1. Introduction
Type 2 diabetes mellitus (T2D) is a metabolic disease characterized by hyperglycemia as a result of impaired insulin secretion and action, estimated to affect 422 million people worldwide [
1]. The two hallmarks of the disease are insulin resistance in peripheral tissues, tightly associated with increased body mass index (BMI), and dysfunction of insulin-producing β-cells found in pancreatic islets. When β-cell failure occurs, these cells cannot compensate for increased insulin demand and cannot keep blood glucose levels within a normal range [
2]. The chronic exposure of β-cells to elevated glucose concentrations (glucotoxicity) as well as the combination of elevated concentrations of glucose and lipids (glucolipotoxicity), are thought to be the main factors responsible for the decline of β-cell function in T2D [
3,
4].
Numerous in vivo and in vitro models have been developed to study T2D relevant β-cell dysfunction [
5]. Rodent models of T2D have provided valuable insights into the regulation of β-cell function and its adaptation to pathological conditions [
6]. However, it is clear that rodent β-cells differ from human β-cells in parameters such as response to different stressors, proliferative capacity under insulin resistance, glucose uptake, kinetics of insulin secretion, cellular composition and architectural distribution, and transcriptional profile [
7]. Various rodent and human cell lines (as well as stem-cell-derived β-like cells) have been used in vitro, but they also present certain limitations such as lack of 3D structure, altered intercellular communication, absent paracrine interactions, and altered proliferation and stress response due to immortalization [
8]. Primary human islets are thus considered to be the best available tool for the study of β-cell function and failure in T2D. However, their experimental use is hindered by heterogeneity in islet size and cellular composition, variable degrees of exocrine tissue contamination (low purity), batch-to-batch differences in functionality [
9], and short ex vivo lifespan due in part to necrosis in the core of large islets [
10]. Previously, we have described how these challenges can be overcome with the use of standardized human islet microtissues, produced by enzymatic dissociation and controlled scaffold-free hanging-drop-based reaggregation of primary islet cells [
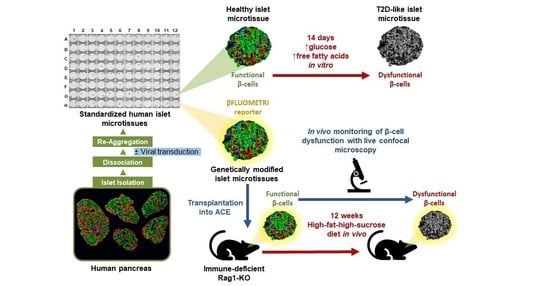
11]. Islet microtissues are uniform in size and cellular composition, and display long-term and stable functionality and viability during in vitro culture, thus could enable disease-modeling under T2D-relevant conditions.
Another historical challenge in studies with primary human islets has been their limited amenability to genetic manipulation. Viral transduction techniques, among the most frequently used methods to induce changes in gene expression, have delivered little success with intact islets as the transduction remains mostly restricted to cells in the periphery of the islets, due to limited accessibility of viral vectors to the inner cells of the 3D structure. Recently, several authors have reported methods for highly efficient viral transduction, including transduction of dissociated islet cells in suspension before pseudoislet formation [
12] or by using a combination of microfluidic flow and transient tissue expansion [
13]. Based on this, the production process of human islet microtissues theoretically opens the possibility to achieve highly efficient genetic manipulation by transducing islet cells while they are dispersed. Thus, islet microtissues are a standardized, functionally robust, and long-lived in vitro human islet model, that can be amenable to genetic manipulation and exploited in many applications for the study of β-cell dysfunction in T2D.
In a previous study we investigated β-cell dysfunction and β-cell insulin resistance by in vivo monitoring of reporter mouse islets transplanted into the anterior chamber of the eye (ACE) of mice exposed to a high-fat-high-sucrose diet (HFHSD) [
14]. Here, following the same approach, we aimed to study the dysfunction of human β-cells by measuring the functional β-cell mass in transduced islet microtissues transplanted into the ACE of diet-induced obese/diabetic mice. To achieve this goal we (1) demonstrated that human islet microtissues are suitable to study glucotoxicity/glucolipotoxicity-induced β-cell dysfunction, (2) established the ideal method for highly efficient viral transduction of islet microtissues, (3) ensured that islet microtissues survive, engraft, and maintain their functionality following transplantation into the ACE of immune-deficient mice, and (4) assessed changes in functional β-cell mass following HFHSD by quantification of glucose-responsive fluorescent reporters introduced to human islet microtissues prior to transplantation.
