
Neoagarooligosaccharide Protects against Hepatic Fibrosis via Inhibition of TGF-β/Smad Signaling Pathway



Abstract
1. Introduction
2. Results
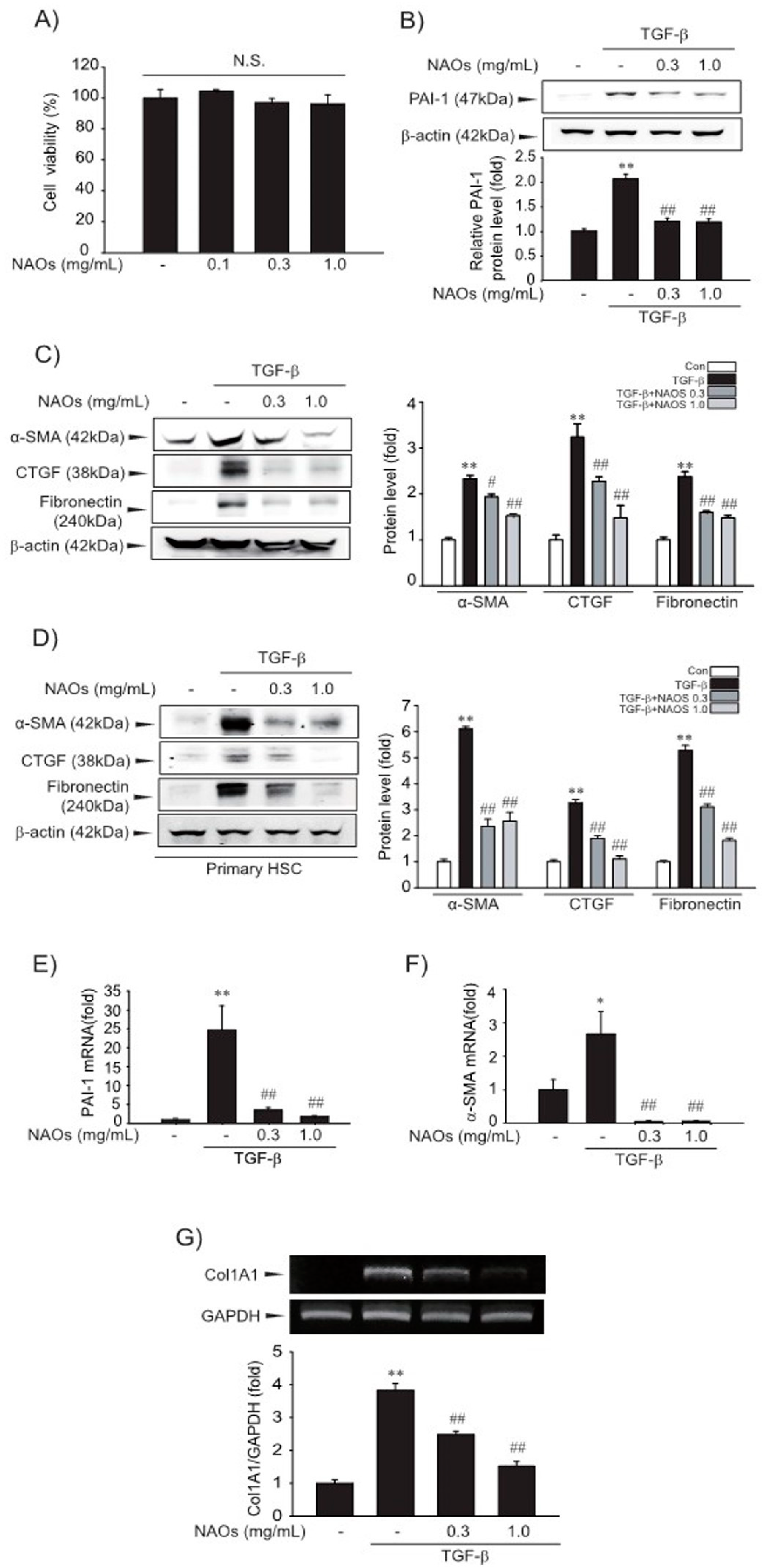
2.1. Suppressive Effect of NAOs on HSCs Activation in Vitro
2.2. Inhibitory Effect of NAOs on TGF-β/Smad Signaling
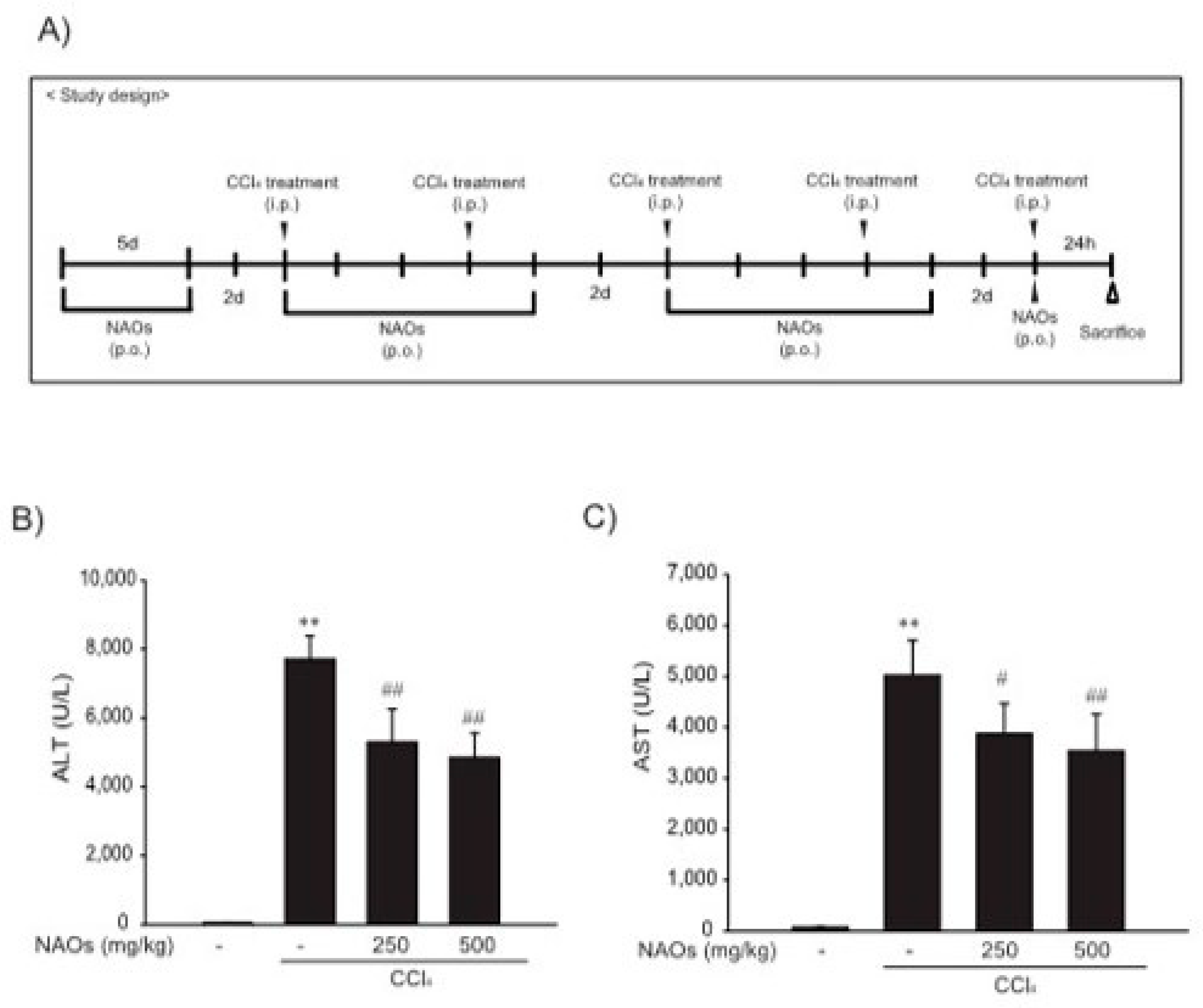
2.3. Suppression of CCl4-Induced Liver Fibrosis by NAOs In Vivo
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of NAOs
4.3. Cell Culture
4.4. Animals
4.5. CCl4-Induced Hepatic Fibrosis
4.6. Blood Chemistry
4.7. Histological Process
4.8. MTT Assay
4.9. Immunoblot Analysis
4.10. RNA Isolation and Real-Time RT-PCR Analysis
4.11. Luciferase Gene Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| α-SMA | smooth muscle actin |
| ALT | Aminotransferase |
| AST | Aspartate aminotransferase |
| CCl4 | Carbon tetrachloride |
| CTGF | Connective tissue growth factor |
| ECM | Extracellular matrix |
| HSCs | Hepatic Stellate Cells |
| NAOs | Neoagarooligosaccharides |
| PAI-1 | Plasminogen activator inhibitor-1 |
| SBEs | Smad-binding elements |
| TGF-β | Transforming growth factor-β |
References
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Zheng, R.-Q.; Wang, Q.-H.; Lu, M.-D.; Xie, S.-B.; Ren, J.; Su, Z.-Z.; Cai, Y.-K.; Yao, J.-L. Liver fibrosis in chronic viral hepatitis: An ultrasonographic study. World J. Gastroenterol. 2003, 9, 2484–2489. [Google Scholar] [CrossRef]
- Hoffmann, C.; Djerir, N.E.H.; Danckaert, A.; Fernandes, J.; Roux, P.; Charrueau, C.; Lachagès, A.-M.; Charlotte, F.; Brocheriou, I.; Clément, K.; et al. Hepatic stellate cell hypertrophy is associated with metabolic liver fibrosis. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Senoo, H.; Mezaki, Y.; Fujiwara, M. The stellate cell system (vitamin A-storing cell system). Anat. Sci. Int. 2017, 92, 387–455. [Google Scholar] [CrossRef]
