
Conventional and Unconventional Mechanisms by which Exocytosis Proteins Oversee β-cell Function and Protection

Abstract
1. Introduction
2. Exocytosis Proteins and β-Cell Function
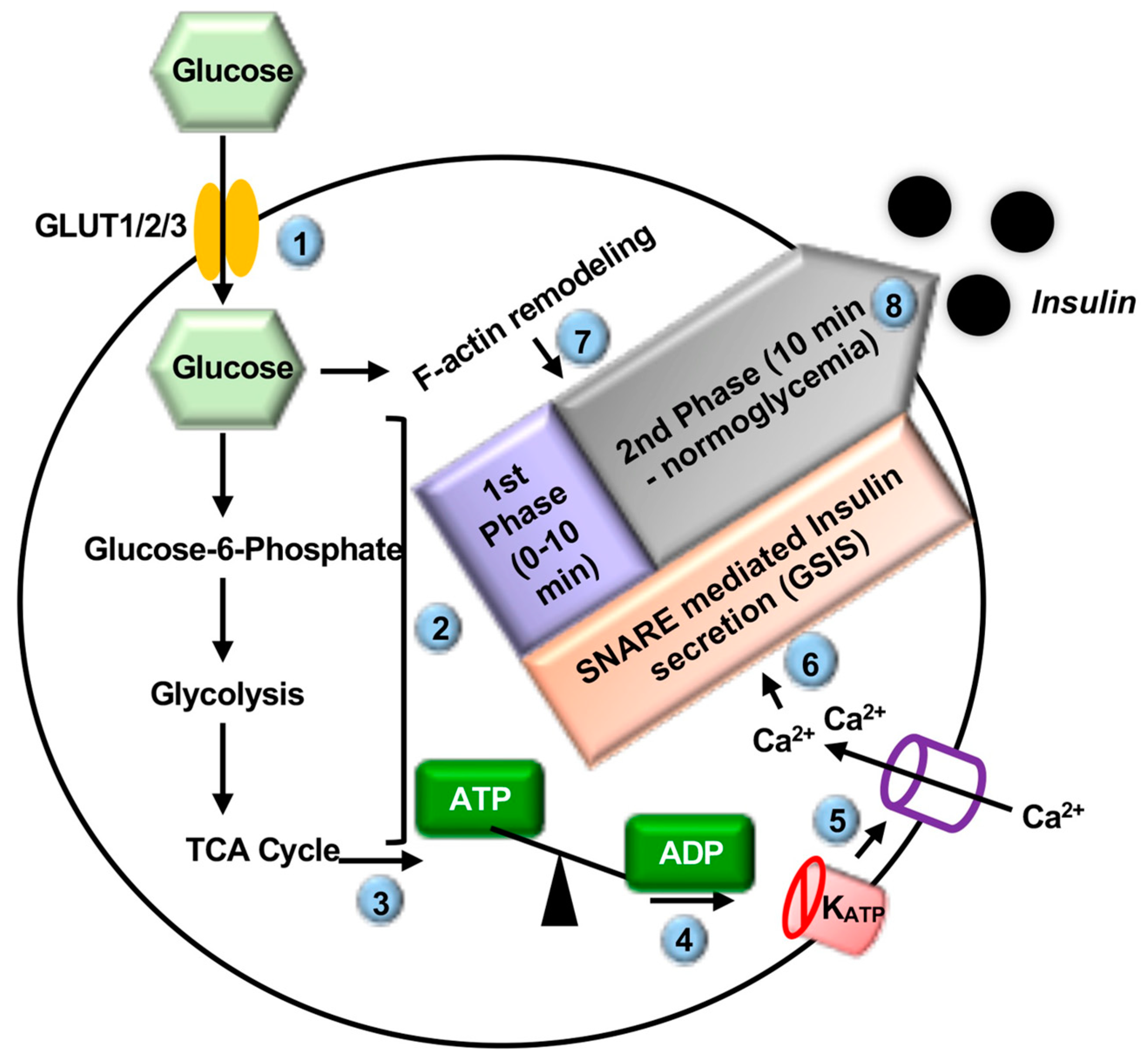
2.1. Introduction to Insulin Secretion
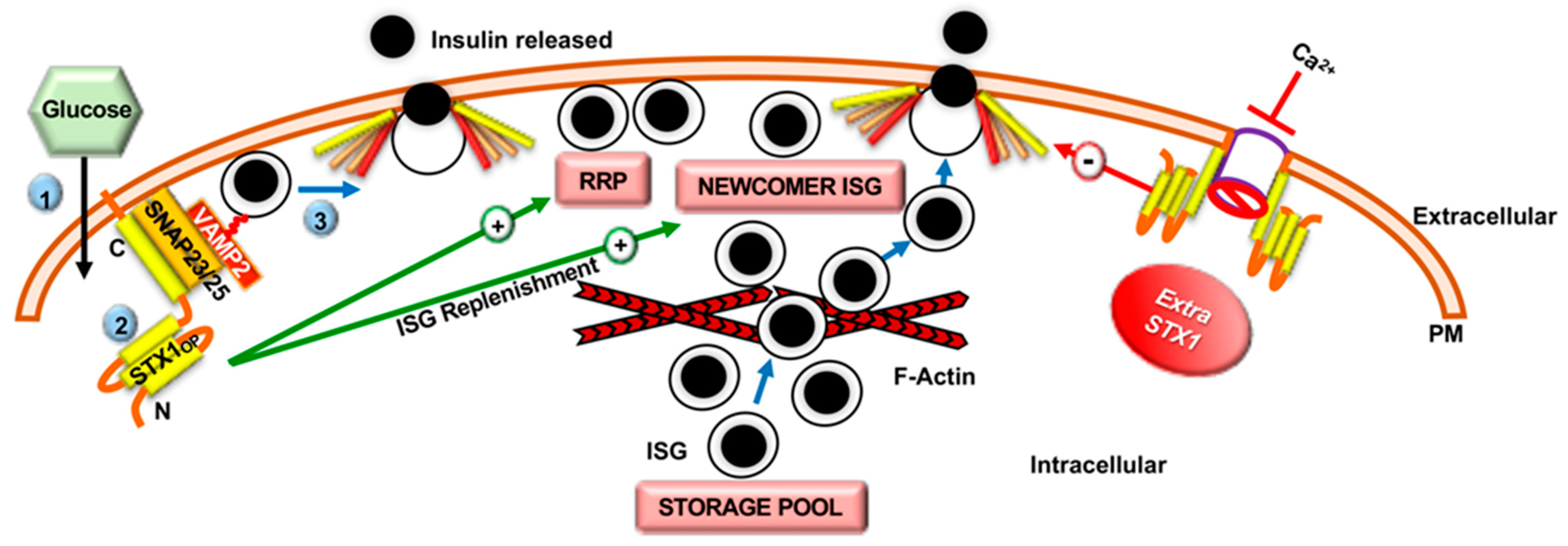
2.2. Role of Syntaxin Proteins in SNARE-Mediated Insulin Secretion
2.3. Role of SNARE-Associated Proteins in Insulin Secretion
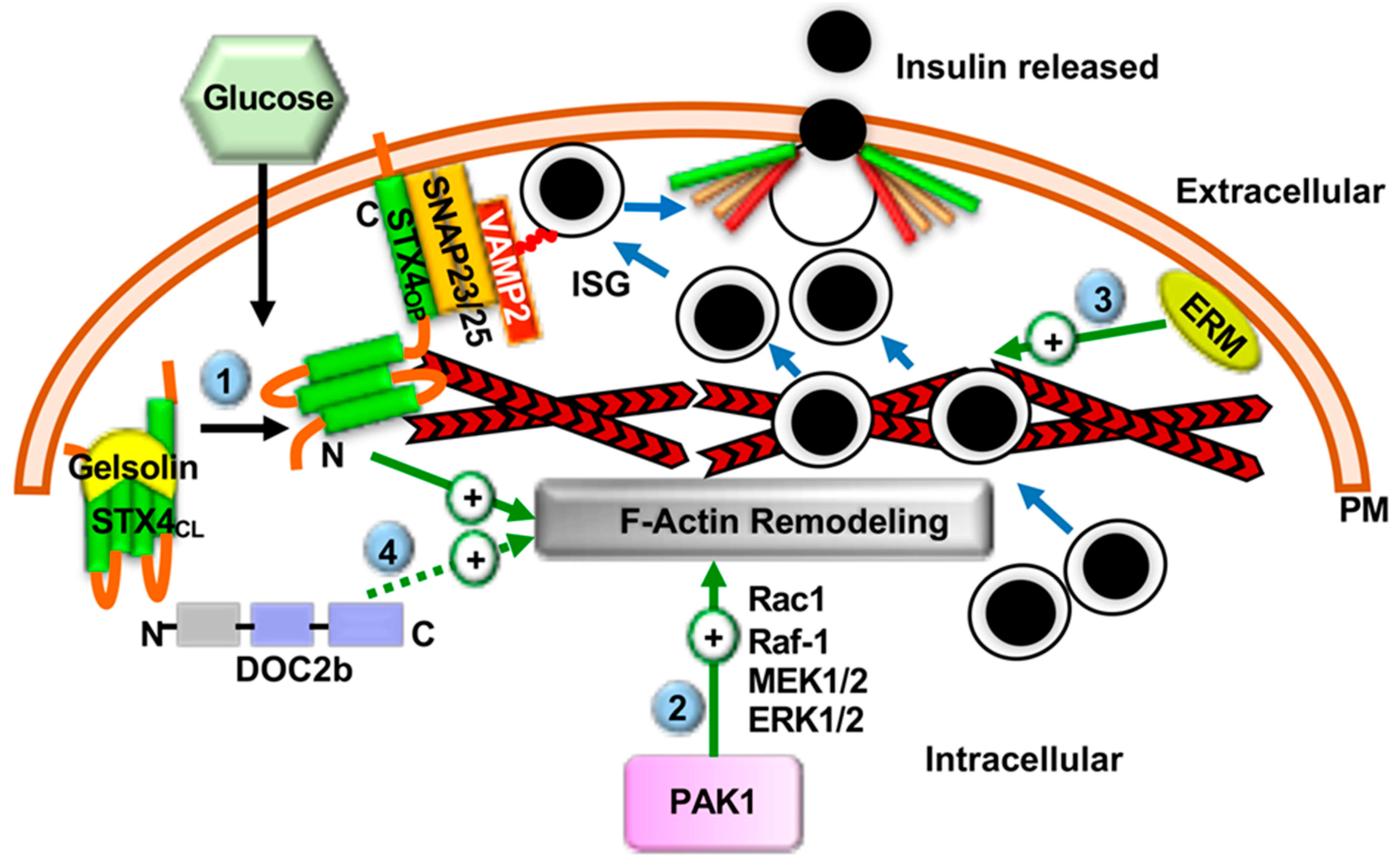
2.4. F-Actin Remodeling in Insulin Secretion
2.5. The Ability of SNARE Proteins to Restore β-Cell Function in Diabetes
3. Exocytosis Proteins and β-Cell Protection
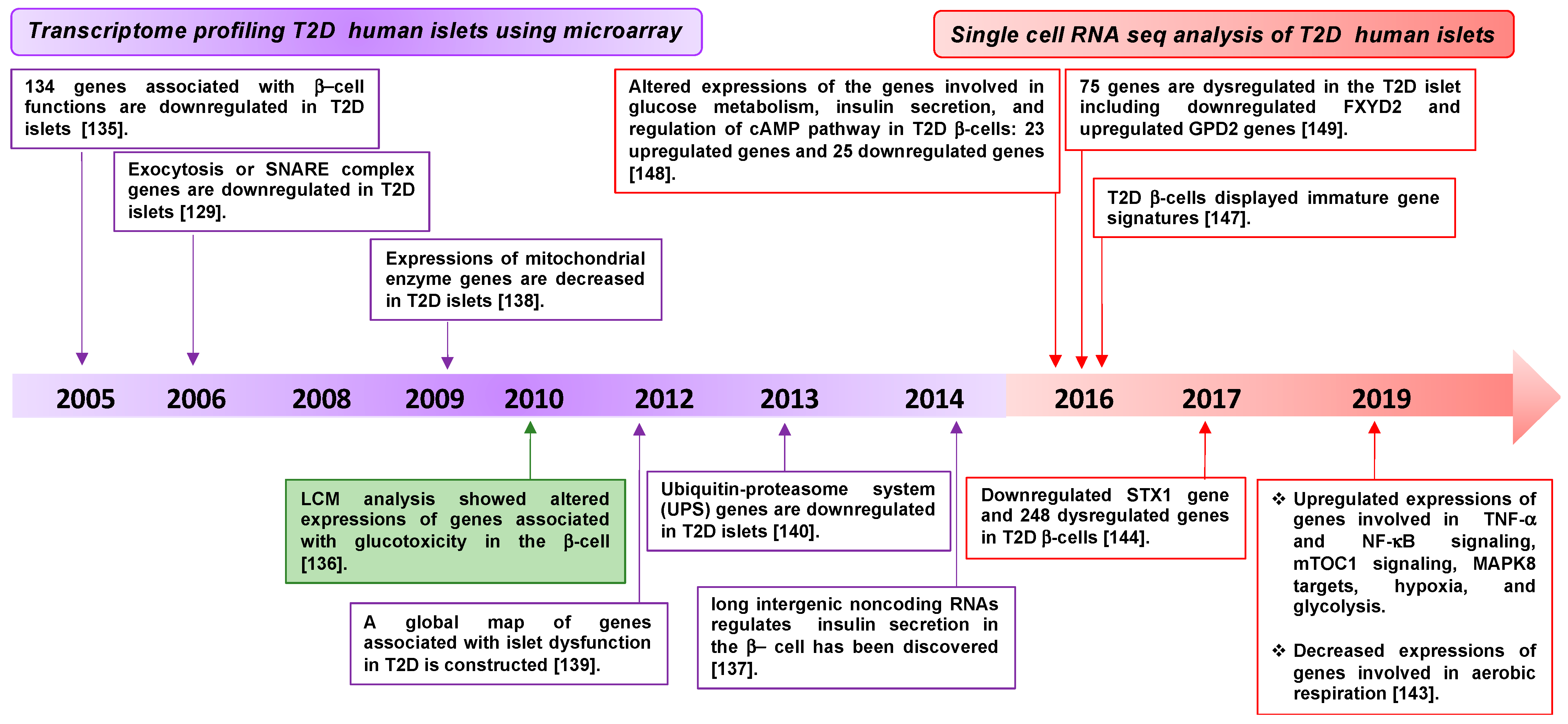
3.1. Diabetes-Associated Genes Revealed by Transcriptomic Profiling of β-Cells
3.2. Regulation of Exocytosis Proteins by Diabetogenic Stressors
3.2.1. Regulation of Exocytosis Proteins and Obesity
3.2.2. Regulation of Exocytosis Proteins and Immune Signaling
3.3. SNARE Proteins Can Modulate Inflammatory Signals in the β-Cell
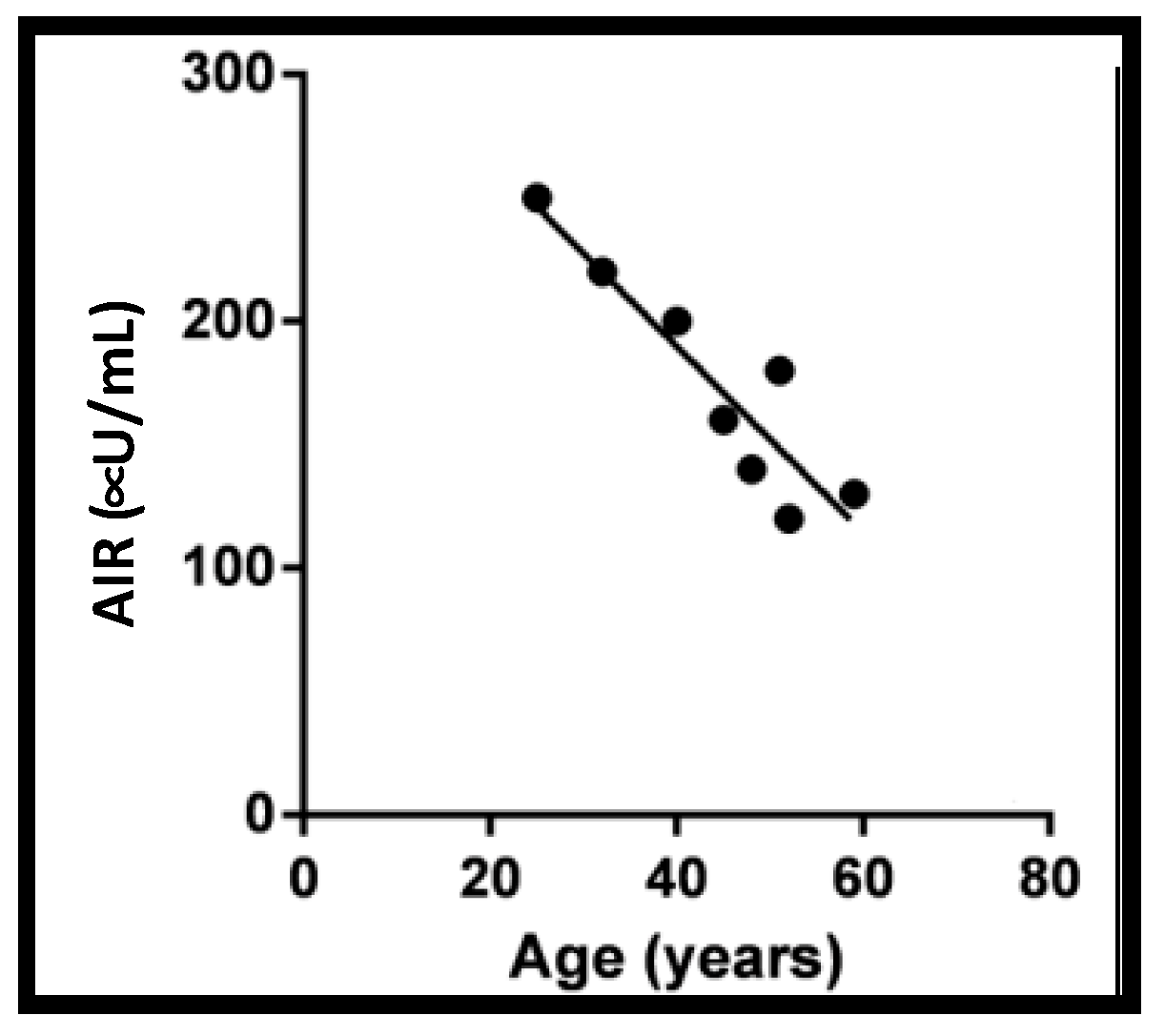
3.4. Exocytosis Proteins and Aging
4. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aslamy, A.; Thurmond, D.C. Exocytosis proteins as novel targets for diabetes prevention and/or remediation? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 312, R739–R752. [Google Scholar] [CrossRef]
- ADA’s Primary Care Advisory Group (PCAG). American Diabetes Association. Standards of Medical Care in Diabetes—2020 Abridged for Primary Care Providers. Clin. Diabetes 2020, 38, 10–38. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Del Guerra, S.; Lupi, R.; Marselli, L.; Masini, M.; Bugliani, M.; Sbrana, S.; Torri, S.; Pollera, M.; Boggi, U.; Mosca, F.; et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 2005, 54, 727–735. [Google Scholar] [CrossRef]
- Hanley, S.C.; Austin, E.; Assouline-Thomas, B.; Kapeluto, J.; Blaichman, J.; Moosavi, M.; Petropavlovskaia, M.; Rosenberg, L. {beta}-Cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology 2010, 151, 1462–1472. [Google Scholar] [CrossRef] [PubMed]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10 (Suppl. 4), 32–42. [Google Scholar] [CrossRef]
- Sakuraba, H.; Mizukami, H.; Yagihashi, N.; Wada, R.; Hanyu, C.; Yagihashi, S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia 2002, 45, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic beta-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef]
- Henquin, J.C.; Rahier, J. Pancreatic alpha cell mass in European subjects with type 2 diabetes. Diabetologia 2011, 54, 1720–1725. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Vidal, J.; Hull, R.L.; Utzschneider, K.M.; Carr, D.B.; Schraw, T.; Scherer, P.E.; Boyko, E.J.; Fujimoto, W.Y.; Kahn, S.E. Progressive loss of beta-cell function leads to worsening glucose tolerance in first-degree relatives of subjects with type 2 diabetes. Diabetes Care 2007, 30, 677–682. [Google Scholar] [CrossRef]
- Utzschneider, K.M.; Prigeon, R.L.; Faulenbach, M.V.; Tong, J.; Carr, D.B.; Boyko, E.J.; Leonetti, D.L.; McNeely, M.J.; Fujimoto, W.Y.; Kahn, S.E. Oral disposition index predicts the development of future diabetes above and beyond fasting and 2-h glucose levels. Diabetes Care 2009, 32, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights into Mechanisms of beta-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef] [PubMed]
- Eastwood, S.L.; Cotter, D.; Harrison, P.J. Cerebellar synaptic protein expression in schizophrenia. Neuroscience 2001, 105, 219–229. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Earle, J.A.; Stary, J.M.; Lee, S.; Sedgewick, J. Altered levels of the synaptosomal associated protein SNAP-25 in hippocampus of subjects with mood disorders and schizophrenia. Neuroreport 2001, 12, 3257–3262. [Google Scholar] [CrossRef]
- Galvez, J.M.; Forero, D.A.; Fonseca, D.J.; Mateus, H.E.; Talero-Gutierrez, C.; Velez-van-Meerbeke, A. Evidence of association between SNAP25 gene and attention deficit hyperactivity disorder in a Latin American sample. Atten. Defic. Hyperact. Disord. 2014, 6, 19–23. [Google Scholar] [CrossRef]
- Kabekkodu, S.P.; Bhat, S.; Radhakrishnan, R.; Aithal, A.; Mascarenhas, R.; Pandey, D.; Rai, L.; Kushtagi, P.; Mundyat, G.P.; Satyamoorthy, K. DNA promoter methylation-dependent transcription of the double C2-like domain beta (DOC2B) gene regulates tumor growth in human cervical cancer. J. Biol. Chem. 2014, 289, 10637–10649. [Google Scholar] [CrossRef]
- Perrotta, C.; Bizzozero, L.; Cazzato, D.; Morlacchi, S.; Assi, E.; Simbari, F.; Zhang, Y.; Gulbins, E.; Bassi, M.T.; Rosa, P.; et al. Syntaxin 4 is required for acid sphingomyelinase activity and apoptotic function. J. Biol. Chem. 2010, 285, 40240–40251. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Ahn, M.; Afelik, S.; Becker, T.C.; Roep, B.O.; Thurmond, D.C. Syntaxin 4 Expression in Pancreatic Beta-Cells Promotes Islet Function and Protects Functional Beta-Cell Mass. Diabetes 2018, 67, 2626–2639. [Google Scholar] [CrossRef]
- Oh, E.; Miller, R.A.; Thurmond, D.C. Syntaxin 4 Overexpression Ameliorates Effects of Aging and High-Fat Diet on Glucose Control and Extends Lifespan. Cell Metab. 2015, 22, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Stull, N.D.; Mirmira, R.G.; Thurmond, D.C. Syntaxin 4 up-regulation increases efficiency of insulin release in pancreatic islets from humans with and without type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2014, 99, E866–E870. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, L.J.; van de Bunt, M.; Braun, M.; Frayn, K.N.; Clark, A.; Gloyn, A.L. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: Implications for understanding genetic association signals at this locus. Mol. Genet. Metab. 2011, 104, 648–653. [Google Scholar] [CrossRef]
- Thorens, B.; Gerard, N.; Deriaz, N. GLUT2 surface expression and intracellular transport via the constitutive pathway in pancreatic beta cells and insulinoma: Evidence for a block in trans-Golgi network exit by brefeldin A. J. Cell Biol. 1993, 123, 1687–1694. [Google Scholar] [CrossRef]