3. Discussion
The development of therapeutic approaches that can completely revert the course of β-cell dysfunction in T2D have been significantly slowed down by limitations of animal and in vitro models. Here we show how long-term in vitro exposure of human islet microtissues to glucotoxic and glucolipotoxic culture can reproducibly induce T2D relevant impairment in β-cell function with increased basal insulin secretion and decreased GSIS, the two hallmarks of β-cell dysfunction in T2D [
15,
16,
17,
18]. These models can now be applied for the discovery of mechanisms leading to loss of β-cell function under glucotoxicity/glucolipotoxicity as well as for the discovery of novel therapeutic agents with potential to prevent or reverse such dysfunction. Moreover, by establishing a method for highly-efficient viral transduction of fluorescent reporters and subsequent transplantation of the transduced islet microtissues, we were able to monitor human β-cell function and mass in vivo and concluded that loss of functional human β-cell mass also occurred in animal models of glucolipotoxicity, such as immune-deficient Rag1-KO mice fed HFHSD.
While the impact of high glucose concentrations (glucotoxicity) on the dysfunction and failure of β-cells in T2D is undisputed, the contribution of lipids (glucolipotoxicity) is more controversial and has recently been questioned [
26]. Our results show that despite glucotoxicity being the main driver of β-cell dysfunction, lipotoxicity alone can increase basal insulin secretion, and therefore, may contribute to the consequent insulin resistance in peripheral tissues. Additionally, we observed that FFAs potentiate the deleterious effects of glucotoxicity on β-cell function, i.e., lower concentrations of glucose impair GSIS.
Another important outcome of our study is the possibility of performing highly efficient genetic manipulation through viral transduction in a model of standard human islets that displays robust and stable functionality, which in turn is scalable and compatible with disease modeling. Decreased insulin secretion, but not insulin content, was observed in the conditions with highest transduction efficiencies indicating a negative impact on protein processing and secretion machinery. Yet, stimulus-secretion coupling mechanisms were partially maintained, as a fold stimulation of insulin secretion higher than 6.7 was obtained in all groups.
Both sugar and fat overconsumption are believed to lead to obesity, insulin resistance, and subsequent development of T2D. In our previous work [
14] we have shown that in rodents, β-cell function is impaired when specific dietary conditions (HFHSD) are brought onto a specific genetic background (B6). We showed that β-cell dysfunction occurs early during T2D progression and is provoked by the combination diet of high fat, which induces lipotoxicity, and sucrose, which triggers increased β-cell workload. Within 8 weeks of HFHSD, B6 mice became obese, developed impaired glucose tolerance and insulin resistance, and despite being hyperinsulinemic displayed non-compensatory insulin release, at least in part, due to reduced expression of syntaxin-1A [
14].
In the current study, we show that human islet microtissues forms stable and well vascularized grafts following transplantation into ACE of Rag1-KO mice. Following HFHSD, the Rag1-KO mice became obese, developed impaired glucose tolerance and insulin resistance, and showed non-compensatory stimulated insulin release despite being hyperinsulinemic. Additionally, the mice developed impaired β-cell function and reduced functional β-cell mass, comparable to B6 islets in B6 mice [
14]. The impairment in β-cell function could already be observed after 4 weeks of diet in mouse islets transplanted into B6 mice, whereas it took 8 weeks of HFHSD to observe similar dysfunction in human islet microtissues transplanted into Rag1-KO mice. In line with the results obtained by exposing islet microtissues to glucolipotoxicity in vitro, the results obtained in islet microtissues transplanted into the ACE of mice prove that human β-cells also become dysfunctional when exposed to glucolipotoxicity in an in vivo situation.
