicts incident heart failure in the community. J. Am. Coll Cardiol. 2012, 60, 1249–1256. [Google Scholar] [CrossRef]
- Perrucci, G.L.; Barbagallo, V.A.; Corliano, M.; Tosi, D.; Santoro, R.; Nigro, P.; Poggio, P.; Bulfamante, G.; Lombardi, F.; Pompilio, G. Integrin alphanubeta5 in vitro inhibition limits pro-fibrotic response in cardiac fibroblasts of spontaneously hypertensive rats. J. Transl. Med. 2018, 16, 352. [Google Scholar] [CrossRef] [PubMed]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef] [PubMed]
- Talele, N.P.; Fradette, J.; Davies, J.E.; Kapus, A.; Hinz, B. Expression of alpha-Smooth Muscle Actin Determines the Fate of Mesenchymal Stromal Cells. Stem Cell Rep. 2015, 4, 1016–1030. [Google Scholar] [CrossRef]
- Li, D.; Liu, Y.; Maruyama, M.; Zhu, W.; Chen, H.; Zhang, W.; Reuter, S.; Lin, S.F.; Haneline, L.S.; Field, L.J.; et al. Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Hum. Mol. Genet. 2011, 20, 4582–4596. [Google Scholar] [CrossRef] [PubMed]
- Dubash, A.D.; Kam, C.Y.; Aguado, B.A.; Patel, D.M.; Delmar, M.; Shea, L.D.; Green, K.J. Plakophilin-2 loss promotes TGF-beta1/p38 MAPK-dependent fibrotic gene expression in cardiomyocytes. J. Cell Biol. 2016, 212, 425–438. [Google Scholar] [CrossRef]
- Moccia, F.; Lodola, F.; Stadiotti, I.; Pilato, C.A.; Bellin, M.; Carugo, S.; Pompilio, G.; Sommariva, E.; Maione, A.S. Calcium as a Key Player in Arrhythmogenic Cardiomyopathy: Adhesion Disorder or Intracellular Alteration? Int. J. Mol. Sci. 2019, 20, 3986. [Google Scholar] [CrossRef]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A.; et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef]
- James, C.A.; Bhonsale, A.; Tichnell, C.; Murray, B.; Russell, S.D.; Tandri, H.; Tedford, R.J.; Judge, D.P.; Calkins, H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J. Am. Coll. Cardiol. 2013, 62, 1290–1297. [Google Scholar] [CrossRef]
- Heinemeier, K.; Langberg, H.; Kjaer, M. Exercise-induced changes in circulating levels of transforming growth factor-beta-1 in humans: Methodological considerations. Eur. J. Appl. Physiol. 2003, 90, 171–177. [Google Scholar] [CrossRef]
- Czarkowska-Paczek, B.; Bartlomiejczyk, I.; Przybylski, J. The serum levels of growth factors: PDGF, TGF-beta and VEGF are increased after strenuous physical exercise. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2006, 57, 189–197. [Google Scholar]
- Todica, A.; Siebermair, J.; Schiller, J.; Zacherl, M.J.; Fendler, W.P.; Massberg, S.; Bartenstein, P.; Cyran, C.C.; Kaab, S.; Hacker, M.; et al. Assessment of right ventricular sympathetic dysfunction in patients with arrhythmogenic right ventricular cardiomyopathy: An (123)I-metaiodobenzylguanidine SPECT/CT study. J. Nucl. Cardiol. 2018, 27, 2402–2409. [Google Scholar] [CrossRef]
- Bhambi, B.; Eghbali, M. Effect of norepinephrine on myocardial collagen gene expression and response of cardiac fibroblasts after norepinephrine treatment. Am. J. Pathol. 1991, 139, 1131–1142. [Google Scholar] [PubMed]
- Castaldi, A.; Zaglia, T.; Di Mauro, V.; Carullo, P.; Viggiani, G.; Borile, G.; Di Stefano, B.; Schiattarella, G.G.; Gualazzi, M.G.; Elia, L.; et al. MicroRNA-133 modulates the beta1-adrenergic receptor transduction cascade. Circ. Res. 2014, 115, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S.; Flesch, M.; Amann, K.; Haeuseler, C.; Kilter, H.; Seeland, U.; Schluter, K.D.; Bohm, M. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1). Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1253–H1262. [Google Scholar] [CrossRef] [PubMed]
- Mak, J.C.; Rousell, J.; Haddad, E.B.; Barnes, P.J. Transforming growth factor-beta1 inhibits beta2-adrenoceptor gene transcription. Naunyn Schmiedebergs Arch. Pharmacol. 2000, 362, 520–525. [Google Scholar] [CrossRef]