- De Vos, A.; Heimberg, H.; Quartier, E.; Huypens, P.; Bouwens, L.; Pipeleers, D.; Schuit, F. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J. Clin. Invest. 1995, 96, 2489–2495. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, A.; Ruz-Maldonado, I.; Liu, B.; Huang, G.C.; Choudhary, P.; Persaud, S.J. Dynamic Profiling of Insulin Secretion and ATP Generation in Isolated Human and Mouse Islets Reveals Differential Glucose Sensitivity. Cell. Physiol. Biochem. 2017, 44, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Harrison, D.E.; Ashcroft, S.J. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 1984, 312, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.L.; Hales, C.N. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature 1984, 311, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Ashcroft, F.M.; Trube, G. Single Ca channel currents in mouse pancreatic B-cells. Pflugers Archiv 1988, 412, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Satin, L.S.; Cook, D.L. Voltage-gated Ca2+ current in pancreatic B-cells. Pflugers Archiv 1985, 404, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Alvarez de Toledo, G.; Montes, M.A.; Montenegro, P.; Borges, R. Phases of the exocytotic fusion pore. FEBS Lett. 2018, 592, 3532–3541. [Google Scholar] [CrossRef] [PubMed]
- Nunemaker, C.S.; Satin, L.S. Episodic hormone secretion: A comparison of the basis of pulsatile secretion of insulin and GnRH. Endocrine 2014, 47, 49–63. [Google Scholar] [CrossRef]
- Song, S.H.; McIntyre, S.S.; Shah, H.; Veldhuis, J.D.; Hayes, P.C.; Butler, P.C. Direct measurement of pulsatile insulin secretion from the portal vein in human subjects. J. Clin. Endocrinol. Metab. 2000, 85, 4491–4499. [Google Scholar] [CrossRef] [PubMed]
- Thurmond, D.C.; Gaisano, H.Y. Recent Insights into Beta-cell Exocytosis in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1310–1325. [Google Scholar] [CrossRef] [PubMed]
- Gaisano, H.Y. Here come the newcomer granules, better late than never. Trends Endocrinol. Metab. 2014, 25, 381–388. [Google Scholar] [CrossRef]
- Barg, S.; Eliasson, L.; Renstrom, E.; Rorsman, P. A subset of 50 secretory granules in close contact with L-type Ca2+ channels accounts for first-phase insulin secretion in mouse beta-cells. Diabetes 2002, 51 (Suppl. 1), S74–S82. [Google Scholar] [CrossRef]
- Daniel, S.; Noda, M.; Straub, S.G.; Sharp, G.W. Identification of the docked granule pool responsible for the first phase of glucose-stimulated insulin secretion. Diabetes 1999, 48, 1686–1690. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, C.S.; Gopel, S.O.; Barg, S.; Galvanovskis, J.; Ma, X.; Salehi, A.; Rorsman, P.; Eliasson, L. Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B-cells. Pflugers Archiv 2002, 444, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Curry, D.L.; Bennett, L.L.; Grodsky, G.M. Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology 1968, 83, 572–584. [Google Scholar] [CrossRef]
- Porte, D., Jr.; Pupo, A.A. Insulin responses to glucose: Evidence for a two pool system in man. J. Clin. Invest. 1969, 48, 2309–2319. [Google Scholar] [CrossRef]
- Cui, J.; Wang, Z.; Cheng, Q.; Lin, R.; Zhang, X.M.; Leung, P.S.; Copeland, N.G.; Jenkins, N.A.; Yao, K.M.; Huang, J.D. Targeted inactivation of kinesin-1 in pancreatic beta-cells in vivo leads to insulin secretory deficiency. Diabetes 2011, 60, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Baumert, M.; Maycox, P.R.; Navone, F.; De Camilli, P.; Jahn, R. Synaptobrevin: An integral membrane protein of 18,000 daltons present in small synaptic vesicles of rat brain. EMBO J. 1989, 8, 379–384. [Google Scholar] [CrossRef]
- Gembal, M.; Gilon, P.; Henquin, J.C. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J. Clin. Invest. 1992, 89, 1288–1295. [Google Scholar] [CrossRef]
- Kiraly-Borri, C.E.; Morgan, A.; Burgoyne, R.D.; Weller, U.; Wollheim, C.B.; Lang, J. Soluble N-ethylmaleimide-sensitive-factor attachment protein and N-ethylmaleimide-insensitive factors are required for Ca2+-stimulated exocytosis of insulin. Biochem. J. 1996, 314 Pt 1, 199–203. [Google Scholar] [CrossRef][Green Version]
- Lang, J. Molecular mechanisms and regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur. J. Biochem. 1999, 259, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Sollner, T.; Bennett, M.K.; Whiteheart, S.W.; Scheller, R.H.; Rothman, J.E. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 1993, 75, 409–418. [Google Scholar] [CrossRef]
- Sollner, T.; Whiteheart, S.W.; Brunner, M.; Erdjument-Bromage, H.; Geromanos, S.; Tempst, P.; Rothman, J.E. SNAP receptors implicated in vesicle targeting and fusion. Nature 1993, 362, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Moya, F.; Gutierrez, L.M.; Reig, J.A.; Soria, B. Role of syntaxin in mouse pancreatic beta cells. Diabetologia 1995, 38, 860–863. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liang, T.; Qin, T.; Xie, L.; Dolai, S.; Zhu, D.; Prentice, K.J.; Wheeler, M.; Kang, Y.; Osborne, L.; Gaisano, H.Y. New Roles of Syntaxin-1A in Insulin Granule Exocytosis and Replenishment. J. Biol. Chem. 2017, 292, 2203–2216. [Google Scholar] [CrossRef] [PubMed]
- Lam, P.P.; Leung, Y.M.; Sheu, L.; Ellis, J.; Tsushima, R.G.; Osborne, L.R.; Gaisano, H.Y. Transgenic mouse overexpressing syntaxin-1A as a diabetes model. Diabetes 2005, 54, 2744–2754. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Watanabe, T.; Fujiwara, T.; Komazaki, S.; Yamaguchi, K.; Tajima, O.; Akagawa, K. HPC-1/syntaxin 1A suppresses exocytosis of PC12 cells. J. Biochem. 1999, 125, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Land, J.; Zhang, H.; Vaidyanathan, V.V.; Sadoul, K.; Niemann, H.; Wollheim, C.B. Transient expression of botulinum neurotoxin C1 light chain differentially inhibits calcium and glucose induced insulin secretion in clonal beta-cells. FEBS Lett. 1997, 419, 13–17. [Google Scholar] [CrossRef]
- Spurlin, B.A.; Thurmond, D.C. Syntaxin 4 facilitates biphasic glucose-stimulated insulin secretion from pancreatic beta-cells. Mol. Endocrinol. 2006, 20, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Spurlin, B.A.; Park, S.Y.; Nevins, A.K.; Kim, J.K.; Thurmond, D.C. Syntaxin 4 transgenic mice exhibit enhanced insulin-mediated glucose uptake in skeletal muscle. Diabetes 2004, 53, 2223–2231. [Google Scholar] [CrossRef]