4. Materials and Methods
4.1. Reaggregated Human Islet Microtissue Production and Culture
InSphero 3D InSight human islet microtissues (InSphero AG, Schlieren, Switzerland) were produced by hanging-drop-based scaffold-free reaggregation of dispersed primary human islets obtained from Prodo Laboratories Inc. (Irvine, CA, USA). Consent was obtained from all next of kin, and there was no information on the identity of the donor for ethical and privacy reasons. For each production, between 10,000 and 20,000 islet equivalents (IEQs) were dispersed in dissociation solution (1992 µL 1X TrypLE Express, 12604013, Thermo Fisher Scientific, Waltham, MA, USA) plus 8 µL DNase I, 10 mg mL−1, to a final concentration of 40 µg mL−1 (10104159001, Sigma-Aldrich, St. Louis, MO, USA) by gentle pipetting at 37 °C. Remaining cell clumps were removed by filtering the cell suspension through a cell strainer (70 µm pore size). Then 2500 cells were seeded into each well of the InSphero Hanging Drop System and cultured for 4.5 days according to manufacturer’s instructions. The primary aggregates were then transferred to an Akura 96 plate to further mature for at least 2 more days before the start of the experiments. All experiments were performed 7–28 days after the start of the reaggregation. Islet microtissues were maintained in 3D InSight Human Islet Maintenance Medium (InSphero AG, Schlieren, Switzerland), renewed every 2–3 days.
4.2. Glucotoxic and Glucolipotoxic Cultures
To produce glucotoxic and glucolipotoxic medium, InSight Human Islet Maintenance Medium (InSphero AG, Schlieren, Switzerland) containing 5.5 mM glucose was supplemented with glucose to adjust the concentration to 8 mM, 11 mM, or 16.7 mM. For the production of the glucolipotoxic medium, separate solutions of sodium oleate (Sigma-Aldrich, Buchs, Switzerland, O7501) and sodium palmitate (P9767, Sigma-Aldrich, St. Louis, MO, USA) conjugated to FFA-free BSA (A8806, Sigma-Aldrich, St. Louis, MO, USA) in a 6:1 ratio (FFA:BSA) were prepared. These solutions were added to InSight Human Islet Maintenance Medium (with adjusted glucose concentrations) to reach a final concentration of 200 µM oleate and 100 µM palmitate. A solution with unconjugated FFA-free BSA was added to the culture medium of the healthy control group (5.5 mM glucose without FFA cocktail) to equilibrate the concentration of BSA (50 µM) in all treatment groups. Islet microtissues were cultured for 14 days in each corresponding medium, which was renewed every 2–3 days.
4.3. Adenoviral Transduction
The adenovirus encoding the β-cell fluorescent metabolic transcriptional-response indicator (βFLUOMETRI) was initially generated in [
14] as follows: pENTR1A.RIP1.DsRed2/rbGK.EGFP/CMV.Cerulean was generated by inserting the RIP1.DsRed2/rbGK.EGFP cassette from pd2.RIP1.DsRed2/rbGK.EGFP into pENTR1A (Thermo Fisher Scientific, Waltham, MA, USA) and adding a CMV.Cerulean cassette downstream of the rbGK.EGFP cassette. The 3 individual expression cassettes are separated by transcription blocker sequences from the pd2EGFP-Promoter (Takara Bio, Kusatsu, Japan). All constructs were verified by DNA sequencing. The RIP1.DsRed2/rbGK.EGFP/CMV.Cerulean cassette was transferred into the promoterless adenovirus plasmid pAd/PL-DEST (Thermo Fisher Scientific, Waltham, MA, USA) by the Gateway technique. The ViraPower Adenoviral Expression System (Thermo Fisher Scientific, Waltham, MA, USA) was used to generate a replication-deficient adenovirus, which was used for transduction of cells and islets.
To transduce before reaggregation, dissociated islet cells were placed in Eppendorf tubes with medium and the corresponding number of viral particles, to a final volume of 1 mL. Tubes were closed with Parafilm, placed inside a Falcon tube, and shaken on a rotator at 37 °C for 1.5 h. Next tubes were centrifuged, supernatants were removed and, following resuspension, cells were seeded in InSphero Hanging Drop System (InSphero AG, Schlieren, Switzerland). Islet microtissue production continued as explained in
Section 4.1.
To transduce during reaggregation, dissociated islet cells were mixed at the required concentration with the corresponding number of viral particles in medium. The cell suspensions containing the adenovirus were seeded in InSphero Hanging Drop System and islet microtissue production continued as explained in
Section 4.1, with the inclusion of two washing steps with InSight Human Islet Maintenance Medium after transferring the islet microtissues to an Akura 96 plate.
