
Novel Genetically Modified Mouse Model to Assess Soman-Induced Toxicity and Medical Countermeasure Efficacy: Human Acetylcholinesterase Knock-in Serum Carboxylesterase Knockout Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Development of GD Exposure Paradigm in KIKO Mice
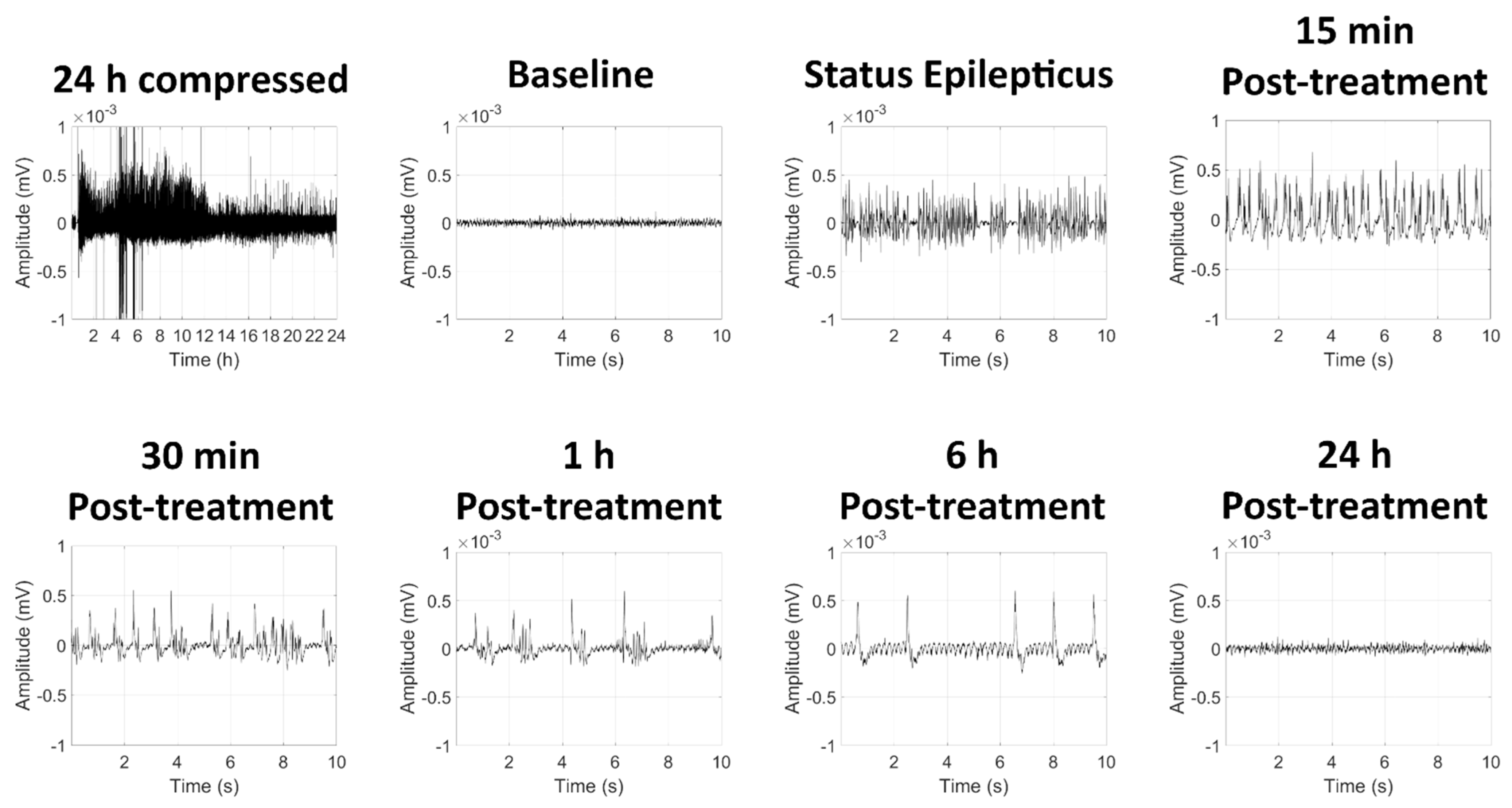
2.1.1. High-Dose GD Exposure Rapidly Induced Seizure in KIKO Mice; Delayed Midazolam Treatment Did Not Halt Seizure Activity
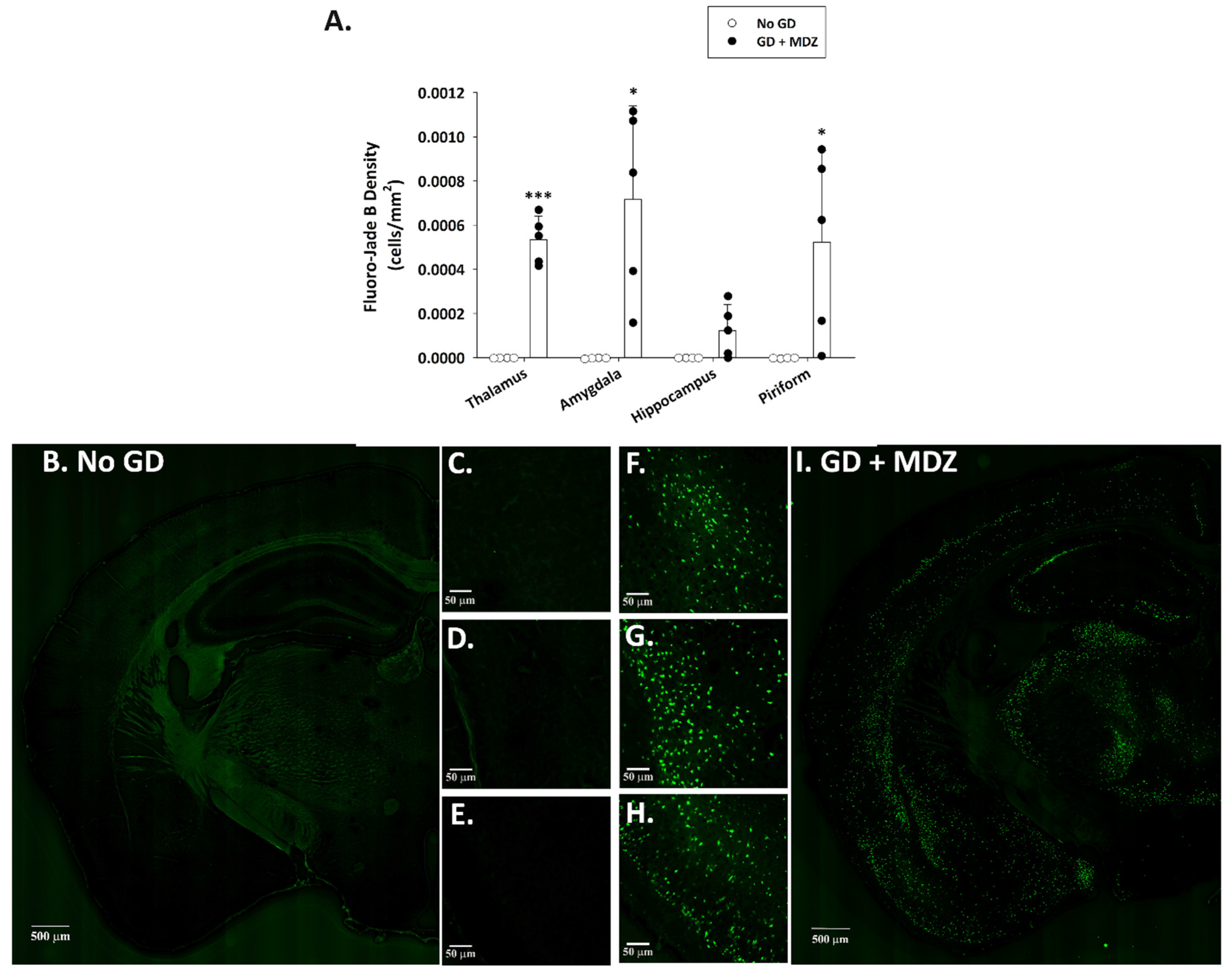
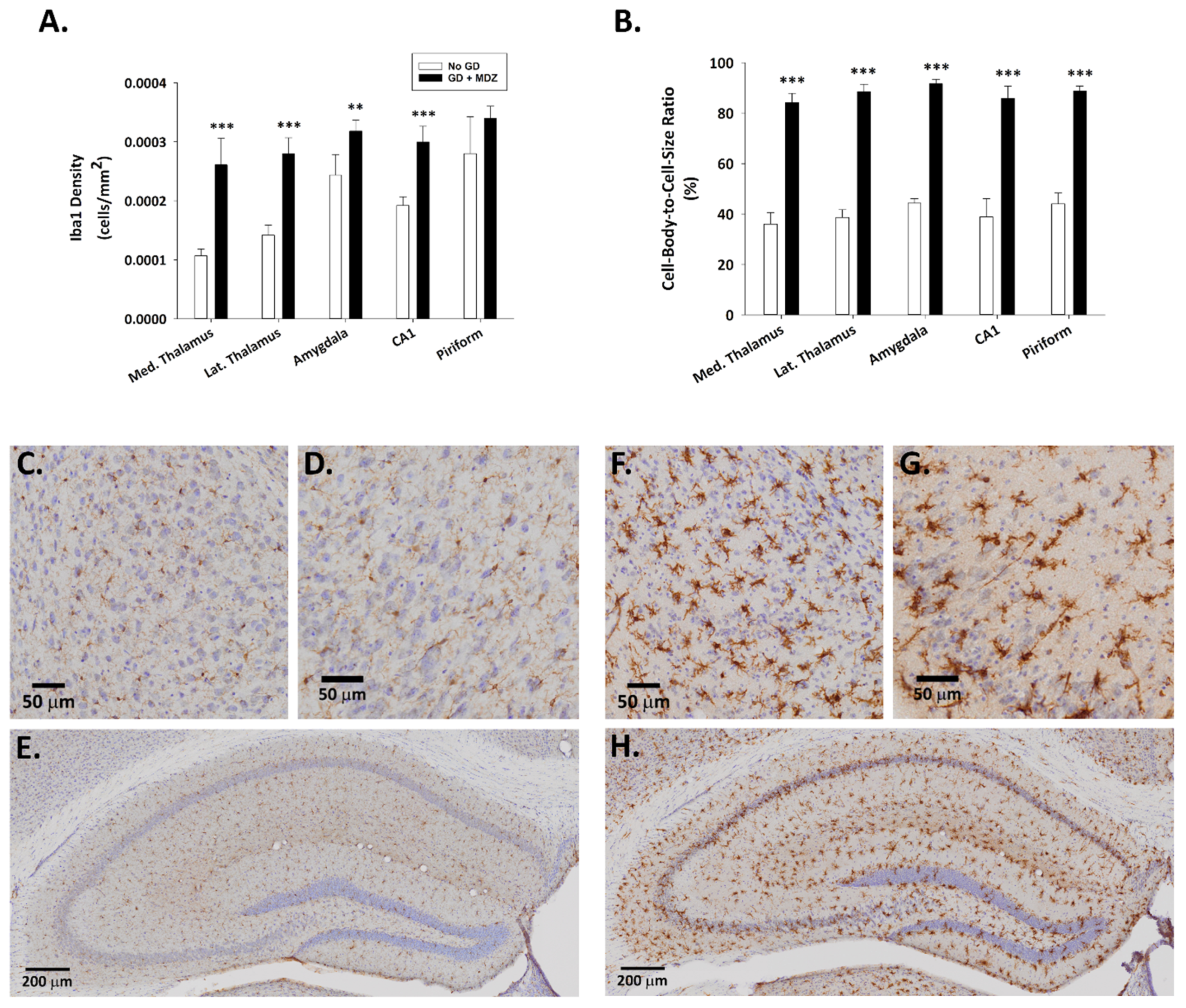
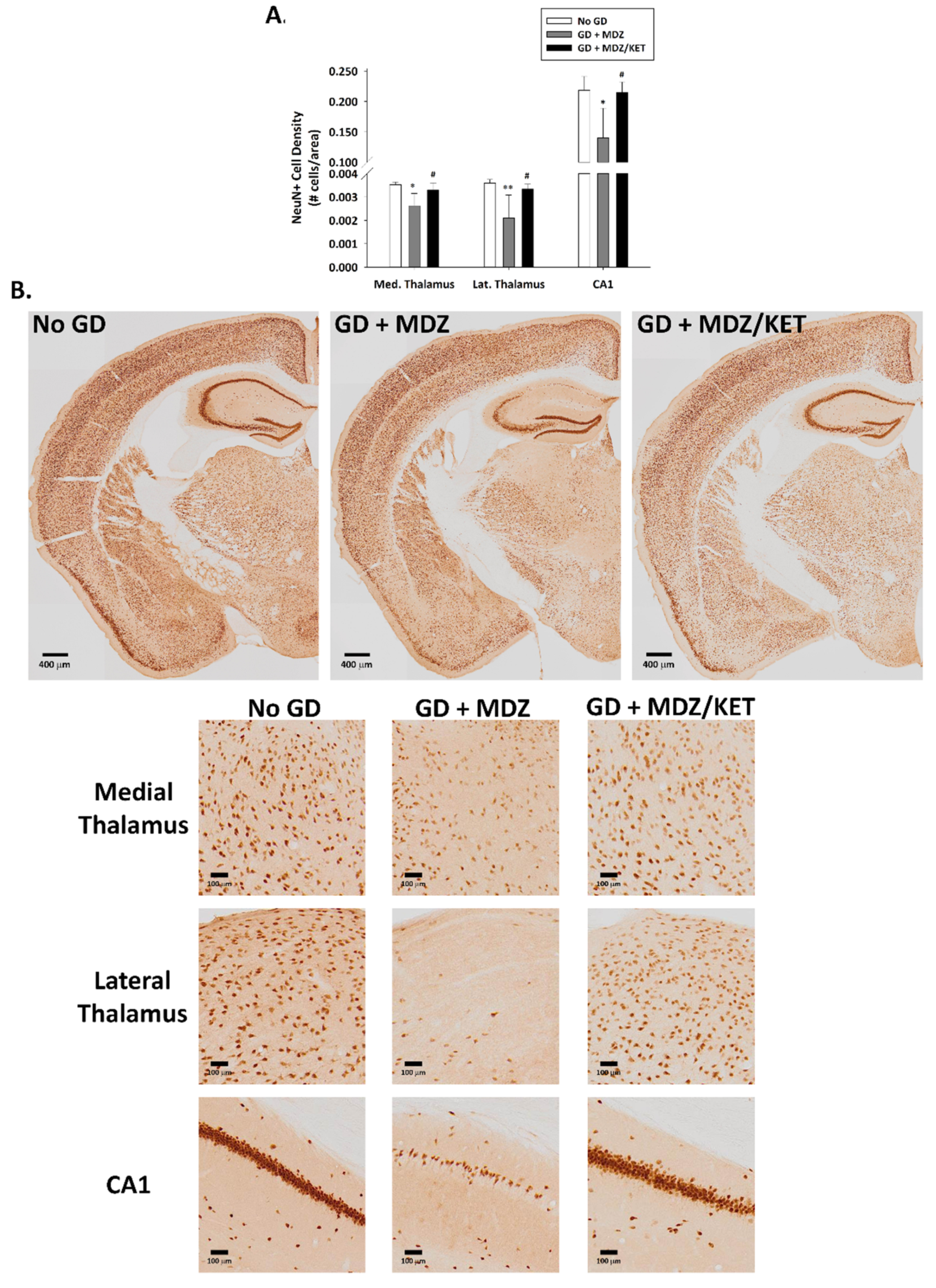
2.1.2. GD Exposure Resulted in Robust Neurodegeneration and Neuroinflammation in Brains of KIKO Mice at 24 h Following GD Exposure and Delayed Midazolam Treatment
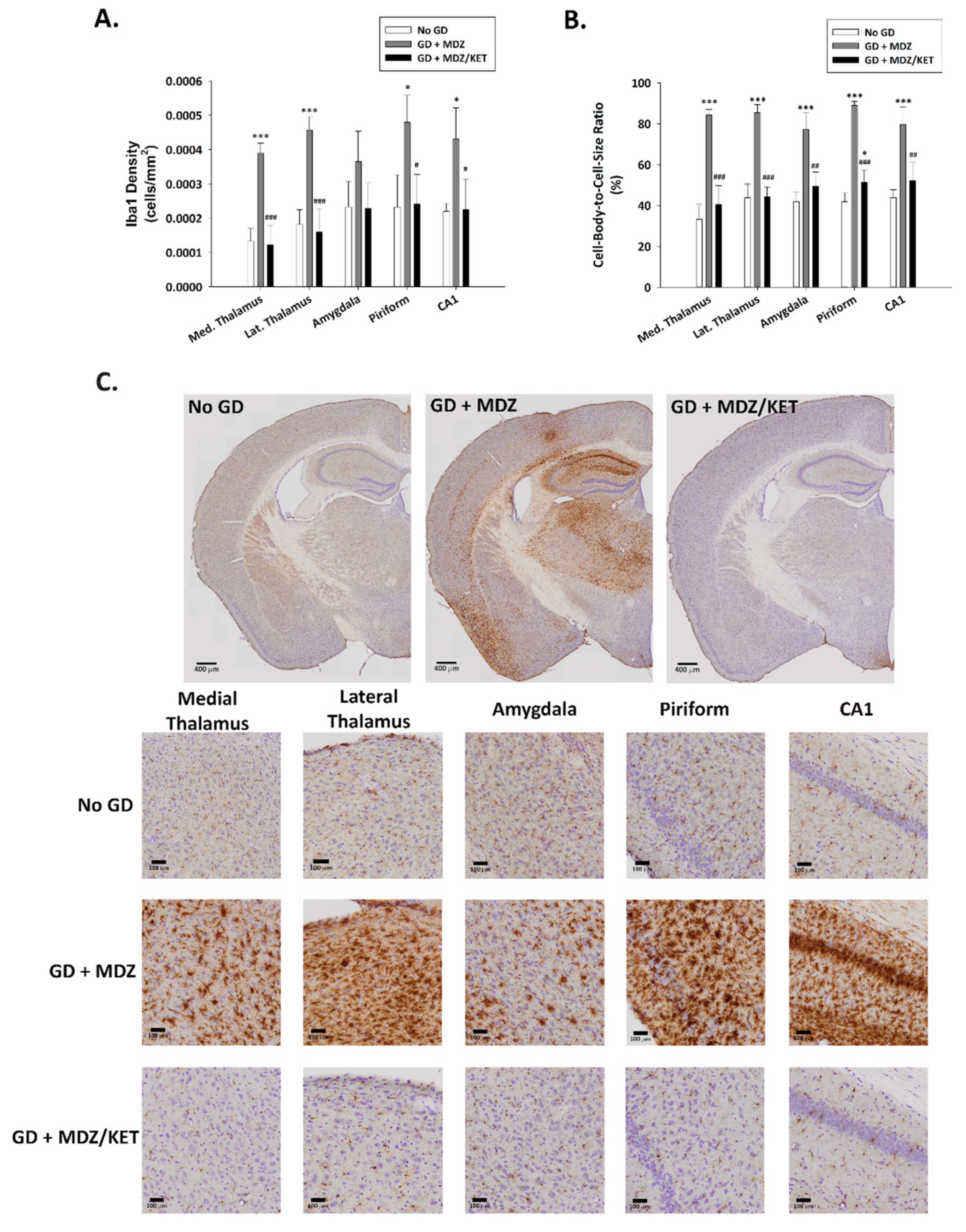
2.2. Evaluation of Ketamine as an Adjunct to Delayed Midazolam Treatment in GD-Exposed KIKO and Es1-/- Mice
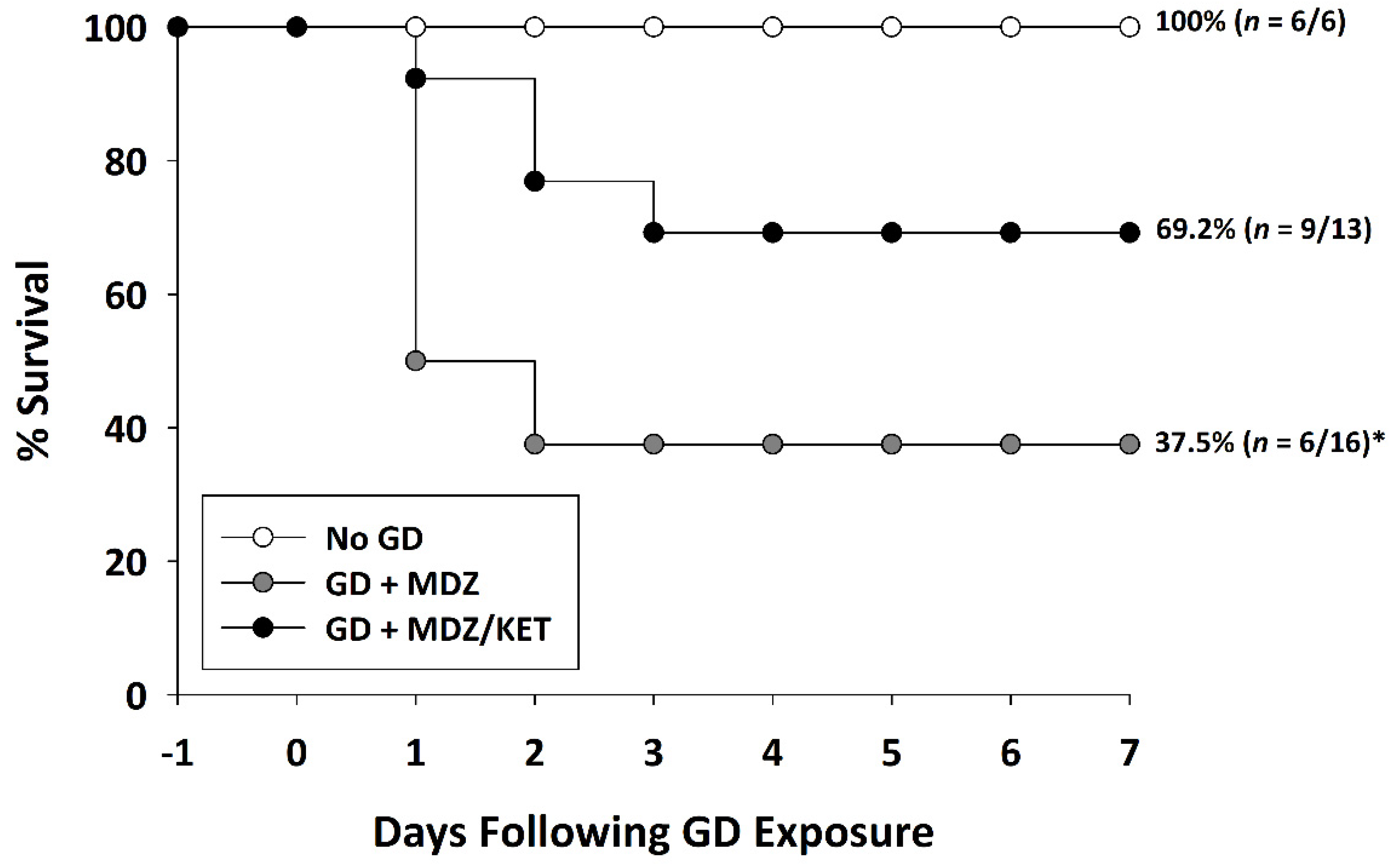
2.2.1. Ketamine Increased Survival in GD-Exposed KIKO and Es1-/- Mice Treated with Midazolam
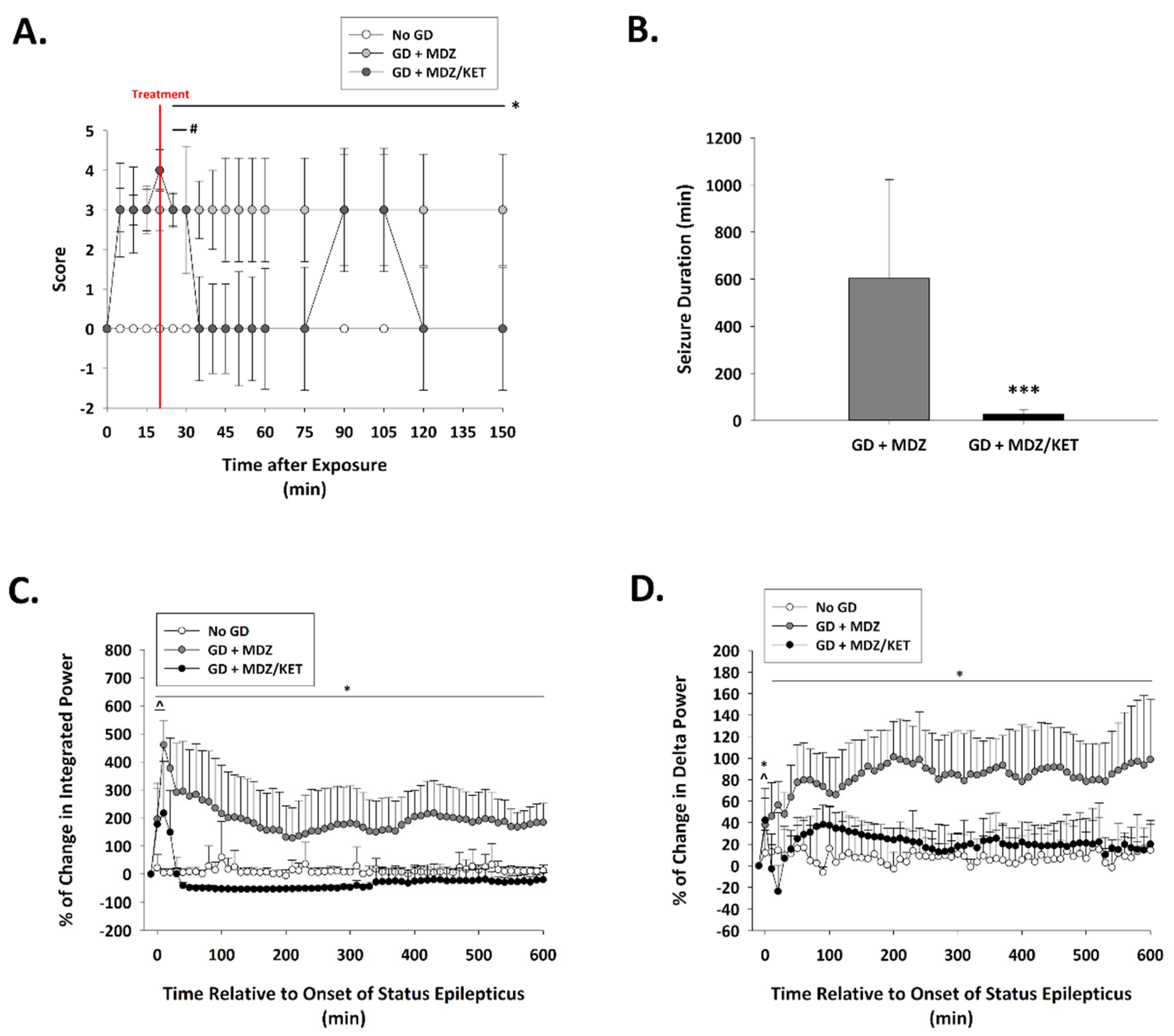
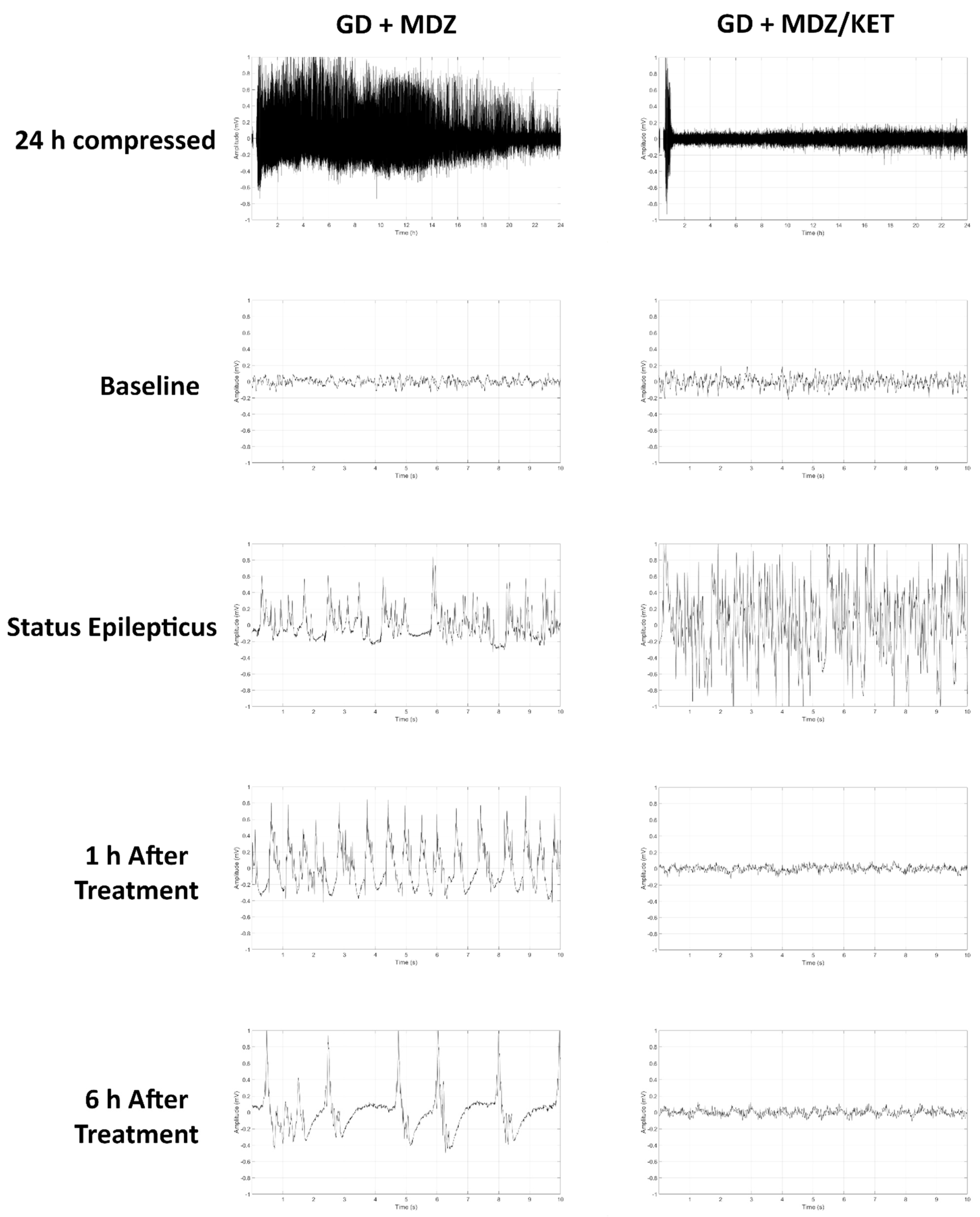
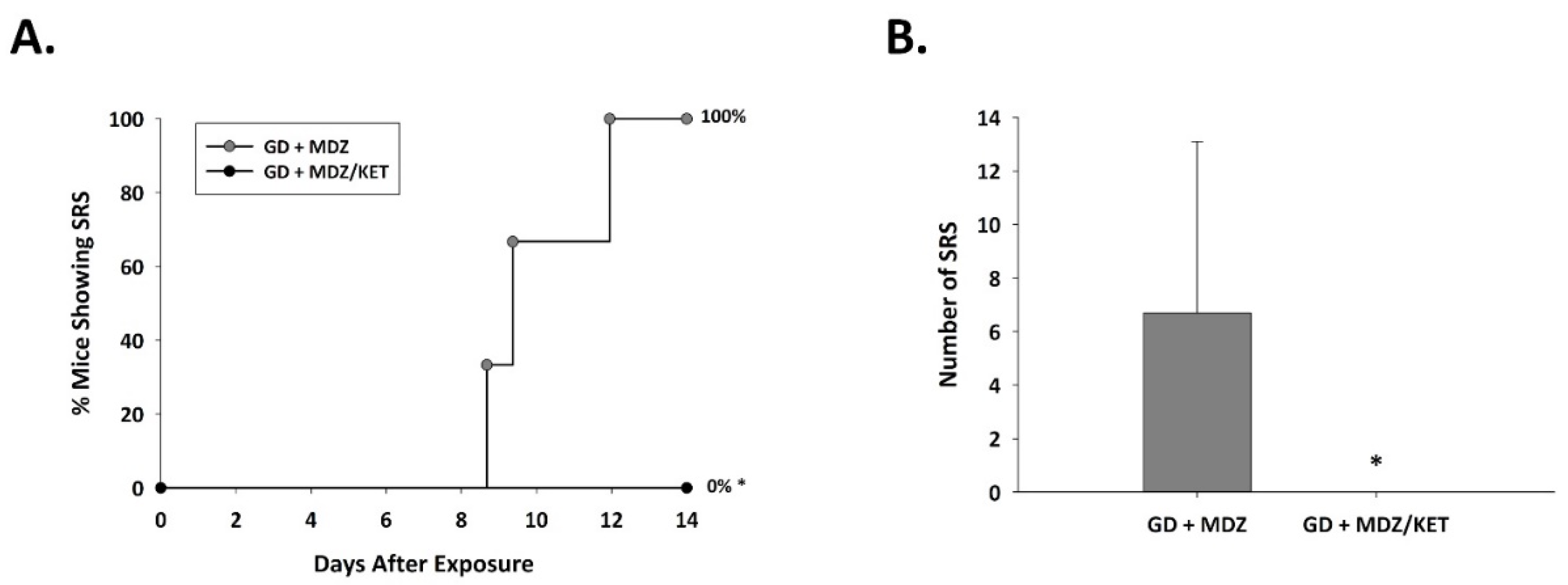
2.2.2. Ketamine in Combination with Midazolam at 15 Min after Seizure Onset Significantly Reduces Behavioral Seizure Severity, Acute Seizure Duration, and Changes in EEG Power in GD-Exposed KIKO Mice, and Prevents the Development of Spontaneous Recurrent Seizures
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Surgeries
4.3. GD Exposures and Treatments
4.3.1. Experiment 1
4.3.2. Experiment 2
4.4. Behavioral and Electroencephalographic (EEG) Seizure Activity
4.5. Brain Tissue Collection and Immunohistochemistry
4.6. Cell Counts
4.7. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Araki, T.; Kasai, K.; Yamasue, H.; Kato, N.; Kudo, N.; Ohtani, T.; Nakagome, K.; Kirihara, K.; Yamada, H.; Abe, O.; et al. Association between lower P300 amplitude and smaller anterior cingulate cortex volume in patients with posttraumatic stress disorder: A study of victims of Tokyo subway sarin attack. Neuroimage 2005, 25, 43–50. [Google Scholar] [CrossRef]