- Casini, A.; Pinzani, M.; Milani, S.; Grappone, C.; Galli, G.; Jezequel, A.M.; Schuppan, D.; Rotella, C.M.; Surrenti, C. Regulation of extracellular matrix synthesis by transforming growth factor beta 1 in human fat-storing cells. Gastroenterology 1993, 105, 245–253. [Google Scholar] [CrossRef]
- Kawarada, Y.; Inoue, Y.; Kawasaki, F.; Fukuura, K.; Sato, K.; Tanaka, T.; Itoh, Y.; Hayashi, H. TGF-β induces p53/Smads complex formation in the PAI-1 promoter to activate transcription. Sci. Rep. 2016, 6, 35483. [Google Scholar] [CrossRef] [PubMed]
- Kamato, D.; Burch, M.; Zhou, Y.; Mohamed, R.; Stow, J.L.; Osman, N.; Zheng, W.; Little, P.J. Individual Smad2 linker region phosphorylation sites determine the expression of proteoglycan and glycosaminoglycan synthesizing genes. Cell. Signal. 2019, 53, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Katoh, D.; Kozuka, Y.; Noro, A.; Ogawa, T.; Imanaka-Yoshida, K.; Yoshida, T. Tenascin-C Induces Phenotypic Changes in Fibroblasts to Myofibroblasts with High Contractility through the Integrin αvβ1/Transforming Growth Factor β/SMAD Signaling Axis in Human. Breast Cancer Am. J. Pathol. 2020, 190, 2123–2135. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Chen, Y.; Suo, L.; Chen, H.; Zhu, L.; Wan, G.; Han, X. Activin a promotes myofibroblast differentiation of endometrial mesenchymal stem cells via STAT3-dependent Smad/CTGF pathway. Cell Commun. Signal. 2019, 17, 45. [Google Scholar] [CrossRef]
- Gao, W.; Lin, P.; Hwang, E.; Wang, Y.; Yan, Z.; Ngo, H.T.; Yi, T. Pterocarpus santalinus L. Regulated Ultraviolet B Irradiation-induced Procollagen Reduction and Matrix Metalloproteinases Expression Through Activation of TGF-β /Smad and Inhibition of the MAPK/AP-1 Pathway in Normal Human Dermal Fibroblasts. Photochem. Photobiol. 2017, 94, 139–149. [Google Scholar] [CrossRef]
