
Activation of KCNQ4 as a Therapeutic Strategy to Treat Hearing Loss


{kind=link}
{kind=link}
Abstract
1. Introduction
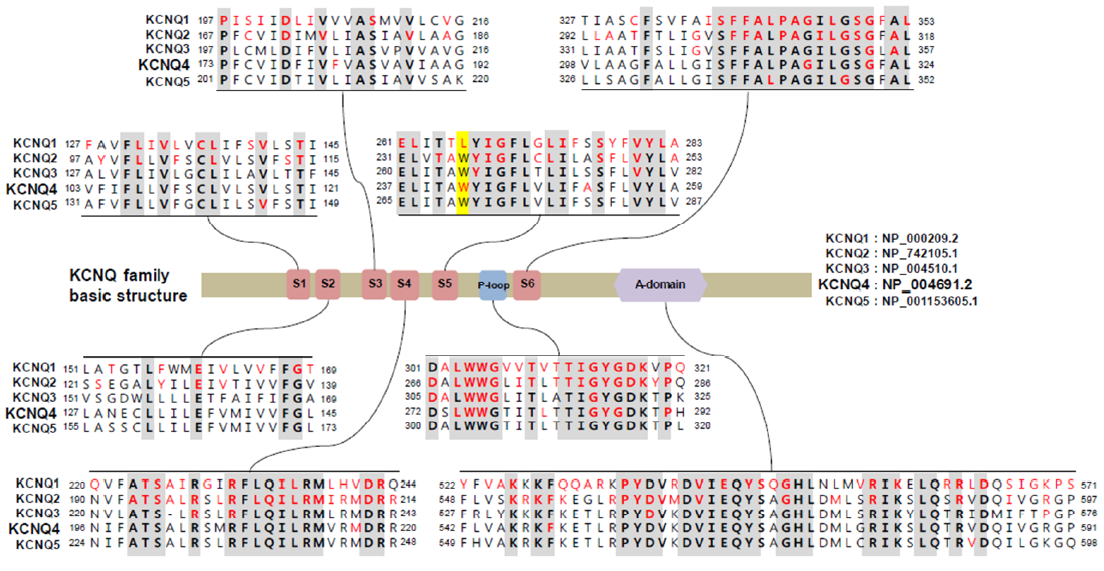
2. KCNQ Potassium Channels
3. Potassium Recycling and KCNQ4 in the Inner Ear
4. Association of KCNQ4 and Noise-Induced Hearing Loss
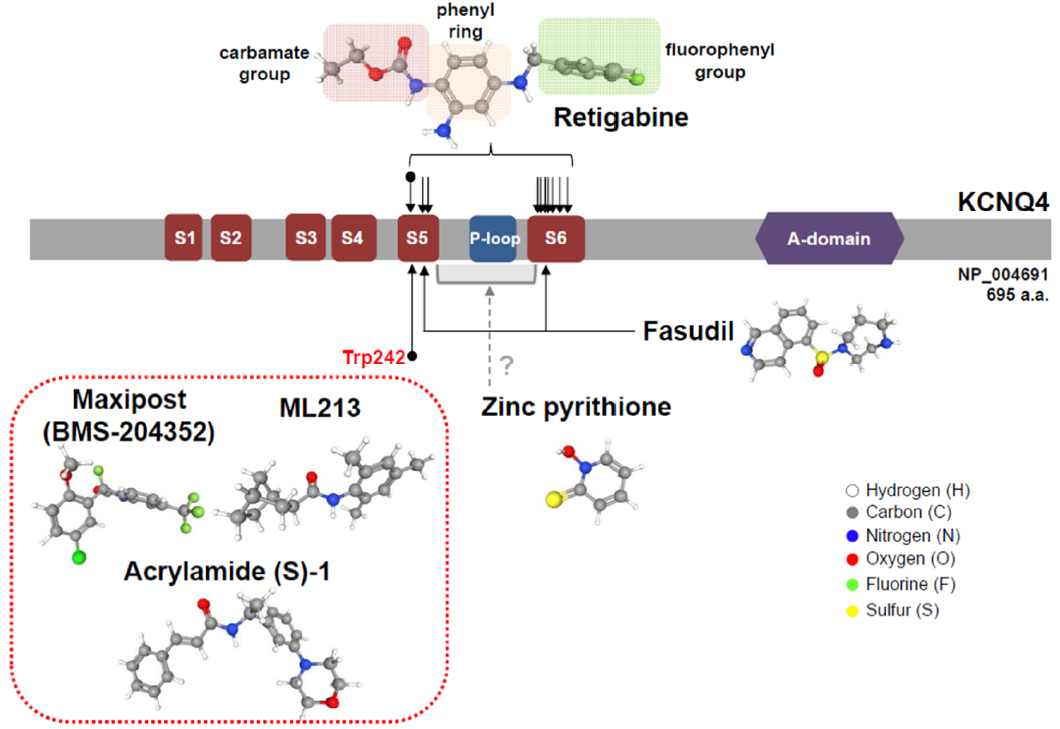
5. KCNQ4 Activators
5.1. Retigabine
5.2. Retigabine Derivatives
5.3. Zinc Pyrithione
5.4. Maxipost
5.5. Acrylamide (S)-1
5.6. Other KCNQ4 Activators
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DFNA2 | deafness nonsyndromic autosomal dominant 2 |
| IHC | Inner hair cell |
| KCNQ | Potassium voltage-gated channel subfamily q |
| OHC | Outer hair cell |
References
- Petit, C.; El-Amraoui, A.; Avan, P. Audition: Hearing and Deafness. In Neuroscience in the 21st Century, 2nd ed.; Springer: New York, NY, USA, 2016. [Google Scholar]
- Cunningham, L.L.; Tucci, D.L. Hearing Loss in Adults. N. Engl. J. Med. 2017, 377, 2465–2473. [Google Scholar] [CrossRef]
- Petit, C.; Levilliers, J.; Hardelin, J.P. Molecular genetics of hearing loss. Annu. Rev. Genet. 2001, 35, 589–646. [Google Scholar] [CrossRef]
- Raviv, D.; Dror, A.A.; Avraham, K.B. Hearing loss: A common disorder caused by many rare alleles. Ann. N. Y. Acad. Sci. 2010, 1214, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Chadha, S.; Cieza, A. World Health Organization and Its Initiative for Ear and Hearing Care. Otolaryngol. Clin. N. Am. 2018, 51, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Kubisch, C.; Schroeder, B.C.; Friedrich, T.; Lutjohann, B.; El-Amraoui, A.; Marlin, S.; Petit, C.; Jentsch, T.J. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 1999, 96, 437–446. [Google Scholar] [CrossRef]
- van Laer, L.; Carlsson, P.I.; Ottschytsch, N.; Bondeson, M.L.; Konings, A.; Vandevelde, A.; Dieltjens, N.; Fransen, E.; Snyders, D.; Borg, E.; et al. The contribution of genes involved in potassium-recycling in the inner ear to noise-induced hearing loss. Hum. Mutat. 2006, 27, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Peixoto Pinheiro, B.; Vona, B.; Löwenheim, H.; Rüttiger, L.; Knipper, M.; Adel, Y. Age-related hearing loss pertaining to potassium ion channels in the cochlea and auditory pathway. Pflüg. Arch. Eur. J. Physiol. 2020. [Google Scholar] [CrossRef]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 2009, 8, 982–1001. [Google Scholar] [CrossRef]
- Maljevic, S.; Wuttke, T.V.; Seebohm, G.; Lerche, H. KV7 channelopathies. Pflüg. Arch. Eur. J. Physiol. 2010, 460, 277–288. [Google Scholar] [CrossRef]
- Schroeder, B.C.; Hechenberger, M.; Weinreich, F.; Kubisch, C.; Jentsch, T.J. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J. Biol. Chem. 2000, 275, 24089–24095. [Google Scholar] [CrossRef]