- Yokoyama, K.; Araki, S.; Murata, K.; Nishikitani, M.; Okumura, T.; Ishimatsu, S.; Takasu, N.; White, R.F. Chronic neurobehavioral effects of Tokyo subway sarin poisoning in relation to posttraumatic stress disorder. Arch. Environ. Health. 1998, 53, 249–256. [Google Scholar] [CrossRef]
- Yamasue, H.; Abe, O.; Kasai, K.; Suga, M.; Iwanami, A.; Yamada, H.; Tochigi, M.; Ohtani, T.; Rogers, M.A.; Sasaki, T.; et al. Human brain structural change related to acute single exposure to sarin. Ann. Neurol. 2007, 61, 37–46. [Google Scholar] [CrossRef]
- Figueiredo, T.H.; Apland, J.P.; Braga, M.F.M.; Marini, A.M. Acute and long-term consequences of exposure to organophosphate nerve agents in humans. Epilepsia 2018, 59 (Suppl. 2), 92–99. [Google Scholar] [CrossRef]
- Jett, D.A.; Sibrizzi, C.A.; Blain, R.B.; Hartman, P.A.; Lein, P.J.; Taylor, K.W.; Rooney, A.A. A national toxicology program systematic review of the evidence for long-term effects after acute exposure to sarin nerve agent. Crit. Rev. Toxicol. 2020, 50, 474–490. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.K.; Wright, L.K.; de Araujo Furtado, M.; Stone, M.F.; Moffett, M.C.; Kelley, N.R.; Bourne, A.R.; Lumeh, W.Z.; Schultz, C.R.; Schwartz, J.E.; et al. Caramiphen edisylate as adjunct to standard therapy attenuates soman-induced seizures and cognitive deficits in rats. Neurotoxicol. Teratol. 2014, 44, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.K.; Wright, L.K.; Stone, M.F.; Schwartz, J.E.; Kelley, N.R.; Moffett, M.C.; Lee, R.B.; Lumley, L.A. The anticholinergic and antiglutamatergic drug caramiphen reduces seizure duration in soman-exposed rats: Synergism with the benzodiazepine diazepam. Toxicol. Appl. Pharmacol. 2012, 259, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.L.; Wright, L.K.; Connis, N.; Lumley, L.A. Characterizing the behavioral effects of nerve agent-induced seizure activity in rats: Increased startle reactivity and perseverative behavior. Pharmacol. Biochem. Behav. 2012, 100, 382–391. [Google Scholar] [CrossRef] [PubMed]
- McDonough, J.H., Jr.; Dochterman, L.W.; Smith, C.D.; Shih, T.M. Protection against nerve agent-induced neuropathology, but not cardiac pathology, is associated with the anticonvulsant action of drug treatment. Neurotoxicology 1995, 16, 123–132. [Google Scholar]
