
Cells Lacking PA200 Adapt to Mitochondrial Dysfunction by Enhancing Glycolysis via Distinct Opa1 Processing

 ,
,  and
and 
Abstract
1. Introduction
2. Results
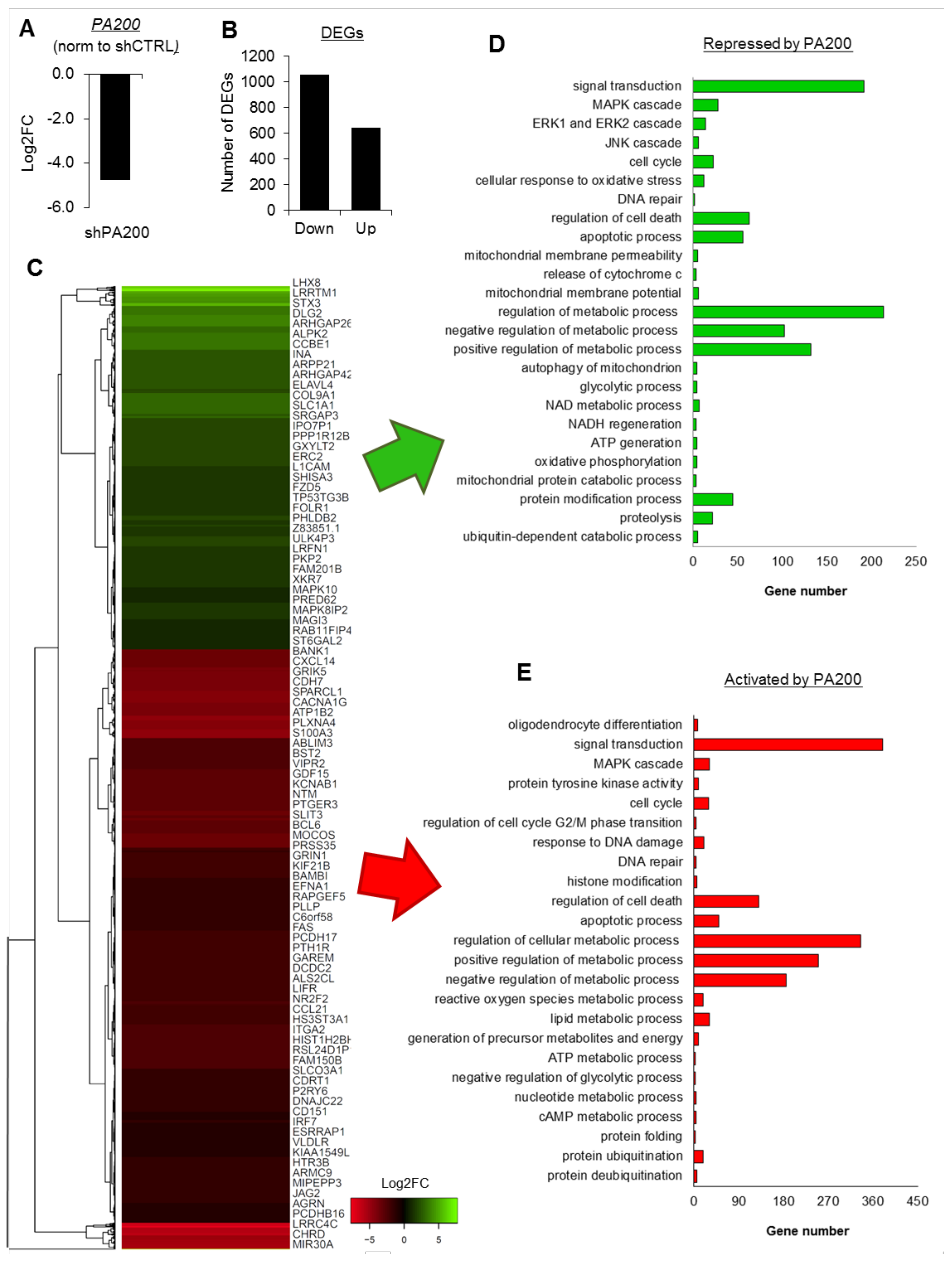