- Xie, L.; Zhu, D.; Dolai, S.; Liang, T.; Qin, T.; Kang, Y.; Xie, H.; Huang, Y.C.; Gaisano, H.Y. Syntaxin-4 mediates exocytosis of pre-docked and newcomer insulin granules underlying biphasic glucose-stimulated insulin secretion in human pancreatic beta cells. Diabetologia 2015, 58, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Spurlin, B.A.; Pessin, J.E.; Thurmond, D.C. Munc18c heterozygous knockout mice display increased susceptibility for severe glucose intolerance. Diabetes 2005, 54, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Spurlin, B.A.; Thomas, R.M.; Nevins, A.K.; Kim, H.J.; Kim, Y.J.; Noh, H.L.; Shulman, G.I.; Kim, J.K.; Thurmond, D.C. Insulin resistance in tetracycline-repressible Munc18c transgenic mice. Diabetes 2003, 52, 1910–1917. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Koo, E.; Kwan, E.; Kang, Y.; Park, S.; Xie, H.; Sugita, S.; Gaisano, H.Y. Syntaxin-3 regulates newcomer insulin granule exocytosis and compound fusion in pancreatic beta cells. Diabetologia 2013, 56, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Dolai, S.; Kang, Y.; Liang, T.; Xie, H.; Qin, T.; Yang, L.; Chen, L.; Gaisano, H.Y. Syntaxin-3 Binds and Regulates Both R- and L-Type Calcium Channels in Insulin-Secreting INS-1 832/13 Cells. PLoS ONE 2016, 11, e0147862. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Xie, L.; Kang, Y.; Dolai, S.; Bondo Hansen, J.; Qin, T.; Xie, H.; Liang, T.; Rubin, D.C.; Osborne, L.; et al. Syntaxin 2 Acts as Inhibitory SNARE for Insulin Granule Exocytosis. Diabetes 2017, 66, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Thurmond, D.C. Munc18c depletion selectively impairs the sustained phase of insulin release. Diabetes 2009, 58, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Xie, L.; Karimian, N.; Liang, T.; Kang, Y.; Huang, Y.C.; Gaisano, H.Y. Munc18c mediates exocytosis of pre-docked and newcomer insulin granules underlying biphasic glucose stimulated insulin secretion in human pancreatic beta-cells. Mol. Metab. 2015, 4, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Tomas, A.; Meda, P.; Regazzi, R.; Pessin, J.E.; Halban, P.A. Munc 18-1 and granuphilin collaborate during insulin granule exocytosis. Traffic 2008, 9, 813–832. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.P.; Gatti, E.; Butler, M.; Burton, J.; De Camilli, P. A rat brain Sec1 homologue related to Rop and UNC18 interacts with syntaxin. Proc. Natl. Acad. Sci. USA 1994, 91, 2003–2007. [Google Scholar] [CrossRef] [PubMed]
- Tamori, Y.; Kawanishi, M.; Niki, T.; Shinoda, H.; Araki, S.; Okazawa, H.; Kasuga, M. Inhibition of insulin-induced GLUT4 translocation by Munc18c through interaction with syntaxin4 in 3T3-L1 adipocytes. J. Biol. Chem. 1998, 273, 19740–19746. [Google Scholar] [CrossRef]
- Tellam, J.T.; Macaulay, S.L.; McIntosh, S.; Hewish, D.R.; Ward, C.W.; James, D.E. Characterization of Munc-18c and syntaxin-4 in 3T3-L1 adipocytes. Putative role in insulin-dependent movement of GLUT-4. J. Biol. Chem. 1997, 272, 6179–6186. [Google Scholar] [CrossRef]
- Thurmond, D.C.; Ceresa, B.P.; Okada, S.; Elmendorf, J.S.; Coker, K.; Pessin, J.E. Regulation of insulin-stimulated GLUT4 translocation by Munc18c in 3T3L1 adipocytes. J. Biol. Chem. 1998, 273, 33876–33883. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Kalwat, M.A.; Kim, M.J.; Verhage, M.; Thurmond, D.C. Munc18-1 regulates first-phase insulin release by promoting granule docking to multiple syntaxin isoforms. J. Biol. Chem. 2012, 287, 25821–25833. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Gandasi, N.R.; Arora, S.; Omar-Hmeadi, M.; Saras, J.; Barg, S. Syntaxin clusters at secretory granules in a munc18-bound conformation. Mol. Biol. Cell 2018, 29, 2700–2708. [Google Scholar] [CrossRef]
- Lam, P.P.; Ohno, M.; Dolai, S.; He, Y.; Qin, T.; Liang, T.; Zhu, D.; Kang, Y.; Liu, Y.; Kauppi, M.; et al. Munc18b is a major mediator of insulin exocytosis in rat pancreatic beta-cells. Diabetes 2013, 62, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Zhang, Y.; Lam, P.P.; Dolai, S.; Liu, Y.; Cai, E.P.; Choi, D.; Schroer, S.A.; Kang, Y.; Allister, E.M.; et al. Dual role of VAMP8 in regulating insulin exocytosis and islet beta cell growth. Cell Metab. 2012, 16, 238–249. [Google Scholar] [CrossRef]
- Oh, E.; Thurmond, D.C. The stimulus-induced tyrosine phosphorylation of Munc18c facilitates vesicle exocytosis. J. Biol. Chem. 2006, 281, 17624–17634. [Google Scholar] [CrossRef]
- Jewell, J.L.; Oh, E.; Bennett, S.M.; Meroueh, S.O.; Thurmond, D.C. The tyrosine phosphorylation of Munc18c induces a switch in binding specificity from syntaxin 4 to Doc2beta. J. Biol. Chem. 2008, 283, 21734–21746. [Google Scholar] [CrossRef] [PubMed]
- Ke, B.; Oh, E.; Thurmond, D.C. Doc2beta is a novel Munc18c-interacting partner and positive effector of syntaxin 4-mediated exocytosis. J. Biol. Chem. 2007, 282, 21786–21797. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, G.; Orita, S.; Maeda, M.; Igarashi, H.; Takai, Y. Molecular cloning of an isoform of Doc2 having two C2-like domains. Biochem. Biophys. Res. Commun. 1995, 217, 1053–1061. [Google Scholar] [CrossRef]
- Verhage, M.; de Vries, K.J.; Roshol, H.; Burbach, J.P.; Gispen, W.H.; Sudhof, T.C. DOC2 proteins in rat brain: Complementary distribution and proposed function as vesicular adapter proteins in early stages of secretion. Neuron 1997, 18, 453–461. [Google Scholar] [CrossRef]
- Li, J.; Cantley, J.; Burchfield, J.G.; Meoli, C.C.; Stockli, J.; Whitworth, P.T.; Pant, H.; Chaudhuri, R.; Groffen, A.J.; Verhage, M.; et al. DOC2 isoforms play dual roles in insulin secretion and insulin-stimulated glucose uptake. Diabetologia 2014, 57, 2173–2182. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, L.; Oh, E.; Yoder, S.M.; Brozinick, J.T.; Kalwat, M.A.; Groffen, A.J.; Verhage, M.; Thurmond, D.C. Doc2b is a key effector of insulin secretion and skeletal muscle insulin sensitivity. Diabetes 2012, 61, 2424–2432. [Google Scholar] [CrossRef]