- Lerche, C.; Scherer, C.R.; Seebohm, G.; Derst, C.; Wei, A.D.; Busch, A.E.; Steinmeyer, K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J. Biol. Chem. 2000, 275, 22395–22400. [Google Scholar] [CrossRef]
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; de Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Vincent, G.M.; Atkinson, D.L.; Keating, M.T. Molecular basis of the long-QT syndrome associated with deafness. N. Engl. J. Med. 1997, 336, 1562–1567. [Google Scholar] [CrossRef] [PubMed]
- Neyroud, N.; Tesson, F.; Denjoy, I.; Leibovici, M.; Donger, C.; Barhanin, J.; Fauré, S.; Gary, F.; Coumel, P.; Petit, C.; et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat. Genet. 1997, 15, 186–189. [Google Scholar] [CrossRef]
- Singh, N.A.; Charlier, C.; Stauffer, D.; DuPont, B.R.; Leach, R.J.; Melis, R.; Ronen, G.M.; Bjerre, I.; Quattlebaum, T.; Murphy, J.V.; et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998, 18, 25–29. [Google Scholar] [CrossRef]
- Biervert, C.; Schroeder, B.C.; Kubisch, C.; Berkovic, S.F.; Propping, P.; Jentsch, T.J.; Steinlein, O.K. A potassium channel mutation in neonatal human epilepsy. Science 1998, 279, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Charlier, C.; Singh, N.A.; Ryan, S.G.; Lewis, T.B.; Reus, B.E.; Leach, R.J.; Leppert, M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat. Genet. 1998, 18, 53–55. [Google Scholar] [CrossRef]
- Lehman, A.; Thouta, S.; Mancini, G.M.S.; Naidu, S.; van Slegtenhorst, M.; McWalter, K.; Person, R.; Mwenifumbo, J.; Salvarinova, R.; Guella, I.; et al. Loss-of-Function and Gain-of-Function Mutations in KCNQ5 Cause Intellectual Disability or Epileptic Encephalopathy. Am. J. Hum. Genet. 2017, 101, 65–74. [Google Scholar] [CrossRef]
- Naito, T.; Nishio, S.-Y.; Iwasa, Y.-I.; Yano, T.; Kumakawa, K.; Abe, S.; Ishikawa, K.; Kojima, H.; Namba, A.; Oshikawa, C.; et al. Comprehensive Genetic Screening of KCNQ4 in a Large Autosomal Dominant Nonsyndromic Hearing Loss Cohort: Genotype-Phenotype Correlations and a Founder Mutation. PLoS ONE 2013, 8, e63231. [Google Scholar]
- Jung, J.; Lin, H.; Koh, Y.I.; Ryu, K.; Lee, J.S.; Rim, J.H.; Choi, H.J.; Lee, H.J.; Kim, H.Y.; Yu, S.; et al. Rare KCNQ4 variants found in public databases underlie impaired channel activity that may contribute to hearing impairment. Exp. Mol. Med. 2019, 51, 99. [Google Scholar] [CrossRef]
- Gao, Y.; Yechikov, S.; Vazquez, A.E.; Chen, D.; Nie, L. Impaired surface expression and conductance of the KCNQ4 channel lead to sensorineural hearing loss. J. Cell. Mol. Med. 2013, 17, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Leitner, M.G.; Feuer, A.; Ebers, O.; Schreiber, D.N.; Halaszovich, C.R.; Oliver, D. Restoration of ion channel function in deafness-causing KCNQ4 mutants by synthetic channel openers. Br. J. Pharmacol. 2012, 165, 2244–2259. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Choi, H.B.; Koh, Y.I.; Rim, J.H.; Choi, H.J.; Kim, S.H.; Lee, J.H.; An, J.; Kim, A.; Lee, J.S.; et al. Whole-exome sequencing identifies two novel mutations in KCNQ4 in individuals with nonsyndromic hearing loss. Sci. Rep. 2018, 8, 16659. [Google Scholar] [CrossRef]