- Shih, T.M.; Duniho, S.M.; McDonough, J.H. Control of nerve agent-induced seizures is critical for neuroprotection and survival. Toxicol. Appl. Pharmacol. 2003, 188, 69–80. [Google Scholar] [CrossRef]
- De Groot, D.M.; Bierman, E.P.; Bruijnzeel, P.L.; Carpentier, P.; Kulig, B.M.; Lallement, G.; Melchers, B.P.; Philippens, I.H.; van Huygevoort, A.H. Beneficial effects of TCP on soman intoxication in guinea pigs: Seizures, brain damage and learning behaviour. J. Appl. Toxicol. 2001, 21 (Suppl. 1), S57–S65. [Google Scholar] [CrossRef] [PubMed]
- De Araujo Furtado, M.; Lumley, L.A.; Robison, C.; Tong, L.C.; Lichtenstein, S.; Yourick, D.L. Spontaneous recurrent seizures after status epilepticus induced by soman in Sprague-Dawley rats. Epilepsia 2010, 51, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Marrero-Rosado, B.; de Araujo Furtado, M.; Schultz, C.R.; Stone, M.; Kundrick, E.; Walker, K.; O’Brien, S.; Du, F.; Lumley, L.A. Soman-induced status epilepticus, epileptogenesis, and neuropathology in carboxylesterase knockout mice treated with midazolam. Epilepsia 2018, 59, 2206–2218. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, D.M.; Brecht, K.M.; O’Neill, B.L. The effect of carboxylesterase inhibition on interspecies differences in soman toxicity. Toxicol. Lett. 1987, 39, 35–42. [Google Scholar] [CrossRef]
- Maxwell, D.M. The specificity of carboxylesterase protection against the toxicity of organophosphorus compounds. Toxicol. Appl. Pharmacol. 1992, 114, 306–312. [Google Scholar] [CrossRef]
- Duysen, E.G.; Koentgen, F.; Williams, G.R.; Timperley, C.M.; Schopfer, L.M.; Cerasoli, D.M.; Lockridge, O. Production of ES1 plasma carboxylesterase knockout mice for toxicity studies. Chem. Res. Toxicol. 2011, 24, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Duysen, E.G.; Cashman, J.R.; Schopfer, L.M.; Nachon, F.; Masson, P.; Lockridge, O. Differential sensitivity of plasma carboxylesterase-null mice to parathion, chlorpyrifos and chlorpyrifos oxon, but not to diazinon, dichlorvos, diisopropylfluorophosphate, cresyl saligenin phosphate, cyclosarin thiocholine, tabun thiocholine, and carbofuran. Chem. Biol. Interact. 2012, 195, 189–198. [Google Scholar]