- Iizuka, K.; Sano, H.; Kawaguchi, H.; Kitabatake, A. Transforming growth factor beta-1 modulates the number of beta-adrenergic receptors in cardiac fibroblasts. J. Mol. Cell. Cardiol. 1994, 26, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, N.; Maguy, A.; De Simone, S.A.; Miragoli, M.; Jousset, F.; Rohr, S. TGF-beta1 (Transforming Growth Factor-beta1) Plays a Pivotal Role in Cardiac Myofibroblast Arrhythmogenicity. Circ. Arrhythm. Electrophysiol. 2017, 10, e004567. [Google Scholar] [CrossRef]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy: Diagnosis, prognosis, and treatment. Heart 2000, 83, 588–595. [Google Scholar] [CrossRef]
- Rusciano, M.R.; Sommariva, E.; Douin-Echinard, V.; Ciccarelli, M.; Poggio, P.; Maione, A.S. CaMKII Activity in the Inflammatory Response of Cardiac Diseases. Int. J. Mol. Sci. 2019, 20, 4374. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 2011, 4, 44–52. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 2017, 136, 549–561. [Google Scholar] [CrossRef]
- Broch, K.; Leren, I.S.; Saberniak, J.; Ueland, T.; Edvardsen, T.; Gullestad, L.; Haugaa, K.H. Soluble ST2 is associated with disease severity in arrhythmogenic right ventricular cardiomyopathy. Biomarkers 2017, 22, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Oz, F.; Onur, I.; Elitok, A.; Ademoglu, E.; Altun, I.; Bilge, A.K.; Adalet, K. Galectin-3 correlates with arrhythmogenic right ventricular cardiomyopathy and predicts the risk of ventricular -arrhythmias in patients with implantable defibrillators. Acta Cardiol. 2017, 72, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Sommariva, E.; Stadiotti, I.; Perrucci, G.L.; Tondo, C.; Pompilio, G. Cell models of arrhythmogenic cardiomyopathy: Advances and opportunities. Dis. Models Mech. 2017, 10, 823–835. [Google Scholar] [CrossRef]
- Pilato, C.A.; Stadiotti, I.; Maione, A.S.; Saverio, V.; Catto, V.; Tundo, F.; Dello Russo, A.; Tondo, C.; Pompilio, G.; Casella, M.; et al. Isolation and Characterization of Cardiac Mesenchymal Stromal Cells from Endomyocardial Bioptic Samples of Arrhythmogenic Cardiomyopathy Patients. J. Vis. Exp. JoVE 2018, 132, 57263. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maione, A.S.; Stadiotti, I.; Pilato, C.A.; Perrucci, G.L.; Saverio, V.; Catto, V.; Vettor, G.; Casella, M.; Guarino, A.; Polvani, G.; et al. Excess TGF-β1 Drives Cardiac Mesenchymal Stromal Cells to a Pro-Fibrotic Commitment in Arrhythmogenic Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 2673. https://doi.org/10.3390/ijms22052673
Maione AS, Stadiotti I, Pilato CA, Perrucci GL, Saverio V, Catto V, Vettor G, Casella M, Guarino A, Polvani G, et al. Excess TGF-β1 Drives Cardiac Mesenchymal Stromal Cells to a Pro-Fibrotic Commitment in Arrhythmogenic Cardiomyopathy. International Journal of Molecular Sciences. 2021; 22(5):2673. https://doi.org/10.3390/ijms22052673
Chicago/Turabian StyleMaione, Angela Serena, Ilaria Stadiotti, Chiara Assunta Pilato, Gianluca Lorenzo Perrucci, Valentina Saverio, Valentina Catto, Giulia Vettor, Michela Casella, Anna Guarino, Gianluca Polvani, and et al. 2021. "Excess TGF-β1 Drives Cardiac Mesenchymal Stromal Cells to a Pro-Fibrotic Commitment in Arrhythmogenic Cardiomyopathy" International Journal of Molecular Sciences 22, no. 5: 2673. https://doi.org/10.3390/ijms22052673
APA StyleMaione, A. S., Stadiotti, I., Pilato, C. A., Perrucci, G. L., Saverio, V., Catto, V., Vettor, G., Casella, M., Guarino, A., Polvani, G., Pompilio, G., & Sommariva, E. (2021). Excess TGF-β1 Drives Cardiac Mesenchymal Stromal Cells to a Pro-Fibrotic Commitment in Arrhythmogenic Cardiomyopathy. International Journal of Molecular Sciences, 22(5), 2673. https://doi.org/10.3390/ijms22052673