- Ramalingam, L.; Oh, E.; Thurmond, D.C. Doc2b enrichment enhances glucose homeostasis in mice via potentiation of insulin secretion and peripheral insulin sensitivity. Diabetologia 2014, 57, 1476–1484. [Google Scholar] [CrossRef]
- Aslamy, A.; Oh, E.; Olson, E.M.; Zhang, J.; Ahn, M.; Moin, A.S.M.; Tunduguru, R.; Salunkhe, V.A.; Veluthakal, R.; Thurmond, D.C. Doc2b Protects beta-Cells Against Inflammatory Damage and Enhances Function. Diabetes 2018, 67, 1332–1344. [Google Scholar] [CrossRef]
- Ramalingam, L.; Lu, J.; Hudmon, A.; Thurmond, D.C. Doc2b serves as a scaffolding platform for concurrent binding of multiple Munc18 isoforms in pancreatic islet beta-cells. Biochem. J. 2014, 464, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Rungger-Brandle, E.; Just, I.; Jonas, J.C.; Aktories, K.; Wollheim, C.B. Effect of disruption of actin filaments by Clostridium botulinum C2 toxin on insulin secretion in HIT-T15 cells and pancreatic islets. Mol. Biol. Cell 1994, 5, 1199–1213. [Google Scholar] [CrossRef] [PubMed]
- Somers, G.; Blondel, B.; Orci, L.; Malaisse, W.J. Motile events in pancreatic endocrine cells. Endocrinology 1979, 104, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.R.; Ludowyke, R.I.; Biden, T.J. A redistribution of actin and myosin IIA accompanies Ca(2+)-dependent insulin secretion. FEBS Lett. 2001, 492, 101–106. [Google Scholar] [CrossRef]
- Orci, L.; Gabbay, K.H.; Malaisse, W.J. Pancreatic beta-cell web: Its possible role in insulin secretion. Science 1972, 175, 1128–1130. [Google Scholar] [CrossRef]
- Kalwat, M.A.; Wiseman, D.A.; Luo, W.; Wang, Z.; Thurmond, D.C. Gelsolin associates with the N terminus of syntaxin 4 to regulate insulin granule exocytosis. Mol. Endocrinol. 2012, 26, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Thurmond, D.C. Mechanisms of biphasic insulin-granule exocytosis—Roles of the cytoskeleton, small GTPases and SNARE proteins. J. Cell Sci. 2009, 122, 893–903. [Google Scholar] [CrossRef]
- Kalwat, M.A.; Thurmond, D.C. Signaling mechanisms of glucose-induced F-actin remodeling in pancreatic islet beta cells. Exp. Mol. Med. 2013, 45, e37. [Google Scholar] [CrossRef]
- Thurmond, D.C.; Gonelle-Gispert, C.; Furukawa, M.; Halban, P.A.; Pessin, J.E. Glucose-stimulated insulin secretion is coupled to the interaction of actin with the t-SNARE (target membrane soluble N-ethylmaleimide-sensitive factor attachment protein receptor protein) complex. Mol. Endocrinol. 2003, 17, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Band, A.M.; Ali, H.; Vartiainen, M.K.; Welti, S.; Lappalainen, P.; Olkkonen, V.M.; Kuismanen, E. Endogenous plasma membrane t-SNARE syntaxin 4 is present in rab11 positive endosomal membranes and associates with cortical actin cytoskeleton. FEBS Lett. 2002, 531, 513–519. [Google Scholar] [CrossRef]
- Jewell, J.L.; Luo, W.; Oh, E.; Wang, Z.; Thurmond, D.C. Filamentous actin regulates insulin exocytosis through direct interaction with Syntaxin 4. J. Biol. Chem. 2008, 283, 10716–10726. [Google Scholar] [CrossRef] [PubMed]
- Nevins, A.K.; Thurmond, D.C. A direct interaction between Cdc42 and vesicle-associated membrane protein 2 regulates SNARE-dependent insulin exocytosis. J. Biol. Chem. 2005, 280, 1944–1952. [Google Scholar] [CrossRef] [PubMed]
- Nagano, F.; Orita, S.; Sasaki, T.; Naito, A.; Sakaguchi, G.; Maeda, M.; Watanabe, T.; Kominami, E.; Uchiyama, Y.; Takai, Y. Interaction of Doc2 with tctex-1, a light chain of cytoplasmic dynein. Implication in dynein-dependent vesicle transport. J. Biol. Chem. 1998, 273, 30065–30068. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, Y.; Okada, S.; Yamada, E.; Pessin, J.E.; Yamada, M. Tctex1d2 Is a Negative Regulator of GLUT4 Translocation and Glucose Uptake. Endocrinology 2015, 156, 3548–3558. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Oh, E.; Merz, K.E.; Aslamy, A.; Veluthakal, R.; Salunkhe, V.A.; Ahn, M.; Tunduguru, R.; Thurmond, D.C. DOC2B promotes insulin sensitivity in mice via a novel KLC1-dependent mechanism in skeletal muscle. Diabetologia 2019, 62, 845–859. [Google Scholar] [CrossRef]
- Daniel, S.; Noda, M.; Cerione, R.A.; Sharp, G.W. A link between Cdc42 and syntaxin is involved in mastoparan-stimulated insulin release. Biochemistry 2002, 41, 9663–9671. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, A.; Metz, S.A. Regulation of guanine-nucleotide binding proteins in islet subcellular fractions by phospholipase-derived lipid mediators of insulin secretion. Biochim. Biophys. Acta 1994, 1222, 360–368. [Google Scholar] [CrossRef]
- Kowluru, A.; Seavey, S.E.; Li, G.; Sorenson, R.L.; Weinhaus, A.J.; Nesher, R.; Rabaglia, M.E.; Vadakekalam, J.; Metz, S.A. Glucose- and GTP-dependent stimulation of the carboxyl methylation of CDC42 in rodent and human pancreatic islets and pure beta cells. Evidence for an essential role of GTP-binding proteins in nutrient-induced insulin secretion. J. Clin. Invest. 1996, 98, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Nevins, A.K.; Thurmond, D.C. Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. Am. J. Physiol. Cell Physiol. 2003, 285, C698–C710. [Google Scholar] [CrossRef]
- Kalwat, M.A.; Yoder, S.M.; Wang, Z.; Thurmond, D.C. A p21-activated kinase (PAK1) signaling cascade coordinately regulates F-actin remodeling and insulin granule exocytosis in pancreatic beta cells. Biochem. Pharmacol. 2013, 85, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Oh, E.; Clapp, D.W.; Chernoff, J.; Thurmond, D.C. Inhibition or ablation of p21-activated kinase (PAK1) disrupts glucose homeostatic mechanisms in vivo. J. Biol. Chem. 2011, 286, 41359–41367. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Oh, E.; Thurmond, D.C. Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J. Biol. Chem. 2007, 282, 9536–9546. [Google Scholar] [CrossRef]
- Ahn, M.; Yoder, S.M.; Wang, Z.; Oh, E.; Ramalingam, L.; Tunduguru, R.; Thurmond, D.C. The p21-activated kinase (PAK1) is involved in diet-induced beta cell mass expansion and survival in mice and human islets. Diabetologia 2016, 59, 2145–2155. [Google Scholar] [CrossRef]