- Kundrick, E.; Marrero-Rosado, B.; Stone, M.; Schultz, C.; Walker, K.; Lee-Stubbs, R.B.; de Araujo Furtado, M.; Lumley, L.A. Delayed midazolam dose effects against soman in male and female plasma carboxylesterase knockout mice. Ann. N. Y. Acad. Sci. 2020, 1479, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Cadieux, C.L. A novel genetically modified mouse model for evaluating nerve agent countermeasures. In Proceedings of the 3rd International CBRNE Research & Innovation Conference, Nantes, France, 20–23 May 2019. [Google Scholar]
- Reinhardt, B.C. Of mice and human. In JSTO in the News; Defense Threat Reduction Agency: Fort Belvoir, VA, USA, 2020; pp. 7–9. [Google Scholar]
- Mazarati, A.M.; Baldwin, R.A.; Sankar, R.; Wasterlain, C.G. Time-dependent decrease in the effectiveness of antiepileptic drugs during the course of self-sustaining status epilepticus. Brain Res. 1998, 814, 179–185. [Google Scholar] [CrossRef]
- Goodkin, H.P.; Liu, X.; Holmes, G.L. Diazepam terminates brief but not prolonged seizures in young, naive rats. Epilepsia 2003, 44, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Apland, J.P.; Aroniadou-Anderjaska, V.; Figueiredo, T.H.; Rossetti, F.; Miller, S.L.; Braga, M.F. The limitations of diazepam as a treatment for nerve agent-induced seizures and neuropathology in rats: Comparison with UBP302. J. Pharmacol. Exp. Ther. 2014, 351, 359–372. [Google Scholar] [CrossRef]
- Niquet, J.; Lumley, L.; Baldwin, R.; Rossetti, F.; Schultz, M.; de Araujo Furtado, M.; Suchomelova, L.; Naylor, D.; Franco-Estrada, I.; Wasterlain, C.G. Early polytherapy for benzodiazepine-refractory status epilepticus. Epilepsy Behav. 2019, 101 Pt B, 106367. [Google Scholar] [CrossRef]
- Niquet, J.; Lumley, L.; Baldwin, R.; Rossetti, F.; Suchomelova, L.; Naylor, D.; Estrada, I.B.F.; Schultz, M.; Furtado, M.A.; Wasterlain, C.G. Rational polytherapy in the treatment of cholinergic seizures. Neurobiol. Dis. 2020, 133, 104537. [Google Scholar] [CrossRef] [PubMed]