- Shin, D.H.; Jung, J.; Koh, Y.I.; Rim, J.H.; Lee, J.S.; Choi, H.J.; Joo, S.Y.; Yu, S.; Cha, D.H.; Lee, S.Y.; et al. A recurrent mutation in KCNQ4 in Korean families with nonsyndromic hearing loss and rescue of the channel activity by KCNQ activators. Hum. Mutat. 2019, 40, 335–346. [Google Scholar] [CrossRef]
- Wangemann, P. K+ cycling and the endocochlear potential. Hear. Res. 2002, 165, 1–9. [Google Scholar] [CrossRef]
- Delmaghani, S.; El-Amraoui, A. Inner Ear Gene Therapies Take Off: Current Promises and Future Challenges. J. Clin. Med. 2020, 9, 2309. [Google Scholar] [CrossRef]
- Pan, B.; Akyuz, N.; Liu, X.-P.; Asai, Y.; Nist-Lund, C.; Kurima, K.; Derfler, B.H.; György, B.; Limapichat, W.; Walujkar, S.; et al. TMC1 Forms the Pore of Mechanosensory Transduction Channels in Vertebrate Inner Ear Hair Cells. Neuron 2018, 99, 736–753.e6. [Google Scholar] [CrossRef] [PubMed]
- Delpire, E.; Lu, J.; England, R.; Dull, C.; Thorne, T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat. Genet. 1999, 22, 192–195. [Google Scholar] [CrossRef]
- Marcus, D.C.; Wu, T.; Wangemann, P.; Kofuji, P. KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am. J. Physiol. Cell Physiol. 2002, 282, C403–C407. [Google Scholar] [CrossRef]
- Dallos, P. Cochlear amplification, outer hair cells and prestin. Curr. Opin. Neurobiol. 2008, 18, 370–376. [Google Scholar] [CrossRef]
- Housley, G.D.; Ashmore, J.F. Ionic currents of outer hair cells isolated from the guinea-pig cochlea. J. Physiol. 1992, 448, 73–98. [Google Scholar] [CrossRef]
- Dallos, P.; Zheng, J.; Cheatham, M.A. Prestin and the cochlear amplifier. J. Physiol. 2006, 576, 37–42. [Google Scholar] [CrossRef]
- Marcotti, W.; Kros, C.J. Developmental expression of the potassium current IK,n contributes to maturation of mouse outer hair cells. J. Physiol. 1999, 520, 653–660. [Google Scholar] [CrossRef]
- Winter, H.; Braig, C.; Zimmermann, U.; Geisler, H.-S.; Fränzer, J.-T.; Weber, T.; Ley, M.; Engel, J.; Knirsch, M.; Bauer, K.; et al. Thyroid hormone receptors TRα1 and TRβ differentially regulate gene expression of Kcnq4 and prestin during final differentiation of outer hair cells. J. Cell Sci. 2006, 119, 2975–2984. [Google Scholar] [CrossRef]
- Boettger, T.; Hübner, C.A.; Maier, H.; Rust, M.B.; Beck, F.X.; Jentsch, T.J. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 2002, 416, 874–878. [Google Scholar] [CrossRef]
- Kharkovets, T.; Hardelin, J.P.; Safieddine, S.; Schweizer, M.; El-Amraoui, A.; Petit, C.; Jentsch, T.J. KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc. Natl. Acad. Sci. USA 2000, 97, 4333–4338. [Google Scholar] [CrossRef]