To transduce after reaggregation, islet microtissues were produced as in
Section 4.1. The Akura plates containing islet microtissues were centrifuged and medium was removed. 70 µL of InSight Human Islet Maintenance Medium containing the corresponding number of viral particles were added to each islet microtissue and the plates were placed in an incubator at 37 °C for 2 h. Then, 2 wash steps with InSight Human Islet Maintenance Medium were performed.
4.4. In Vitro Insulin Secretion Assay and Quantification of Insulin, ATP, and Caspase 3/7 Activity
To prepare the islet microtissues for static GSIS, culture medium corresponding to the last 48–72 h in culture was removed and stored for the analysis of chronic insulin secretion. Islet microtissues were washed twice with 70 µL Krebs-Ringer Hepes-bicarbonate Buffer (KRHB; 131 mM NaCl, 4.8 mM KCl, 1.3 mM CaCl2, 25 mM Hepes, 1.2 mMm KH2PO4, 1.2 mM MgSO4, 0.5% BSA) containing 2.8 mM glucose and equilibrated for 1 h in the same solution. GSIS was performed in the Akura 96 plate in 50 µL KRHB containing different glucose concentrations for 2 h. The supernatant was collected for ELISA analysis. After GSIS, the microtissues were lysed to analyze total ATP content using CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA with protease inhibitor cocktail G6521 from the same company) and a microplate reader (Infinite M1000, TECAN, Männedorf, Switzerland). The lysates were then used for assessment of total insulin content. Alternatively, the microtissues were lysed to analyze caspase 3/7 activity using Caspase-Glo® 3/7 Assay System (G8091, Promega, Madison, WI, USA). After proper dilutions in KRHB were performed, total and secreted insulin was quantified using Stellux Chemi Human Insulin ELISA (80-INSHU-CH10, Alpco, Salem, NH, USA). The obtained insulin and ATP values are shown unnormalized, expressed as ng of insulin or pmol of ATP per islet microtissue.
4.5. Immunofluorescence Confocal Microscopy and Image Analysis for Quantification of Adenoviral Transduction Efficiency and Homogeneity
Islet microtissues were treated with nuclear dye DRAQ5 for 2 h prior to fixation. Then, microtissues were fixed for 30 min in 2% paraformaldehyde (PFA) at RT. PFA was removed, microtissues were washed twice with PBS and kept in PBS with 0.05% azide until imaging.
Image acquisition and analysis was performed by Visikol (Hampton, NJ, USA). DRAQ5 treated microtissues were optically cleared with the Visikol® HISTO™ system (Visikol, NJ, Hampton, USA) and were imaged by 4-channel high content confocal microscopy using a CX7 LZR (Thermo Fisher Scientific, Waltham, MA, USA) in a 384-well glass bottom plates (CellCarrier-384 Ultra Microplates, PerkinElmer, Waltham, MA, USA, 6057300) with 5 µm Z-steps for the following fluorophores: Cerulean (Excitation/Emission max 435/475 nm), eGFP (Excitation/Emission max 488/510 nm), DsRed2 (Excitation/Emission max 556/584 nm), and DRAQ5 (Excitation/Emission max 647/681 nm).
Nuclei were detected, segmented, and counted using a customized CellProfiler 2.2.0 (
www.cellprofiler.org, Cambridge, MA, USA) pipeline. Coordinates (X,Y,Z) of nuclei were used to extract intensity values of CFP, EGFP, and DsRed2 channels for each nucleus. Transduction efficiency was calculated as a percentage by dividing the number of CFP positive cells by the total number of DRAQ5 nuclei in each islet microtissue. Transduction homogeneity was evaluated by calculating the transduction efficiency in three predefined regions of each islet. These regions were defined as follows: the radius of each nuclear point to the manually selected center-point of the spheroid was calculated. Radii distances were normalized by dividing by the maximum radius obtained for the microtissue. Nuclei were grouped into three bins based on normalized radius from center: bin 1, inner core (r0 = 0–0.333), bin 2, middle third (r0 = 0.334–0.666) and bin 3, outer third (0.666–1.0).