- Wang, B.; Lin, H.; Li, X.; Lu, W.; Kim, J.B.; Xu, A.; Cheng, K.K.Y. The adaptor protein APPL2 controls glucose-stimulated insulin secretion via F-actin remodeling in pancreatic beta-cells. Proc. Natl. Acad. Sci. USA 2020, 117, 28307–28315. [Google Scholar] [CrossRef]
- Asahara, S.; Shibutani, Y.; Teruyama, K.; Inoue, H.Y.; Kawada, Y.; Etoh, H.; Matsuda, T.; Kimura-Koyanagi, M.; Hashimoto, N.; Sakahara, M.; et al. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia 2013, 56, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Luo, R.; Kowluru, A.; Li, G. Novel regulation by Rac1 of glucose- and forskolin-induced insulin secretion in INS-1 beta-cells. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E818–E827. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.K.; Lam, K.S.; Wu, D.; Wang, Y.; Sweeney, G.; Hoo, R.L.; Zhang, J.; Xu, A. APPL1 potentiates insulin secretion in pancreatic beta cells by enhancing protein kinase Akt-dependent expression of SNARE proteins in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 8919–8924. [Google Scholar] [CrossRef] [PubMed]
- Niggli, V.; Andreoli, C.; Roy, C.; Mangeat, P. Identification of a phosphatidylinositol-4,5-bisphosphate-binding domain in the N-terminal region of ezrin. FEBS Lett. 1995, 376, 172–176. [Google Scholar] [CrossRef]
- Ponuwei, G.A. A glimpse of the ERM proteins. J. Biomed. Sci. 2016, 23, 35. [Google Scholar] [CrossRef] [PubMed]
- Turunen, O.; Wahlstrom, T.; Vaheri, A. Ezrin has a COOH-terminal actin-binding site that is conserved in the ezrin protein family. J. Cell Biol. 1994, 126, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.P.; Turner, J.R.; Philipson, L.H. Glucose-induced ERM protein activation and translocation regulates insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E772–E785. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fu, J.; Githaka, J.M.; Dai, X.; Plummer, G.; Suzuki, K.; Spigelman, A.F.; Bautista, A.; Kim, R.; Greitzer-Antes, D.; Fox, J.E.M.; et al. A glucose-dependent spatial patterning of exocytosis in human beta-cells is disrupted in type 2 diabetes. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Ferri, G.; Digiacomo, L.; Lavagnino, Z.; Occhipinti, M.; Bugliani, M.; Cappello, V.; Caracciolo, G.; Marchetti, P.; Piston, D.W.; Cardarelli, F. Insulin secretory granules labelled with phogrin-fluorescent proteins show alterations in size, mobility and responsiveness to glucose stimulation in living beta-cells. Sci. Rep. 2019, 9, 2890. [Google Scholar] [CrossRef]
- Makhmutova, M.; Liang, T.; Gaisano, H.; Caicedo, A.; Almaca, J. Confocal Imaging of Neuropeptide Y-pHluorin: A Technique to Visualize Insulin Granule Exocytosis in Intact Murine and Human Islets. J. Vis. Exp. 2017. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Sawada, W.; Noguchi, J.; Watanabe, S.; Ucar, H.; Hayashi-Takagi, A.; Yagishita, S.; Ohno, M.; Tokumaru, H.; Kasai, H. Two-photon fluorescence lifetime imaging of primed SNARE complexes in presynaptic terminals and beta cells. Nat. Commun. 2015, 6, 8531. [Google Scholar] [CrossRef] [PubMed]
- Huypens, P.; Ling, Z.; Pipeleers, D.; Schuit, F. Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia 2000, 43, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Ahren, B. GLP-1 for type 2 diabetes. Exp. Cell Res. 2011, 317, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, P.; Chepurny, O.G.; Holz, G.G. Regulation of glucose homeostasis by GLP-1. Prog. Mol. Biol. Transl. Sci. 2014, 121, 23–65. [Google Scholar] [CrossRef]
- Meloni, A.R.; DeYoung, M.B.; Lowe, C.; Parkes, D.G. GLP-1 receptor activated insulin secretion from pancreatic beta-cells: Mechanism and glucose dependence. Diabetes Obes. Metab. 2013, 15, 15–27. [Google Scholar] [CrossRef]
- Kwan, E.P.; Gaisano, H.Y. Glucagon-like peptide 1 regulates sequential and compound exocytosis in pancreatic islet beta-cells. Diabetes 2005, 54, 2734–2743. [Google Scholar] [CrossRef] [PubMed]
- Kwan, E.P.; Xie, L.; Sheu, L.; Ohtsuka, T.; Gaisano, H.Y. Interaction between Munc13-1 and RIM is critical for glucagon-like peptide-1 mediated rescue of exocytotic defects in Munc13-1 deficient pancreatic beta-cells. Diabetes 2007, 56, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Dolz, M.; Movassat, J.; Bailbe, D.; Le Stunff, H.; Giroix, M.H.; Fradet, M.; Kergoat, M.; Portha, B. cAMP-secretion coupling is impaired in diabetic GK/Par rat beta-cells: A defect counteracted by GLP-1. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E797–E806. [Google Scholar] [CrossRef]
- Kong, X.; Yan, D.; Sun, J.; Wu, X.; Mulder, H.; Hua, X.; Ma, X. Glucagon-like peptide 1 stimulates insulin secretion via inhibiting RhoA/ROCK signaling and disassembling glucotoxicity-induced stress fibers. Endocrinology 2014, 155, 4676–4685. [Google Scholar] [CrossRef] [PubMed]
- Quinault, A.; Gausseres, B.; Bailbe, D.; Chebbah, N.; Portha, B.; Movassat, J.; Tourrel-Cuzin, C. Disrupted dynamics of F-actin and insulin granule fusion in INS-1 832/13 beta-cells exposed to glucotoxicity: Partial restoration by glucagon-like peptide 1. Biochim. Biophys. Acta 2016, 1862, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Wei, S.; Petersen, N.; Ali, Y.; Wang, X.; Bacaj, T.; Rorsman, P.; Hong, W.; Sudhof, T.C.; Han, W. Synaptotagmin-7 phosphorylation mediates GLP-1-dependent potentiation of insulin secretion from beta-cells. Proc. Natl. Acad. Sci. USA 2015, 112, 9996–10001. [Google Scholar] [CrossRef] [PubMed]
- Gleizes, C.; Kreutter, G.; Abbas, M.; Kassem, M.; Constantinescu, A.A.; Boisrame-Helms, J.; Yver, B.; Toti, F.; Kessler, L. beta cell membrane remodelling and procoagulant events occur in inflammation-driven insulin impairment: A GLP-1 receptor dependent and independent control. J. Cell. Mol. Med. 2016, 20, 231–242. [Google Scholar] [CrossRef]
- Vikman, J.; Svensson, H.; Huang, Y.C.; Kang, Y.; Andersson, S.A.; Gaisano, H.Y.; Eliasson, L. Truncation of SNAP-25 reduces the stimulatory action of cAMP on rapid exocytosis in insulin-secreting cells. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E452–E461. [Google Scholar] [CrossRef]