- Mammano, F.; Ashmore, J.F. Differential expression of outer hair cell potassium currents in the isolated cochlea of the guinea-pig. J. Physiol. 1996, 496, 639–646. [Google Scholar] [CrossRef]
- Beisel, K.W.; Nelson, N.C.; Delimont, D.C.; Fritzsch, B. Longitudinal gradients of KCNQ4 expression in spiral ganglion and cochlear hair cells correlate with progressive hearing loss in DFNA211Published on the World Wide Web on 13 September 2000. Mol. Brain Res. 2000, 82, 137–149. [Google Scholar] [CrossRef]
- Oliver, D.; Knipper, M.; Derst, C.; Fakler, B. Resting potential and submembrane calcium concentration of inner hair cells in the isolated mouse cochlea are set by KCNQ-type potassium channels. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 2141–2149. [Google Scholar] [CrossRef]
- Beisel, K.W.; Rocha-Sanchez, S.M.; Morris, K.A.; Nie, L.; Feng, F.; Kachar, B.; Yamoah, E.N.; Fritzsch, B. Differential expression of KCNQ4 in inner hair cells and sensory neurons is the basis of progressive high-frequency hearing loss. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 9285–9293. [Google Scholar] [CrossRef]
- Kharkovets, T.; Dedek, K.; Maier, H.; Schweizer, M.; Khimich, D.; Nouvian, R.; Vardanyan, V.; Leuwer, R.; Moser, T.; Jentsch, T.J. Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J. 2006, 25, 642–652. [Google Scholar] [CrossRef]
- Carignano, C.; Barila, E.P.; Rías, E.I.; Dionisio, L.; Aztiria, E.; Spitzmaul, G. Inner Hair Cell and Neuron Degeneration Contribute to Hearing Loss in a DFNA2-Like Mouse Model. Neuroscience 2019, 410, 202–216. [Google Scholar] [CrossRef]
- Rüttiger, L.; Sausbier, M.; Zimmermann, U.; Winter, H.; Braig, C.; Engel, J.; Knirsch, M.; Arntz, C.; Langer, P.; Hirt, B.; et al. Deletion of the Ca2+-activated potassium (BK) alpha-subunit but not the BKbeta1-subunit leads to progressive hearing loss. Proc. Natl. Acad. Sci. USA 2004, 101, 12922–12927. [Google Scholar] [CrossRef]
- Selyanko, A.A.; Hadley, J.K.; Wood, I.C.; Abogadie, F.C.; Jentsch, T.J.; Brown, D.A. Inhibition of KCNQ1–4 potassium channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. J. Physiol. 2000, 522 Pt 3, 349–355. [Google Scholar] [CrossRef]
- Alberti, P.W.; Symons, F.; Hyde, M.L. Occupational hearing loss. The significance of asymmetrical hearing thresholds. Acta Otolaryngol. 1979, 87, 255–263. [Google Scholar] [CrossRef]
- World Health Organization. Hearing Loss Due to Recreational Exposure to Loud Sounds: A Review; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Konings, A.; Laer, L.V.; Camp, G.V. Genetic Studies on Noise-Induced Hearing Loss: A Review. Ear Hear. 2009, 30, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Pawelczyk, M.; van Laer, L.; Fransen, E.; Rajkowska, E.; Konings, A.; Carlsson, P.I.; Borg, E.; van Camp, G.; Sliwinska-Kowalska, M. Analysis of gene polymorphisms associated with K ion circulation in the inner ear of patients susceptible and resistant to noise-induced hearing loss. Ann. Hum. Genet. 2009, 73 Pt 4, 411–421. [Google Scholar] [CrossRef]