- Lumley, L.; Miller, D.; Muse, W.T.; Marrero-Rosado, B.; de Araujo Furtado, M.; Stone, M.; McGuire, J.; Whalley, C. Neurosteroid and benzodiazepine combination therapy reduces status epilepticus and long-term effects of whole-body sarin exposure in rats. Epilepsia Open 2019, 4, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Naylor, D.E.; Liu, H.; Wasterlain, C.G. Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J. Neurosci. 2005, 25, 7724–7733. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S. Mechanism-based novel antidotes for organophosphate neurotoxicity. Curr. Opin. Toxicol. 2019, 14, 35–45. [Google Scholar] [CrossRef]
- Niquet, J.; Baldwin, R.; Norman, K.; Suchomelova, L.; Lumley, L.; Wasterlain, C.G. Midazolam-ketamine dual therapy stops cholinergic status epilepticus and reduces Morris water maze deficits. Epilepsia 2016, 57, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Lumley, L.A.; Rossetti, F.; de Araujo Furtado, M.; Marrero-Rosado, B.; Schultz, C.R.; Schultz, M.K.; Niquet, J.; Wasterlain, C.G. Dataset of EEG power integral, spontaneous recurrent seizure and behavioral responses following combination drug therapy in soman-exposed rats. Data Brief 2019, 27, 104629. [Google Scholar] [CrossRef]
- Marrero-Rosado, B.M.; de Araujo Furtado, M.; Kundrick, E.R.; Walker, K.A.; Stone, M.F.; Schultz, C.R.; Nguyen, D.A.; Lumley, L.A. Ketamine as adjunct to midazolam treatment following soman-induced status epilepticus reduces seizure severity, epileptogenesis, and brain pathology in plasma carboxylesterase knockout mice. Epilepsy Behav. 2020, 111, 107229. [Google Scholar] [CrossRef]
- Lewine, J.D.; Weber, W.; Gigliotti, A.; McDonald, J.D.; Doyle-Eisele, M.; Bangera, N.; Paulson, K.; Magcalas, C.; Hamilton, D.A.; Garcia, E.; et al. Addition of ketamine to standard-of-care countermeasures for acute organophosphate poisoning improves neurobiological outcomes. Neurotoxicology 2018, 69, 37–46. [Google Scholar] [CrossRef]
- Dorandeu, F.; Carpentier, P.; Baubichon, D.; Four, E.; Bernabe, D.; Burckhart, M.F.; Lallement, G. Efficacy of the ketamine-atropine combination in the delayed treatment of soman-induced status epilepticus. Brain Res. 2005, 1051, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Dorandeu, F.; Baille, V.; Mikler, J.; Testylier, G.; Lallement, G.; Sawyer, T.; Carpentier, P. Protective