- Yang, J.H.; Kim, S.C.; Kim, K.M.; Jang, C.H.; Cho, S.S.; Kim, S.J.; Ku, S.K.; Cho, I.J.; Ki, S.H. Isorhamnetin attenuates liver fibrosis by inhibiting TGF-β/Smad signaling and relieving oxidative stress. Eur. J. Pharmacol. 2016, 783, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; Jia, L.; Xiao, S. Dicliptera Chinensis polysaccharides target TGF-β/Smad pathway and inhibit stellate cells activation in rats with dimethylnitrosamine-induced hepatic fibrosis. Cell Mol. Biol. 2016, 62, 99–103. [Google Scholar] [PubMed]
- Yang, J.H.; Cho, S.S.; Kim, K.M.; Kim, J.Y.; Kim, E.J.; Park, E.Y.; Lee, J.H.; Ki, S.H. Neoagarooligosaccharides enhance the level and efficiency of LDL receptor and improve cholesterol homeostasis. J. Funct. Foods 2017, 38, 529–539. [Google Scholar] [CrossRef]
- Yang, J.H.; Na, C.-S.; Cho, S.S.; Kim, K.M.; Lee, J.H.; Chen, X.-Q.; Ku, S.K.; Cho, I.J.; Kim, E.J.; Lee, J.H.; et al. Hepatoprotective Effect of Neoagarooligosaccharide via Activation of Nrf2 and Enhanced Antioxidant Efficacy. Biol. Pharm. Bull. 2020, 43, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.J.; Lee, J.-H.; Kim, E.J.; Yang, H.J.; Park, J.-S.; Hong, S.-K. Toxicological evaluation of neoagarooligosaccharides prepared by enzymatic hydrolysis of agar. Regul. Toxicol. Pharmacol. 2017, 90, 9–21. [Google Scholar] [CrossRef]
- Scholten, D.; Trebicka, J.; Liedtke, C.; Weiskirchen, R. The carbon tetrachloride model in mice. Lab. Anim. 2015, 49, 4–11. [Google Scholar] [CrossRef]
- Dong, S.; Chen, Q.-L.; Song, Y.-N.; Sun, Y.; Wei, B.; Li, X.-Y.; Hu, Y.-Y.; Liu, P.; Su, S.-B. Mechanisms of CCl4-induced liver fibrosis with combined transcriptomic and proteomic analysis. J. Toxicol. Sci. 2016, 41, 561–572. [Google Scholar] [CrossRef]
- Carpino, G.; Morini, S.; Corradini, S.G.; Franchitto, A.; Merli, M.; Siciliano, M.; Gentili, F.; Muda, A.O.; Berloco, P.B.; Rossi, M. Alpha-SMA expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Dig. Liver Dis. 2005, 37, 349–356. [Google Scholar] [CrossRef]