- Sliwinska-Kowalska, M.; Pawelczyk, M. Contribution of genetic factors to noise-induced hearing loss: A human studies review. Mutat. Res. 2013, 752, 61–65. [Google Scholar] [CrossRef]
- Guo, H.; Ding, E.; Sheng, R.; Cheng, J.; Cai, W.; Guo, J.; Wang, N.; Zhang, H.; Zhu, B. Genetic variation in KCNQ4 gene is associated with susceptibility to noise-induced hearing loss in a Chinese population. Environ. Toxicol. Pharm. 2018, 63, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Marchetta, P.; Möhrle, D.; Eckert, P.; Reimann, K.; Wolter, S.; Tolone, A.; Lang, I.; Wolters, M.; Feil, R.; Engel, J.; et al. Guanylyl Cyclase A/cGMP Signaling Slows Hidden, Age- and Acoustic Trauma-Induced Hearing Loss. Front. Aging Neurosci. 2020, 12, 83. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.; Braig, C.; Rüttiger, L.; Kuhn, S.; Zimmermann, U.; Blin, N.; Sausbier, M.; Kalbacher, H.; Münkner, S.; Rohbock, K.; et al. Two classes of outer hair cells along the tonotopic axis of the cochlea. Neuroscience 2006, 143, 837–849. [Google Scholar] [CrossRef]
- Xiong, Q.; Gao, Z.; Wang, W.; Li, M. Activation of Kv7 (KCNQ) voltage-gated potassium channels by synthetic compounds. Trends Pharmacol. Sci. 2008, 29, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Rostock, A.; Tober, C.; Rundfeldt, C.; Bartsch, R.; Engel, J.; Polymeropoulos, E.E.; Kutscher, B.; Löscher, W.; Hönack, D.; White, H.S.; et al. D-23129: A new anticonvulsant with a broad spectrum activity in animal models of epileptic seizures. Epilepsy Res. 1996, 23, 211–223. [Google Scholar] [CrossRef]
- Rundfeldt, C. The new anticonvulsant retigabine (D-23129) acts as an opener of K+ channels in neuronal cells. Eur. J. Pharmacol. 1997, 336, 243–249. [Google Scholar] [CrossRef]
- Tober, C.; Rostock, A.; Rundfeldt, C.; Bartsch, R. D-23129: A potent anticonvulsant in the amygdala kindling model of complex partial seizures. Eur. J. Pharmacol. 1996, 303, 163–169. [Google Scholar] [CrossRef]
- French, J.A.; Abou-Khalil, B.W.; Leroy, R.F.; Yacubian, E.M.T.; Shin, P.; Hall, S.; Mansbach, H.; Nohria, V. Randomized, double-blind, placebo-controlled trial of ezogabine (retigabine) in partial epilepsy. Neurology 2011, 76, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Gunthorpe, M.J.; Large, C.H.; Sankar, R. The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia 2012, 53, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Tatulian, L.; Delmas, P.; Abogadie, F.C.; Brown, D.A. Activation of Expressed KCNQ Potassium Currents and Native Neuronal M-Type Potassium Currents by the Anti-Convulsant Drug Retigabine. J. Neurosci. 2001, 21, 5535–5545. [Google Scholar] [CrossRef] [PubMed]