effects of S+ ketamine and atropine against lethality and brain damage during soman-induced status epilepticus in guinea-pigs. Toxicology 2007, 234, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, D.G. Neuroprotective effect of ketamine administered after status epilepticus onset. Epilepsia 1995, 36, 186–195. [Google Scholar] [CrossRef]
- Johnson, E.A.; Kan, R.K. The acute phase response and soman-induced status epilepticus: Temporal, regional and cellular changes in rat brain cytokine concentrations. J. Neuroinflammation 2010, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Ikegaya, Y.; Koyama, R. The effects of microglia- and astrocyte-derived factors on neurogenesis in health and disease. Eur. J. Neurosci. 2020. [Google Scholar] [CrossRef]
- Luo, C.; Koyama, R.; Ikegaya, Y. Microglia engulf viable newborn cells in the epileptic dentate gyrus. Glia. 2016, 64, 1508–1517. [Google Scholar] [CrossRef]
- Dorandeu, F.; Dhote, F.; Barbier, L.; Baccus, B.; Testylier, G. Treatment of status epilepticus with ketamine, are we there yet? CNS Neurosci. Ther. 2013, 19, 411–427. [Google Scholar] [CrossRef]
- Ballough, G.P.; Newmark, J.; Levine, E.S.; Filbert, M.G. Neuroprotection as a treatment for nerve agent survivors. In Medical Aspects of Chemical Warfare; Tuorinsky, S.D., Ed.; Department of the Army, Office of The Surgeon General and Borden Institute: Washington, DC, USA, 2008; pp. 221–242. [Google Scholar]
- Martin, B.S.; Kapur, J. A combination of ketamine and diazepam synergistically controls refractory status epilepticus induced by cholinergic stimulation. Epilepsia 2008, 49, 248–255. [Google Scholar] [CrossRef]
- Philippens, I.H.; Melchers, B.P.; de Groot, D.M.; Wolthuis, O.L. Behavioral performance, brain histology, and EEG sequela after immediate combined atropine/diazepam treatment of soman-intoxicated rats. Pharmacol. Biochem. Behav. 1992, 42, 711–719. [Google Scholar] [CrossRef]
- McDonough, J.H., Jr.; Clark, T.R.; Slone, T.W., Jr.; Zoeffel, D.; Brown, K.; Kim, S.; Smith, C.D. Neural lesions in the rat and their relationship to EEG delta activity following seizures induced by the nerve agent soman. Neurotoxicology 1998, 19, 381–391. [Google Scholar]