- Andersson, S.A.; Olsson, A.H.; Esguerra, J.L.; Heimann, E.; Ladenvall, C.; Edlund, A.; Salehi, A.; Taneera, J.; Degerman, E.; Groop, L.; et al. Reduced insulin secretion correlates with decreased expression of exocytotic genes in pancreatic islets from patients with type 2 diabetes. Mol. Cell. Endocrinol. 2012, 364, 36–45. [Google Scholar] [CrossRef]
- Gaisano, H.Y.; Ostenson, C.G.; Sheu, L.; Wheeler, M.B.; Efendic, S. Abnormal expression of pancreatic islet exocytotic soluble N-ethylmaleimide-sensitive factor attachment protein receptors in Goto-Kakizaki rats is partially restored by phlorizin treatment and accentuated by high glucose treatment. Endocrinology 2002, 143, 4218–4226. [Google Scholar] [CrossRef] [PubMed]
- Nagamatsu, S.; Nakamichi, Y.; Yamamura, C.; Matsushima, S.; Watanabe, T.; Ozawa, S.; Furukawa, H.; Ishida, H. Decreased expression of t-SNARE, syntaxin 1, and SNAP-25 in pancreatic beta-cells is involved in impaired insulin secretion from diabetic GK rat islets: Restoration of decreased t-SNARE proteins improves impaired insulin secretion. Diabetes 1999, 48, 2367–2373. [Google Scholar] [CrossRef]
- Ostenson, C.G.; Gaisano, H.; Sheu, L.; Tibell, A.; Bartfai, T. Impaired gene and protein expression of exocytotic soluble N-ethylmaleimide attachment protein receptor complex proteins in pancreatic islets of type 2 diabetic patients. Diabetes 2006, 55, 435–440. [Google Scholar] [CrossRef]
- Tsunoda, K.; Sanke, T.; Nakagawa, T.; Furuta, H.; Nanjo, K. Single nucleotide polymorphism (D68D, T to C) in the syntaxin 1A gene correlates to age at onset and insulin requirement in Type II diabetic patients. Diabetologia 2001, 44, 2092–2097. [Google Scholar] [CrossRef] [PubMed]
- Aslamy, A.; Oh, E.; Ahn, M.; Moin, A.S.M.; Chang, M.; Duncan, M.; Hacker-Stratton, J.; El-Shahawy, M.; Kandeel, F.; DiMeglio, L.A.; et al. Exocytosis Protein DOC2B as a Biomarker of Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 1966–1976. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.P.; Choi, Y.; Wang, P.; Davis, D.B.; Rabaglia, M.E.; Oler, A.T.; Stapleton, D.S.; Argmann, C.; Schueler, K.L.; Edwards, S.; et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 2008, 18, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Kwan, E.P.; Xie, L.; Sheu, L.; Nolan, C.J.; Prentki, M.; Betz, A.; Brose, N.; Gaisano, H.Y. Munc13-1 deficiency reduces insulin secretion and causes abnormal glucose tolerance. Diabetes 2006, 55, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Salunkhe, V.A.; Veluthakal, R.; Kahn, S.E.; Thurmond, D.C. Novel approaches to restore beta cell function in prediabetes and type 2 diabetes. Diabetologia 2018, 61, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Gunton, J.E.; Kulkarni, R.N.; Yim, S.; Okada, T.; Hawthorne, W.J.; Tseng, Y.H.; Roberson, R.S.; Ricordi, C.; O’Connell, P.J.; Gonzalez, F.J.; et al. Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell 2005, 122, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Marselli, L.; Thorne, J.; Dahiya, S.; Sgroi, D.C.; Sharma, A.; Bonner-Weir, S.; Marchetti, P.; Weir, G.C. Gene expression profiles of Beta-cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PLoS ONE 2010, 5, e11499. [Google Scholar] [CrossRef] [PubMed]
- Fadista, J.; Vikman, P.; Laakso, E.O.; Mollet, I.G.; Esguerra, J.L.; Taneera, J.; Storm, P.; Osmark, P.; Ladenvall, C.; Prasad, R.B.; et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 13924–13929. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.J.; Longacre, M.J.; Langberg, E.C.; Tibell, A.; Kendrick, M.A.; Fukao, T.; Ostenson, C.G. Decreased levels of metabolic enzymes in pancreatic islets of patients with type 2 diabetes. Diabetologia 2009, 52, 1087–1091. [Google Scholar] [CrossRef]
- Taneera, J.; Lang, S.; Sharma, A.; Fadista, J.; Zhou, Y.; Ahlqvist, E.; Jonsson, A.; Lyssenko, V.; Vikman, P.; Hansson, O.; et al. A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab. 2012, 16, 122–134. [Google Scholar] [CrossRef]
- Bugliani, M.; Liechti, R.; Cheon, H.; Suleiman, M.; Marselli, L.; Kirkpatrick, C.; Filipponi, F.; Boggi, U.; Xenarios, I.; Syed, F.; et al. Microarray analysis of isolated human islet transcriptome in type 2 diabetes and the role of the ubiquitin-proteasome system in pancreatic beta cell dysfunction. Mol. Cell. Endocrinol. 2013, 367, 1–10. [Google Scholar] [CrossRef]
- Blodgett, D.M.; Redick, S.D.; Harlan, D.M. Surprising Heterogeneity of Pancreatic Islet Cell Subsets. Cell Syst. 2016, 3, 330–332. [Google Scholar] [CrossRef] [PubMed]
- Baron, M.; Veres, A.; Wolock, S.L.; Faust, A.L.; Gaujoux, R.; Vetere, A.; Ryu, J.H.; Wagner, B.K.; Shen-Orr, S.S.; Klein, A.M.; et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst. 2016, 3, 346–360.e4. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Weng, C.; Li, H.; Tao, R.; Mai, W.; Liu, X.; Lu, L.; Lai, S.; Duan, Q.; Alvarez, C.; et al. Single-Cell Heterogeneity Analysis and CRISPR Screen Identify Key Beta-Cell-Specific Disease Genes. Cell Rep. 2019, 26, 3132–3144.e7. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, N.; George, J.; Bolisetty, M.; Kursawe, R.; Sun, L.; Sivakamasundari, V.; Kycia, I.; Robson, P.; Stitzel, M.L. Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes. Genome Res. 2017, 27, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Klughammer, J.; Farlik, M.; Penz, T.; Spittler, A.; Barbieux, C.; Berishvili, E.; Bock, C.; Kubicek, S. Single-cell transcriptomes reveal characteristic features of human pancreatic islet cell types. EMBO Rep. 2016, 17, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Muraro, M.J.; Dharmadhikari, G.; Grun, D.; Groen, N.; Dielen, T.; Jansen, E.; van Gurp, L.; Engelse, M.A.; Carlotti, F.; de Koning, E.J.; et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016, 3, 385–394.e3. [Google Scholar] [CrossRef]
- Wang, Y.J.; Schug, J.; Won, K.J.; Liu, C.; Naji, A.; Avrahami, D.; Golson, M.L.; Kaestner, K.H. Single-Cell Transcriptomics of the Human Endocrine Pancreas. Diabetes 2016, 65, 3028–3038. [Google Scholar] [CrossRef]