- Cordero-Espinoza, L.; Huch, M. The balancing act of the liver: Tissue regeneration versus fibrosis. J. Clin. Investig. 2018, 128, 85–96. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic fibrosis—Overview. Toxicology 2008, 254, 120–129. [Google Scholar] [CrossRef]
- Geervliet, E.; Bansal, R. Matrix Metalloproteinases as Potential Biomarkers and Therapeutic Targets in Liver Diseases. Cells 2020, 9, 1212. [Google Scholar] [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.S.; Mokhtari, R.B.; El Hout, Y.; Azadi, M.A.; Alauddin, M.; Yeger, H.; Farhat, W.A. TGF-β1 induces EMT reprogramming of porcine bladder urothelial cells into collagen producing fibroblasts-like cells in a Smad2/Smad3-dependent manner. J. Cell Commun. Signal. 2013, 8, 39–58. [Google Scholar] [CrossRef]
- Huang, F.; Chen, Y.-G. Regulation of TGF-β receptor activity. Cell Biosci. 2012, 2, 9. [Google Scholar] [CrossRef]
- Frederick, J.P.; Liberati, N.T.; Waddell, D.S.; Shi, Y.; Wang, X.-F. Transforming Growth Factor β-Mediated Transcriptional Repression of c-myc Is Dependent on Direct Binding of Smad3 to a Novel Repressive Smad Binding Element. Mol. Cell. Biol. 2004, 24, 2546–2559. [Google Scholar] [CrossRef] [PubMed]
- Al-Tamimi, J.; Alhazza, I.M.; Al-Khalifa, M.; Metwalli, A.; Rady, A.; Ebaid, H. Potential effects of samsum ant, Brachyponera sennaarensis, venom on TNF-α/NF-κB mediated inflammation in CCl4-toxicity in vivo. Lipids Health Dis. 2016, 15, 198. [Google Scholar] [CrossRef]
- Goodla, L.; Manubolu, M.; Pathakoti, K.; Jayakumar, T.; Sheu, J.-R.; Fraker, M.; Tchounwou, P.B.; Poondamalli, P.R. Protective Effects of Ammannia baccifera against CCl4-Induced Oxidative Stress in Rats. Int. J. Environ. Res. Public Health 2019, 16, 1440. [Google Scholar] [CrossRef]