- Carpentier, P.; Foquin, A.; Dorandeu, F.; Lallement, G. Delta activity as an early indicator for soman-induced brain damage: A review. Neurotoxicology 2001, 22, 299–315. [Google Scholar] [CrossRef]
- Müller, C.J.; Gröticke, I.; Bankstahl, M.; Löscher, W. Behavioral and cognitive alterations, spontaneous seizures, and neuropathology developing after a pilocarpine-induced status epilepticus in C57BL/6 mice. Experimental Neurology 2009, 219, 284–297. [Google Scholar] [CrossRef]
- Dhote, F.; Carpentier, P.; Barbier, L.; Peinnequin, A.; Baille, V.; Pernot, F.; Testylier, G.; Beaup, C.; Foquin, A.; Dorandeu, F. Combinations of ketamine and atropine are neuroprotective and reduce neuroinflammation after a toxic status epilepticus in mice. Toxicol. Appl. Pharmacol. 2012, 259, 195–209. [Google Scholar] [CrossRef]
- Wang, C.Q.; Ye, Y.; Chen, F.; Han, W.C.; Sun, J.M.; Lu, X.; Guo, R.; Cao, K.; Zheng, M.J.; Liao, L.C. Posttraumatic administration of a sub-anesthetic dose of ketamine exerts neuroprotection via attenuating inflammation and autophagy. Neuroscience 2017, 343, 30–38. [Google Scholar] [CrossRef]
- Chang, E.I.; Zarate, M.A.; Rabaglino, M.B.; Richards, E.M.; Arndt, T.J.; Keller-Wood, M.; Wood, C.E. Ketamine decreases inflammatory and immune pathways after transient hypoxia in late gestation fetal cerebral cortex. Physiol Rep. 2016, 4, e12741. [Google Scholar] [CrossRef] [PubMed]
- National Research Council. Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2010. [Google Scholar]
- Silbergleit, R.; Durkalski, V.; Lowenstein, D.; Conwit, R.; Pancioli, A.; Palesch, Y.; Barsan, W.; Investigators, N. Intramuscular versus intravenous therapy for prehospital status epilepticus. N. Engl. J. Med. 2012, 366, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Nissinen, J.; Lukasiuk, K.; Pitkanen, A. Is mossy fiber sprouting present at the time of the first spontaneous seizures in rat experimental temporal lobe epilepsy? Hippocampus 2001, 11, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.M.; Raine, L.; Fanger, H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: A comparison between ABC and unlabeled antibody (PAP) procedures. J. Histochem. Cytochem. 