4.6. Immunofluorescence Confocal Microscopy and Image Analysis for Quantification of Proliferation and Apoptosis
Islet microtissues were washed twice with PBS and fixed for 30 min in 2% PFA at RT. PFA was removed, microtissues were washed twice with PBS and kept in PBS with 0.5% azide until immunofluorescent staining.
Staining, image acquisition and analysis was performed by Visikol (Hampton, NJ, USA). Islet microtissues were permeabilized with 0.2% Triton X-100 in PBS, followed by incubation in Penetration Buffer containing 0.2% Triton X-100 and 20% DMSO for 1 h each. The tissues were then blocked with a 6% donkey serum in PBS buffer solution for 1 h at 37 °C. The primary antibodies used for immunolabeling were Rabbit Anti-Ki67 (RB-1510, Thermo Fisher Scientific, Waltham, MA, USA) at a 1:200 dilution, Guinea Pig Anti-PDX1 (ab47308, Abcam, Cambridge, UK) at a 1:100 dilution, and Mouse Anti-Caspase3 (MA5-11516, Thermo Fisher Scientific, Waltham, MA, USA) at a 1:150 dilution in Antibody Buffer containing 3% donkey serum in PBS buffer. Tissues were washed 3 times between primary and secondary labeling steps with washing buffer composed of 0.2% Tween in PBS. The secondary antibody dilutions used were 1:200 for Goat Anti-Guinea Pig Alexa 488, Anti-Rabbit Alexa 568, and Anti-Mouse Alexa 674, along with DAPI at a 1:1000 dilution (all from Thermo Fisher Scientific, Waltham, MA, USA). Tissues were washed and transferred to the film-bottomed Akura™ 384 plate (InSphero AG, Switzerland), and cleared with Visikol® HISTO™ system (Visikol, NJ, Hampton, USA).
High content imaging of islet microtissues was performed using a CX7-LZR (Thermo Fisher Scientific, Waltham, MA, USA) platform. Twenty optical sections were acquired at 5 µm intervals for each well/channel with 20X magnification. Image stacks of the individual microtissues were quantified using customized CellProfiler 2.2.0 (
www.cellprofiler.org, Cambridge, MA, USA) pipelines.
4.7. Animals and Diet
Male NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) and male B6.129S7-Rag1tm1Mom/J (Rag1-KO) mice were purchased at 2 months of age from The Jackson Laboratory (Bar Harbor, ME, USA). After delivery, mice were left to adapt to the animal core facility for 1 week before the start of the experiment. All mice were group-housed at a 12/12 h dark-light-cycle with free access to food and water. If not otherwise indicated, the mice received a normal chow diet (R70, Lantmännen, Klimstad, Sweden). Starting at month 3, NSG and Rag1-KO mice received a HFHSD, consisting of a solid HFD (60% kcal from fat, TD.06414, Envigo, Venray, Netherlands) and 32% sucrose solved in tap water, for 16 weeks. A control diet was given to the control group: Managed formulation purified ingredient diet (#5P76, LabDiet, St. Louis, MO, USA) and tap water. The control diet given to the control group differs from the R70 diet all animals received before the diet intervention. Consequently, all the animals underwent a change of diet. All experiments were performed in accordance with the Karolinska Institutet’s guidelines for the care and use of animals in research and were approved by the institute’s Animal Ethics Committee.
4.8. Transplantation of Pancreatic Islets into the ACE
Islets microtissues were transplanted into the ACE of 2 months old NSG and Rag1-KO mice using a technique described previously [
22]. Briefly, under anesthesia, islets of Langerhans were transplanted into the ACE using a glass cannula after generating a puncture in the cornea with a 27-gauge needle. Great care was taken to avoid bleeding and damage to the iris. Mice were injected subcutaneously with Temgesic (0.1 mL/kg, RB Pharmaceuticals Limited, Berkshire, UK) for postoperative analgesia.
4.9. In Vivo Imaging of Intraocular Islet Grafts and Image Analysis
Islet grafts were imaged in vivo beginning 1 month after transplantation and at 4, 8 and 12 weeks after start of diet intervention. An upright laser scanning confocal microscope (Leica TCSSP5, LEICA Microsystems, Wetzlar, Germany) equipped with a long-distance water-dipping objective (Leica HXC-APO10×/0.30 NA) and a custom-built stereotaxic head holder allowing positioning of the mouse eye containing the engrafted islets toward the objective was used. Viscotears (Théa Nordic AB, Örebro, Sweden) was used as an immersion liquid between the eye and the objective, and isoflurane was used to anesthetize the mice during in vivo imaging. Islets were imaged as 3D-stacks with 3 µm step-size. After imaging, the mice were allowed to recover from anesthesia and an IPGTT was performed. At 4 h post glucose injection, a second imaging was performed using the same setting as described above to obtain the 4 h-image set.