- Yang, J.H.; Kim, K.M.; Cho, S.S.; Shin, S.M.; Ka, S.O.; Na, C.-S.; Park, B.H.; Jegal, K.H.; Kim, J.K.; Ku, S.K.; et al. Inhibitory Effect of Sestrin 2 on Hepatic Stellate Cell Activation and Liver Fibrosis. Antioxid. Redox Signal. 2019, 31, 243–259. [Google Scholar] [CrossRef]
- Raven, A.; Lu, W.-Y.; Man, T.Y.; Ferreira-Gonzalez, S.; O’Duibhir, E.; Dwyer, B.J.; Thomson, J.P.; Meehan, R.R.; Bogorad, R.; Koteliansky, V.; et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 2017, 547, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Ki, S.H.; Yang, J.H.; Ku, S.K.; Kim, S.C.; Kim, Y.W.; Cho, I.J. Red ginseng extract protects against carbon tetrachloride-induced liver fibrosis. J. Ginseng Res. 2013, 37, 45–53. [Google Scholar] [CrossRef]
- Knockaert, L.; Berson, A.; Ribault, C.; Prost, P.-E.; Fautrel, A.; Pajaud, J.; Lepage, S.; Lucas-Clerc, C.; Bégué, J.-M.; Fromenty, B.; et al. Carbon tetrachloride-mediated lipid peroxidation induces early mitochondrial alterations in mouse liver. Lab. Investig. 2011, 92, 396–410. [Google Scholar] [CrossRef]
- Tipoe, G.L.; Leung, T.M.; Liong, E.C.; Lau, T.Y.H.; Fung, M.L.; Nanji, A.A. Epigallocatechin-3-gallate (EGCG) reduces liver inflammation, oxidative stress and fibrosis in carbon tetrachloride (CCl4)-induced liver injury in mice. Toxicology 2010, 273, 45–52. [Google Scholar] [CrossRef]
- Shin, B.Y.; Jin, S.H.; Cho, I.J.; Ki, S.H. Nrf2-ARE pathway regulates induction of Sestrin-2 expression. Free. Radic. Biol. Med. 2012, 53, 834–841. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups Index (Unit) | Controls | CCl4 with NAOs | ||
|---|---|---|---|---|
| Vehicle (n = 5) | CCl4 (n = 5) | 250 mg/kg (n = 5) | 500 mg/kg (n = 5) | |
| Hepatic Staging Scores (Max = 6) | 0.30 ± 0.48 | 4.40 ± 0.52 ** | 3.00 ± 1.05 ## | 2.10 ± 0.57 ## |
| Degenerative regions (%/mm2) | 2.08 ± 1.18 | 54.43 ± 13.37 ** | 29.61 ± 10.41 ## | 18.13 ± 10.50 ## |
| Degenerative hepatocytes (cells/1000 hepatocytes) | 17.20 ± 11.48 | 436.70 ± 127.05 ** | 206.00 ± 109.82 ## | 126.50 ± 85.64 ## |