- Xin, Y.; Kim, J.; Okamoto, H.; Ni, M.; Wei, Y.; Adler, C.; Murphy, A.J.; Yancopoulos, G.D.; Lin, C.; Gromada, J. RNA Sequencing of Single Human Islet Cells Reveals Type 2 Diabetes Genes. Cell Metab. 2016, 24, 608–615. [Google Scholar] [CrossRef]
- Segerstolpe, A.; Palasantza, A.; Eliasson, P.; Andersson, E.M.; Andreasson, A.C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef]
- Hou, J.; Li, Z.; Zhong, W.; Hao, Q.; Lei, L.; Wang, L.; Zhao, D.; Xu, P.; Zhou, Y.; Wang, Y.; et al. Temporal Transcriptomic and Proteomic Landscapes of Deteriorating Pancreatic Islets in Type 2 Diabetic Rats. Diabetes 2017, 66, 2188–2200. [Google Scholar] [CrossRef]
- Enge, M.; Arda, H.E.; Mignardi, M.; Beausang, J.; Bottino, R.; Kim, S.K.; Quake, S.R. Single-Cell Analysis of Human Pancreas Reveals Transcriptional Signatures of Aging and Somatic Mutation Patterns. Cell 2017, 171, 321–330.e14. [Google Scholar] [CrossRef]
- Torrejon-Escribano, B.; Escoriza, J.; Montanya, E.; Blasi, J. Glucose-dependent changes in SNARE protein levels in pancreatic beta-cells. Endocrinology 2011, 152, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Nagao, M.; Esguerra, J.L.S.; Asai, A.; Ofori, J.K.; Edlund, A.; Wendt, A.; Sugihara, H.; Wollheim, C.B.; Oikawa, S.; Eliasson, L. Potential Protection Against Type 2 Diabetes in Obesity Through Lower CD36 Expression and Improved Exocytosis in beta-Cells. Diabetes 2020, 69, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Krus, U.; King, B.C.; Nagaraj, V.; Gandasi, N.R.; Sjolander, J.; Buda, P.; Garcia-Vaz, E.; Gomez, M.F.; Ottosson-Laakso, E.; Storm, P.; et al. The complement inhibitor CD59 regulates insulin secretion by modulating exocytotic events. Cell Metab. 2014, 19, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Hu, W.; Shahsafaei, A.; Song, W.; Dobarro, M.; Sukhova, G.K.; Bronson, R.R.; Shi, G.P.; Rother, R.P.; Halperin, J.A.; et al. Complement regulator CD59 protects against atherosclerosis by restricting the formation of complement membrane attack complex. Circ. Res. 2009, 104, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Golec, E.; Rosberg, R.; Zhang, E.; Renstrom, E.; Blom, A.M.; King, B.C. A cryptic non-GPI-anchored cytosolic isoform of CD59 controls insulin exocytosis in pancreatic beta-cells by interaction with SNARE proteins. FASEB J. 2019, 33, 12425–12434. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Vaidya, A.; Sahoo, R.; Goldfine, A.; Herring, N.; Bry, L.; Chorev, M.; Halperin, J.A. Glycation of the complement regulatory protein CD59 is a novel biomarker for glucose handling in humans. J. Clin. Endocrinol. Metab. 2014, 99, E999–E1006. [Google Scholar] [CrossRef] [PubMed]
- Boni-Schnetzler, M.; Meier, D.T. Islet inflammation in type 2 diabetes. Semin. Immunopathol. 2019, 41, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Volund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Sauter, N.S.; Thienel, C.; Plutino, Y.; Kampe, K.; Dror, E.; Traub, S.; Timper, K.; Bedat, B.; Pattou, F.; Kerr-Conte, J.; et al. Angiotensin II induces interleukin-1beta-mediated islet inflammation and beta-cell dysfunction independently of vasoconstrictive effects. Diabetes 2015, 64, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.; Biollaz, G.; et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef] [PubMed]
- Kamata, K.; Mizukami, H.; Inaba, W.; Tsuboi, K.; Tateishi, Y.; Yoshida, T.; Yagihashi, S. Islet amyloid with macrophage migration correlates with augmented beta-cell deficits in type 2 diabetic patients. Amyloid 2014, 21, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Islet-associated macrophages in type 2 diabetes. Diabetologia 2009, 52, 1686–1688. [Google Scholar] [CrossRef] [PubMed]
- Ortis, F.; Naamane, N.; Flamez, D.; Ladriere, L.; Moore, F.; Cunha, D.A.; Colli, M.L.; Thykjaer, T.; Thorsen, K.; Orntoft, T.F.; et al. Cytokines interleukin-1beta and tumor necrosis factor-alpha regulate different transcriptional and alternative splicing networks in primary beta-cells. Diabetes 2010, 59, 358–374. [Google Scholar] [CrossRef] [PubMed]
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. Eur. Cardiol. 2019, 14, 50–59. [Google Scholar] [CrossRef]
- Eldor, R.; Yeffet, A.; Baum, K.; Doviner, V.; Amar, D.; Ben-Neriah, Y.; Christofori, G.; Peled, A.; Carel, J.C.; Boitard, C.; et al. Conditional and specific NF-kappaB blockade protects pancreatic beta cells from diabetogenic agents. Proc. Natl. Acad. Sci. USA 2006, 103, 5072–5077. [Google Scholar] [CrossRef] [PubMed]
- Malle, E.K.; Zammit, N.W.; Walters, S.N.; Koay, Y.C.; Wu, J.; Tan, B.M.; Villanueva, J.E.; Brink, R.; Loudovaris, T.; Cantley, J.; et al. Nuclear factor kappaB-inducing kinase activation as a mechanism of pancreatic beta cell failure in obesity. J. Exp. Med. 2015, 212, 1239–1254. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Veluthakal, R.O.E.; Ahn, M.; Chatterjee-Bhowmick, D.; Thurmond, D.C. Syntaxin 4 mediates NF-kB signaling and chemokine ligand expression via specific interaction with IkBβ. Diabetes 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Perrotta, C.; Cervia, D.; Di Renzo, I.; Moscheni, C.; Bassi, M.T.; Campana, L.; Martelli, C.; Catalani, E.; Giovarelli, M.; Zecchini, S.; et al. Nitric Oxide Generated by Tumor-Associated Macrophages Is Responsible for Cancer Resistance to Cisplatin and Correlated with Syntaxin 4 and Acid Sphingomyelinase Inhibition. Front. Immunol. 2018, 9, 1186. [Google Scholar] [CrossRef] [PubMed]
- Palmer, Z.J.; Duncan, R.R.; Johnson, J.R.; Lian, L.Y.; Mello, L.V.; Booth, D.; Barclay, J.W.; Graham, M.E.; Burgoyne, R.D.; Prior, I.A.; et al. S-nitrosylation of syntaxin 1 at Cys(145) is a regulatory switch controlling Munc18-1 binding. Biochem. J. 2008, 413, 479–491. [Google Scholar] [CrossRef]
- Wiseman, D.A.; Kalwat, M.A.; Thurmond, D.C. Stimulus-induced S-nitrosylation of Syntaxin 4 impacts insulin granule exocytosis. J. Biol. Chem. 2011, 286, 16344–16354. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. National Diabetes Statistics Report, 2020; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2020; pp. 1–21.