- Schenzer, A.; Friedrich, T.; Pusch, M.; Saftig, P.; Jentsch, T.J.; Grötzinger, J.; Schwake, M. Molecular Determinants of KCNQ (Kv7) K+ Channel Sensitivity to the Anticonvulsant Retigabine. J. Neurosci. 2005, 25, 5051–5060. [Google Scholar] [CrossRef]
- Friedman, A.K.; Juarez, B.; Ku, S.M.; Zhang, H.; Calizo, R.C.; Walsh, J.J.; Chaudhury, D.; Zhang, S.; Hawkins, A.; Dietz, D.M.; et al. KCNQ channel openers reverse depressive symptoms via an active resilience mechanism. Nat. Commun. 2016, 7, 11671. [Google Scholar] [CrossRef]
- Fretwell, L.V.; Woolard, J. Cardiovascular responses to retigabine in conscious rats—Under normotensive and hypertensive conditions. Br. J. Pharm. 2013, 169, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Iwata, M.; Tsuchimori, N.; Matsumoto, T. Activation of peripheral KCNQ channels attenuates inflammatory pain. Mol. Pain 2014, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Korsgaard, M.P.G.; Hartz, B.P.; Brown, W.D.; Ahring, P.K.; Strøbæk, D.; Mirza, N.R. Anxiolytic Effects of Maxipost (BMS-204352) and Retigabine via Activation of Neuronal Kv7 Channels. J. Pharmacol. Exp. Ther. 2005, 314, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Redrobe, J.P.; Nielsen, A.N. Effects of neuronal Kv7 potassium channel activators on hyperactivity in a rodent model of mania. Behav. Brain Res. 2009, 198, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Brickel, N.; Gandhi, P.; VanLandingham, K.; Hammond, J.; DeRossett, S. The urinary safety profile and secondary renal effects of retigabine (ezogabine): A first-in-class antiepileptic drug that targets KCNQ (Kv7) potassium channels. Epilepsia 2012, 53, 606–612. [Google Scholar] [CrossRef]
- Wainger, B.J.; Macklin, E.A.; Vucic, S.; McIlduff, C.E.; Paganoni, S.; Maragakis, N.J.; Bedlack, R.; Goyal, N.A.; Rutkove, S.B.; Lange, D.J.; et al. Effect of Ezogabine on Cortical and Spinal Motor Neuron Excitability in Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. JAMA Neurol. 2021, 78, 186–196. [Google Scholar] [CrossRef]
- van Rijn, C.M.; van Bree, E.W. Synergy between retigabine and GABA in modulating the convulsant site of the GABAA receptor complex. Eur. J. Pharm. 2003, 464, 95–100. [Google Scholar] [CrossRef]
- Schroder, R.L.; Jespersen, T.; Christophersen, P.; Strobaek, D.; Jensen, B.S.; Olesen, S.P. KCNQ4 channel activation by BMS-204352 and retigabine. Neuropharmacology 2001, 40, 888–898. [Google Scholar] [CrossRef]
- Li, T.; Wu, K.; Yue, Z.; Wang, Y.; Zhang, F.; Shen, H. Structural Basis for the Modulation of Human KCNQ4 by Small-Molecule Drugs. Mol. Cell 2020, 81, 25–37. [Google Scholar] [CrossRef]
- Wuttke, T.V.; Seebohm, G.; Bail, S.; Maljevic, S.; Lerche, H. The new anticonvulsant retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol. Pharm. 2005, 67, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Craciun, L.C.; Mirshahi, T.; Rohács, T.; Lopes, C.M.B.; Jin, T.; Logothetis, D.E. PIP2 Activates KCNQ Channels, and Its Hydrolysis Underlies Receptor-Mediated Inhibition of M Currents. Neuron 2003, 37, 963–975. [Google Scholar] [CrossRef]