1981, 29, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Hovens, I.B.; Nyakas, C.; Schoemaker, R.G. A novel method for evaluating microglial activation using ionized calcium-binding adaptor protein-1 staining: Cell body to cell size ratio. Neuroimmunol. Neuroinflammation 2014, 1, 82–88. [Google Scholar] [CrossRef]
- Tynan, R.J.; Naicker, S.; Hinwood, M.; Nalivaiko, E.; Buller, K.M.; Pow, D.V.; Day, T.A.; Walker, F.R. Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav. Immun. 2010, 24, 1058–1068. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marrero-Rosado, B.M.; Stone, M.F.; de Araujo Furtado, M.; Schultz, C.R.; Cadieux, C.L.; Lumley, L.A. Novel Genetically Modified Mouse Model to Assess Soman-Induced Toxicity and Medical Countermeasure Efficacy: Human Acetylcholinesterase Knock-in Serum Carboxylesterase Knockout Mice. Int. J. Mol. Sci. 2021, 22, 1893. https://doi.org/10.3390/ijms22041893
Marrero-Rosado BM, Stone MF, de Araujo Furtado M, Schultz CR, Cadieux CL, Lumley LA. Novel Genetically Modified Mouse Model to Assess Soman-Induced Toxicity and Medical Countermeasure Efficacy: Human Acetylcholinesterase Knock-in Serum Carboxylesterase Knockout Mice. International Journal of Molecular Sciences. 2021; 22(4):1893. https://doi.org/10.3390/ijms22041893
Chicago/Turabian StyleMarrero-Rosado, Brenda M., Michael F. Stone, Marcio de Araujo Furtado, Caroline R. Schultz, C. Linn Cadieux, and Lucille A. Lumley. 2021. "Novel Genetically Modified Mouse Model to Assess Soman-Induced Toxicity and Medical Countermeasure Efficacy: Human Acetylcholinesterase Knock-in Serum Carboxylesterase Knockout Mice" International Journal of Molecular Sciences 22, no. 4: 1893. https://doi.org/10.3390/ijms22041893
APA StyleMarrero-Rosado, B. M., Stone, M. F., de Araujo Furtado, M., Schultz, C. R., Cadieux, C. L., & Lumley, L. A. (2021). Novel Genetically Modified Mouse Model to Assess Soman-Induced Toxicity and Medical Countermeasure Efficacy: Human Acetylcholinesterase Knock-in Serum Carboxylesterase Knockout Mice. International Journal of Molecular Sciences, 22(4), 1893. https://doi.org/10.3390/ijms22041893