- Chow, H.M.; Shi, M.; Cheng, A.; Gao, Y.; Chen, G.; Song, X.; So, R.W.L.; Zhang, J.; Herrup, K. Age-related hyperinsulinemia leads to insulin resistance in neurons and cell-cycle-induced senescence. Nat. Neurosci. 2019, 22, 1806–1819. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.K.; Kontopantelis, E.; Emsley, R.; Buchan, I.; Sattar, N.; Rutter, M.K.; Ashcroft, D.M. Life Expectancy and Cause-Specific Mortality in Type 2 Diabetes: A Population-Based Cohort Study Quantifying Relationships in Ethnic Subgroups. Diabetes Care 2017, 40, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Leal, J.; Gray, A.M.; Clarke, P.M. Development of life-expectancy tables for people with type 2 diabetes. Eur. Heart J. 2009, 30, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; van Haaren, M.; Mruk, M.; Lee, T.B., Jr.; Crawford, C.; Hollister-Lock, J.; Sullivan, B.A.; Johnson, J.W.; Ebrahimi, A.; Dreyfuss, J.M.; et al. beta Cell Aging Markers Have Heterogeneous Distribution and Are Induced by Insulin Resistance. Cell Metab. 2017, 25, 898–910.e5. [Google Scholar] [CrossRef] [PubMed]
- Westacott, M.J.; Farnsworth, N.L.; St Clair, J.R.; Poffenberger, G.; Heintz, A.; Ludin, N.W.; Hart, N.J.; Powers, A.C.; Benninger, R.K.P. Age-Dependent Decline in the Coordinated Ca(2+) and Insulin Secretory Dynamics in Human Pancreatic Islets. Diabetes 2017, 66, 2436–2445. [Google Scholar] [CrossRef] [PubMed]
- Niclauss, N.; Bosco, D.; Morel, P.; Demuylder-Mischler, S.; Brault, C.; Milliat-Guittard, L.; Colin, C.; Parnaud, G.; Muller, Y.D.; Giovannoni, L.; et al. Influence of donor age on islet isolation and transplantation outcome. Transplantation 2011, 91, 360–366. [Google Scholar] [CrossRef]
- Scaglia, L.; Cahill, C.J.; Finegood, D.T.; Bonner-Weir, S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 1997, 138, 1736–1741. [Google Scholar] [CrossRef]
- Gregg, B.E.; Moore, P.C.; Demozay, D.; Hall, B.A.; Li, M.; Husain, A.; Wright, A.J.; Atkinson, M.A.; Rhodes, C.J. Formation of a human beta-cell population within pancreatic islets is set early in life. J. Clin. Endocrinol. Metab. 2012, 97, 3197–3206. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Spinas, G.A.; Lehmann, R.; Sergeev, P.; Weber, M.; Fontana, A.; Kaiser, N.; Donath, M.Y. Glucose induces beta-cell apoptosis via upregulation of the Fas receptor in human islets. Diabetes 2001, 50, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B., Jr.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of beta Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142.e4. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.J.; Shah, A.; Ntranos, V.; Van Gool, F.; Atkinson, M.; Bhushan, A. Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab. 2019, 29, 1045–1060.e10. [Google Scholar] [CrossRef]
- Chandra, T.; Ewels, P.A.; Schoenfelder, S.; Furlan-Magaril, M.; Wingett, S.W.; Kirschner, K.; Thuret, J.Y.; Andrews, S.; Fraser, P.; Reik, W. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015, 10, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Helman, A.; Klochendler, A.; Azazmeh, N.; Gabai, Y.; Horwitz, E.; Anzi, S.; Swisa, A.; Condiotti, R.; Granit, R.Z.; Nevo, Y.; et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med. 2016, 22, 412–420. [Google Scholar] [CrossRef]
- Vaughan, S.; Jat, P.S. Deciphering the role of nuclear factor-kappaB in cellular senescence. Aging (Albany NY) 2011, 3, 913–919. [Google Scholar] [CrossRef]
- Flores, R.R.; Zhou, L.; Robbins, P.D. Expression of IL-2 in beta cells by AAV8 gene transfer in pre-diabetic NOD mice prevents diabetes through activation of FoxP3-positive regulatory T cells. Gene Ther. 2014, 21, 715–722. [Google Scholar] [CrossRef]
- Flotte, T.R.; Trapnell, B.C.; Humphries, M.; Carey, B.; Calcedo, R.; Rouhani, F.; Campbell-Thompson, M.; Yachnis, A.T.; Sandhaus, R.A.; McElvaney, N.G.; et al. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing alpha1-antitrypsin: Interim results. Hum. Gene Ther. 2011, 22, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, M.S.; Kuo, H.T.; Bunnapradist, S. Outcomes of simultaneous pancreas-kidney transplantation in type 2 diabetic recipients. Clin. J. Am. Soc. Nephrol. 2011, 6, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Bahendeka, S.; Sahay, R.; Ghosh, S.; Md, F.; Orabi, A.; Ramaiya, K.; Al Shammari, S.; Shrestha, D.; Shaikh, K.; et al. Consensus Recommendations on Sulfonylurea and Sulfonylurea Combinations in the Management of Type 2 Diabetes Mellitus—International Task Force. Indian J. Endocrinol. Metab. 2018, 22, 132–157. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Tendal, B.; Mustafa, R.A.; Vandvik, P.O.; Li, S.; Hao, Q.; Tunnicliffe, D.; Ruospo, M.; Natale, P.; Saglimbene, V.; et al. Sodium-glucose cotransporter protein-2 (SGLT-2) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists for type 2 diabetes: Systematic review and network meta-analysis of randomised controlled trials. BMJ 2021, 372, m4573. [Google Scholar] [CrossRef] [PubMed]
- Thielen, L.; Chen, J.; Xu, G.; Jing, G.; Grayson, T.; Jo, S.; Shalev, A. Novel Small Molecule TXNIP Inhibitor Protects Against Diabetes. Diabetes 2018, 67, 87-OR. [Google Scholar] [CrossRef]
- Thielen, L.; Shalev, A. Diabetes pathogenic mechanisms and potential new therapies based upon a novel target called TXNIP. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 75–80. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Technique | Genes Identified | Reference |
|---|---|---|---|
| Human T2D islets | Microarray and qRT-PCR | Dysregulation of 370 genes (134 genes downregulated associated with β-cell function [e.g., HNF4a, IR, IRS2, AKT2, ARNT]). | [135] |
| Human T2D islets | Microarray and qRT-PCR | Decreased expression of exocytotic SNARE complex proteins (STX1A, SNAP25, VAMP2, Munc18-1, Munc13-1, Synaptophysin). | [129] |
| Human T2D islets | Microarray and qRT-PCR | Decreased expression of metabolic enzymes (mitochondrial enzymes). | [138] |
| Human T2D pancreas | Microarray of islets captured by LCM | Increased expression of genes associated with glucotoxicity. Decreased expression of exocytotic protein, SNAP25. | [136] |
| Human T2D islets | Microarray, qRT-PCR and SNP array | A global map of genes associated with islet dysfunction. Decreased expression of genes related to insulin secretion (KCNJ11, WFS1, SLCA2A, JAZF1, G6PC2). | [139] |
| Human T2D islets | Microarray, qRT-PCR | Downregulation of ubiquitin-proteasome system genes. | [140] |
| Human T2D islets | RNA-seq | Decreased expression of long intergenic noncoding RNAs (LOC283177) which positively associate with insulin exocytosis. | [137] |
| Human T2D islets | Single-cell RNA-seq | Dysregulation of 48 genes which are involved in sensing and metabolism of glucose and regulating cAMP pathways, correlated with insulin secretion. | [148] |
| Human T2D islets | Single-cell RNA-seq | Dysregulation of 75 genes; downregulation of FXYD2 (unlike the findings in ref 4) and upregulation of GPD2. | [149] |
| Human T2D islets | Single-cell RNA-seq | A more immature gene signature found in T2D β-cells | [147] |
| Human T2D islets | Single-cell RNA-seq | Downregulation of 248 genes in T2D β-cells, including STX1A. | [144] |
| GK T2D rat | RNA-seq and TMT-based proteomics | Downregulation of STX1A and STXBP1. | [150] |
| Human T2D islets | Single-cell RNA-seq | T2D β-cells with increased TNF-α signaling via NF-κB, MAPK8 targets, mTOC1 signaling, hypoxia, glycolysis, proteasome pathway and decreased aerobic cellular respiration compared to non-diabetic β-cells. | [143] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatterjee Bhowmick, D.; Ahn, M.; Oh, E.; Veluthakal, R.; Thurmond, D.C. Conventional and Unconventional Mechanisms by which Exocytosis Proteins Oversee β-cell Function and Protection. Int. J. Mol. Sci. 2021, 22, 1833. https://doi.org/10.3390/ijms22041833
Chatterjee Bhowmick D, Ahn M, Oh E, Veluthakal R, Thurmond DC. Conventional and Unconventional Mechanisms by which Exocytosis Proteins Oversee β-cell Function and Protection. International Journal of Molecular Sciences. 2021; 22(4):1833. https://doi.org/10.3390/ijms22041833
Chicago/Turabian StyleChatterjee Bhowmick, Diti, Miwon Ahn, Eunjin Oh, Rajakrishnan Veluthakal, and Debbie C. Thurmond. 2021. "Conventional and Unconventional Mechanisms by which Exocytosis Proteins Oversee β-cell Function and Protection" International Journal of Molecular Sciences 22, no. 4: 1833. https://doi.org/10.3390/ijms22041833
APA StyleChatterjee Bhowmick, D., Ahn, M., Oh, E., Veluthakal, R., & Thurmond, D. C. (2021). Conventional and Unconventional Mechanisms by which Exocytosis Proteins Oversee β-cell Function and Protection. International Journal of Molecular Sciences, 22(4), 1833. https://doi.org/10.3390/ijms22041833