- Zaydman, M.A.; Silva, J.R.; Delaloye, K.; Li, Y.; Liang, H.; Larsson, H.P.; Shi, J.; Cui, J. Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc. Natl. Acad. Sci. USA 2013, 110, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, A.M.; Chen, G.-D.; Salvi, R. Potassium ion channel openers, Maxipost and Retigabine, protect against peripheral salicylate ototoxicity in rats. Hear. Res. 2015, 327, 1–8. [Google Scholar] [CrossRef]
- Wang, L.; Qiao, G.-H.; Hu, H.-N.; Gao, Z.-B.; Nan, F.-J. Discovery of Novel Retigabine Derivatives as Potent KCNQ4 and KCNQ5 Channel Agonists with Improved Specificity. ACS Med. Chem. Lett. 2019, 10, 27–33. [Google Scholar] [CrossRef]
- Liu, R.; Tzounopoulos, T.; Wipf, P. Synthesis and Optimization of Kv7 (KCNQ) Potassium Channel Agonists: The Role of Fluorines in Potency and Selectivity. ACS Med. Chem. Lett. 2019, 10, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Dalby-Brown, W.; Jessen, C.; Hougaard, C.; Jensen, M.L.; Jacobsen, T.A.; Nielsen, K.S.; Erichsen, H.K.; Grunnet, M.; Ahring, P.K.; Christophersen, P.; et al. Characterization of a novel high-potency positive modulator of Kv7 channels. Eur. J. Pharmacol. 2013, 709, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Jepps, T.A.; Bentzen, B.H.; Stott, J.B.; Povstyan, O.V.; Sivaloganathan, K.; Dalby-Brown, W.; Greenwood, I.A. Vasorelaxant effects of novel Kv 7.4 channel enhancers ML213 and NS15370. Br. J. Pharmacol. 2014, 171, 4413–4424. [Google Scholar] [CrossRef] [PubMed]
- Marks, R.; Pearse, A.D.; Walker, A.P. The effects of a shampoo containing zinc pyrithione on the control of dandruff. Br. J. Derm. 1985, 112, 415–422. [Google Scholar] [CrossRef]
- Xiong, Q.; Sun, H.; Li, M. Zinc pyrithione-mediated activation of voltage-gated KCNQ potassium channels rescues epileptogenic mutants. Nat. Chem. Biol. 2007, 3, 287–296. [Google Scholar] [CrossRef]
- Xiong, Q.; Sun, H.; Zhang, Y.; Nan, F.; Li, M. Combinatorial augmentation of voltage-gated KCNQ potassium channels by chemical openers. Proc. Natl. Acad. Sci. USA 2008, 105, 3128–3133. [Google Scholar] [CrossRef] [PubMed]
- Hewawasam, P.; Gribkoff, V.K.; Pendri, Y.; Dworetzky, S.I.; Meanwell, N.A.; Martinez, E.; Boissard, C.G.; Post-Munson, D.J.; Trojnacki, J.T.; Yeleswaram, K.; et al. The synthesis and characterization of BMS-204352 (MaxiPost™) and related 3-fluorooxindoles as openers of maxi-K potassium channels. Bioorg. Med. Chem. Lett. 2002, 12, 1023–1026. [Google Scholar] [CrossRef]