β-cell fluorescent metabolic transcriptional response indicator (βFLUOMETRI): Cerulean fluorescence was excited at 405 nm and detected at 460 to 490 nm. EGFP fluorescence was excited at 488 nm and detected at 505 to 535 nm. DsRed2 fluorescence was excited at 561 nm and detected at 580 to 650 nm. Backscatter signal from the 561 nm excitation was collected at 555 to 565 nm. Channel crosstalk was avoided by using in-between-lines sequential imaging combining CFP (Cerulean), backscatter, and DsRed2 signal and separating the EGFP signal.
Image analysis for in vitro experiments was performed as described in [
14] using ImageJ. Average fluorescence intensities for each cell were determined for t = 60 min (start) and t = 240 min. Images derived from in vivo imaging were analyzed using Leica LAS software (Leica Microsystems). For each analyzed cell, fluorescence intensity for all 3 fluorescent dyes (CFP, EGFP, DsRed2) was determined before glucose stimulation and 4 h after glucose stimulation. To calculate the change in promoter activity after glucose stimulation as readout for β-cell functionality, fluorescence intensities for each cell were determined at the beginning of the experiment and 4 h after glucose stimulation. Cells were identified using the DsRed2-fluorescence signal thereby ensuring the analysis of β-cells. Promoter activation was calculated as follows: ((EGFP/DsRed2
4h-Background
4h)/(CFP
4h-Background
4h))/((EGFP/DsRed2
start-Background
start)/(CFP
start-Background
start)). To determine functional β-cell mass as a percentage of glucose responsive cells, cells with a promoter activation >1.15 were considered responsive.
Imaging of vascularization: for blood vessel imaging mice were injected intravenously with 100 μL TRITC-labelled dextran (2.000.000 MW, 5 mg/mL, Thermo Fisher Scientific, Waltham, MA, USA). TRITC fluorescence was excited at 555 nm and detected at 580 to 620 nm. Backscatter signal from the 633 nm excitation was collected at 630 to 640 nm.
4.10. IPGTT
To determine glucose tolerance, blood glucose levels were measured in mice fasted for 6 h (or together with the imaging after overnight fasting) at basal state (0 min), 5, 10, 30, 60, and 120 min after intraperitoneal glucose injection (2 g/kg body weight, dissolved in PBS). The results were depicted as area under the curve (AUC) of the IPGTT. Glucose concentrations were measured using the Accu-Chek Aviva monitoring system (Roche, Basel, Switzerland).
4.11. IPITT
To measure whole body insulin resistance, the IPITT was performed determining glucose blood levels after an insulin challenge. Blood glucose concentration was measured in mice fasted for 6 h (basal state, 0 min). Then, mice were intraperitoneally injected with insulin (0.25 U/kg body weight, diluted in PBS, Novo Nordisk, Bagsværd, Denmark) followed by intraperitoneal glucose administration (1 g/kg body weight) and blood glucose concentrations were determined at 15, 30, 60, 90, and 120 min after glucose injection. The results were depicted as AUC of the IPITT.
4.12. Body Weight and Fasting Blood Glucose
Body weight and fasting blood glucose were measured after 6 h fasting time or overnight fasting.
4.13. Plasma Biology
Blood samples were collected at different time points during the IPGTT, centrifuged at 5000× g during 30 min for plasma obtention and preserved at −20 °C until use. Ultrasensitive mouse enzyme-linked immunosorbent assay (ELISA) kits (CrystalChem, Elk Grove Village, IL, USA) were used to analyze plasma insulin and C-Peptide.
4.14. Statistics
GraphPad Prism 9 (GraphPad, San Diego, CA, USA), Origin 2015 64 Bit (OriginLab, Northampton, MA, USA) and Microsoft Office Excel (Microsoft, Redmont, WA, USA) were used for statistical analysis.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}