| Inflammatory cells (cells/1000 hepatocytes) | 24.80 ± 14.37 | 320.40 ± 89.63 ** | 168.00 ± 69.00 ## | 112.00 ± 22.39 ## |
| Sirius red-stained collagen occupied regions (%/mm2) | 1.75 ± 1.29 | 29.26 ± 10.69 ** | 15.13 ± 7.51 ## | 10.00 ± 4.25 ## |
| Mean α-SMA immunoreactive cell numbers | 8.00 ± 5.73 | 192.40 ± 55.07 ** | 88.80 ± 28.96 ## | 48.00 ± 17.02 ## |
| Mean PAI-1 immunoreactive cell numbers | 82.40 ± 25.68 | 660.80 ± 127.15 ** | 315.20 ± 125.71 ## | 235.00 ± 113.28 ## |
| Mean p-Smad3 immunoreactive cell numbers | 17.60 ± 13.43 | 340.80 ± 110.16 ** | 137.60 ± 32.11 ## | 59.60 ± 22.78 ## |
| Mean TGF-β1 immunoreactive cell numbers | 14.20 ± 11.72 | 375.50 ± 105.59 ** | 159.40 ± 48.34 ## | 73.80 ± 26.72 ## |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.H.; Ku, S.K.; Cho, I.J.; Lee, J.H.; Na, C.-S.; Ki, S.H. Neoagarooligosaccharide Protects against Hepatic Fibrosis via Inhibition of TGF-β/Smad Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 2041. https://doi.org/10.3390/ijms22042041
Yang JH, Ku SK, Cho IJ, Lee JH, Na C-S, Ki SH. Neoagarooligosaccharide Protects against Hepatic Fibrosis via Inhibition of TGF-β/Smad Signaling Pathway. International Journal of Molecular Sciences. 2021; 22(4):2041. https://doi.org/10.3390/ijms22042041
Chicago/Turabian StyleYang, Ji Hye, Sae Kwang Ku, IL Je Cho, Je Hyeon Lee, Chang-Su Na, and Sung Hwan Ki. 2021. "Neoagarooligosaccharide Protects against Hepatic Fibrosis via Inhibition of TGF-β/Smad Signaling Pathway" International Journal of Molecular Sciences 22, no. 4: 2041. https://doi.org/10.3390/ijms22042041
APA StyleYang, J. H., Ku, S. K., Cho, I. J., Lee, J. H., Na, C.-S., & Ki, S. H. (2021). Neoagarooligosaccharide Protects against Hepatic Fibrosis via Inhibition of TGF-β/Smad Signaling Pathway. International Journal of Molecular Sciences, 22(4), 2041. https://doi.org/10.3390/ijms22042041