- Lobarinas, E.; Dalby-Brown, W.; Stolzberg, D.; Mirza, N.R.; Allman, B.L.; Salvi, R. Effects of the potassium ion channel modulators BMS-204352 Maxipost and its R-enantiomer on salicylate-induced tinnitus in rats. Physiol. Behav. 2011, 104, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Bentzen, B.H.; Schmitt, N.; Calloe, K.; Dalby Brown, W.; Grunnet, M.; Olesen, S.P. The acrylamide (S)-1 differentially affects Kv7 (KCNQ) potassium channels. Neuropharmacology 2006, 51, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Boissard, C.G.; Greco, C.; Gribkoff, V.K.; Harden, D.G.; He, H.; L’Heureux, A.; Kang, S.H.; Kinney, G.G.; Knox, R.J.; et al. (S)-N-[1-(3-morpholin-4-ylphenyl)ethyl]-3-phenylacrylamide: An orally bioavailable KCNQ2 opener with significant activity in a cortical spreading depression model of migraine. J. Med. Chem. 2003, 46, 3197–3200. [Google Scholar] [CrossRef] [PubMed]
- Blom, S.M.; Rottländer, M.; Kehler, J.; Bundgaard, C.; Schmitt, N.; Jensen, H.S. From pan-reactive KV7 channel opener to subtype selective opener/inhibitor by addition of a methyl group. PLoS ONE 2014, 9, e100209. [Google Scholar] [CrossRef]
- Landoulsi, Z.; Miceli, F.; Palmese, A.; Amoresano, A.; Marino, G.; El Ayeb, M.; Taglialatela, M.; Benkhalifa, R. Subtype-Selective Activation of Kv7 Channels by AaTXKβ(2–64), a Novel Toxin Variant from the Androctonus australis Scorpion Venom. Mol. Pharmacol. 2013, 84, 763–773. [Google Scholar] [CrossRef]
- Zhang, X.; An, H.; Li, J.; Zhang, Y.; Liu, Y.; Jia, Z.; Zhang, W.; Chu, L.; Zhang, H. Selective activation of vascular Kv 7.4/Kv 7.5 K+ channels by fasudil contributes to its vasorelaxant effect. Br. J. Pharmacol. 2016, 173, 3480–3491. [Google Scholar] [CrossRef]
- Yu, H.; Wu, M.; Townsend, S.D.; Zou, B.; Long, S.; Daniels, J.S.; McManus, O.B.; Li, M.; Lindsley, C.W.; Hopkins, C.R. Discovery, Synthesis, and Structure–Activity Relationship of a Series of N-Aryl-bicyclo [2.2.1]heptane-2-carboxamides: Characterization of ML213 as a Novel KCNQ2 and KCNQ4 Potassium Channel Opener. ACS Chem. Neurosci. 2011, 2, 572–577. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rim, J.H.; Choi, J.Y.; Jung, J.; Gee, H.Y. Activation of KCNQ4 as a Therapeutic Strategy to Treat Hearing Loss. Int. J. Mol. Sci. 2021, 22, 2510. https://doi.org/10.3390/ijms22052510
Rim JH, Choi JY, Jung J, Gee HY. Activation of KCNQ4 as a Therapeutic Strategy to Treat Hearing Loss. International Journal of Molecular Sciences. 2021; 22(5):2510. https://doi.org/10.3390/ijms22052510
Chicago/Turabian StyleRim, John Hoon, Jae Young Choi, Jinsei Jung, and Heon Yung Gee. 2021. "Activation of KCNQ4 as a Therapeutic Strategy to Treat Hearing Loss" International Journal of Molecular Sciences 22, no. 5: 2510. https://doi.org/10.3390/ijms22052510
APA StyleRim, J. H., Choi, J. Y., Jung, J., & Gee, H. Y. (2021). Activation of KCNQ4 as a Therapeutic Strategy to Treat Hearing Loss. International Journal of Molecular Sciences, 22(5), 2510. https://doi.org/10.3390/ijms22052510