2.1. PA200 Regulates Transcription of Genes Involved in the Key Cellular Processes
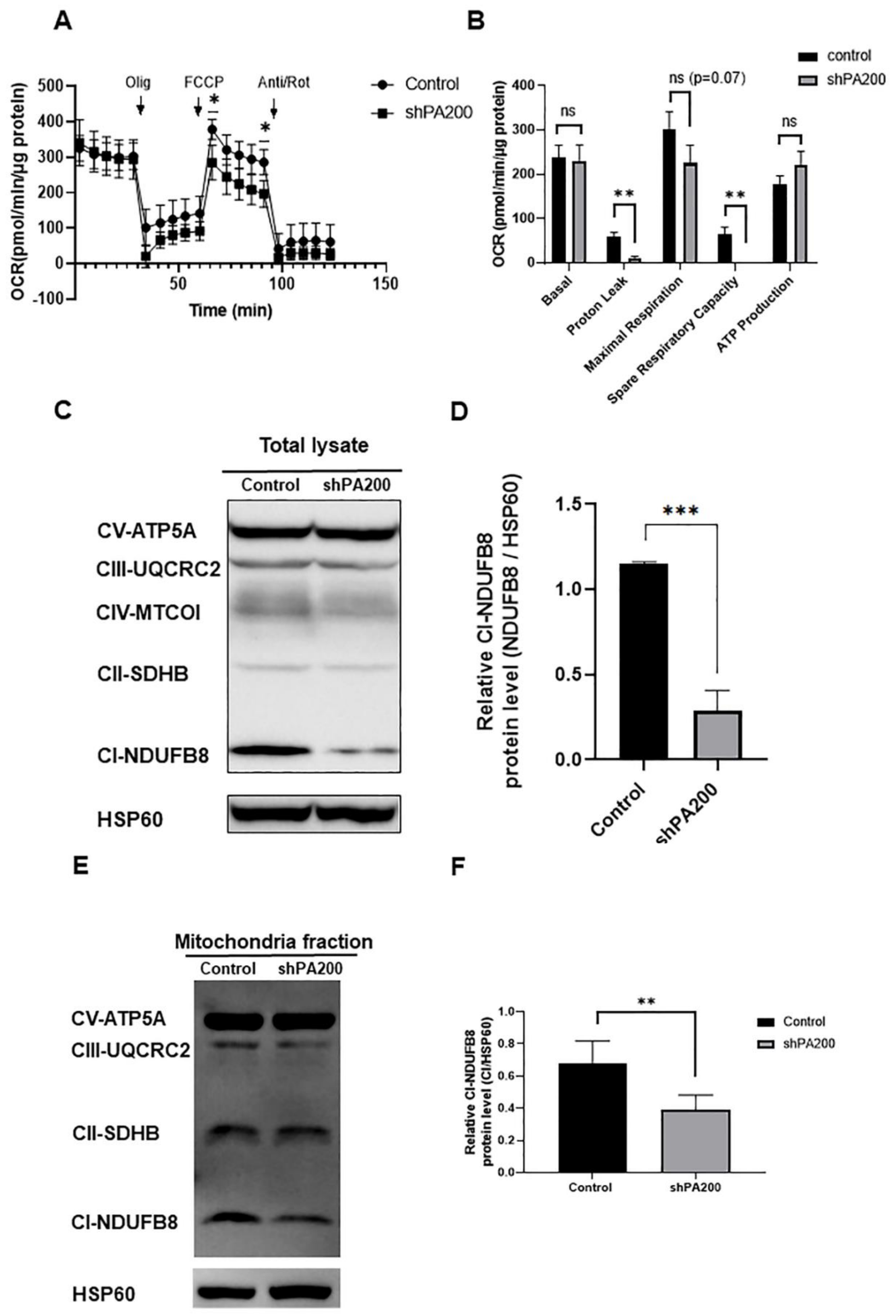
2.2. Mitochondrial Stress Assay Indicates Mitochondrial Dysfunction in shPA200 Cells
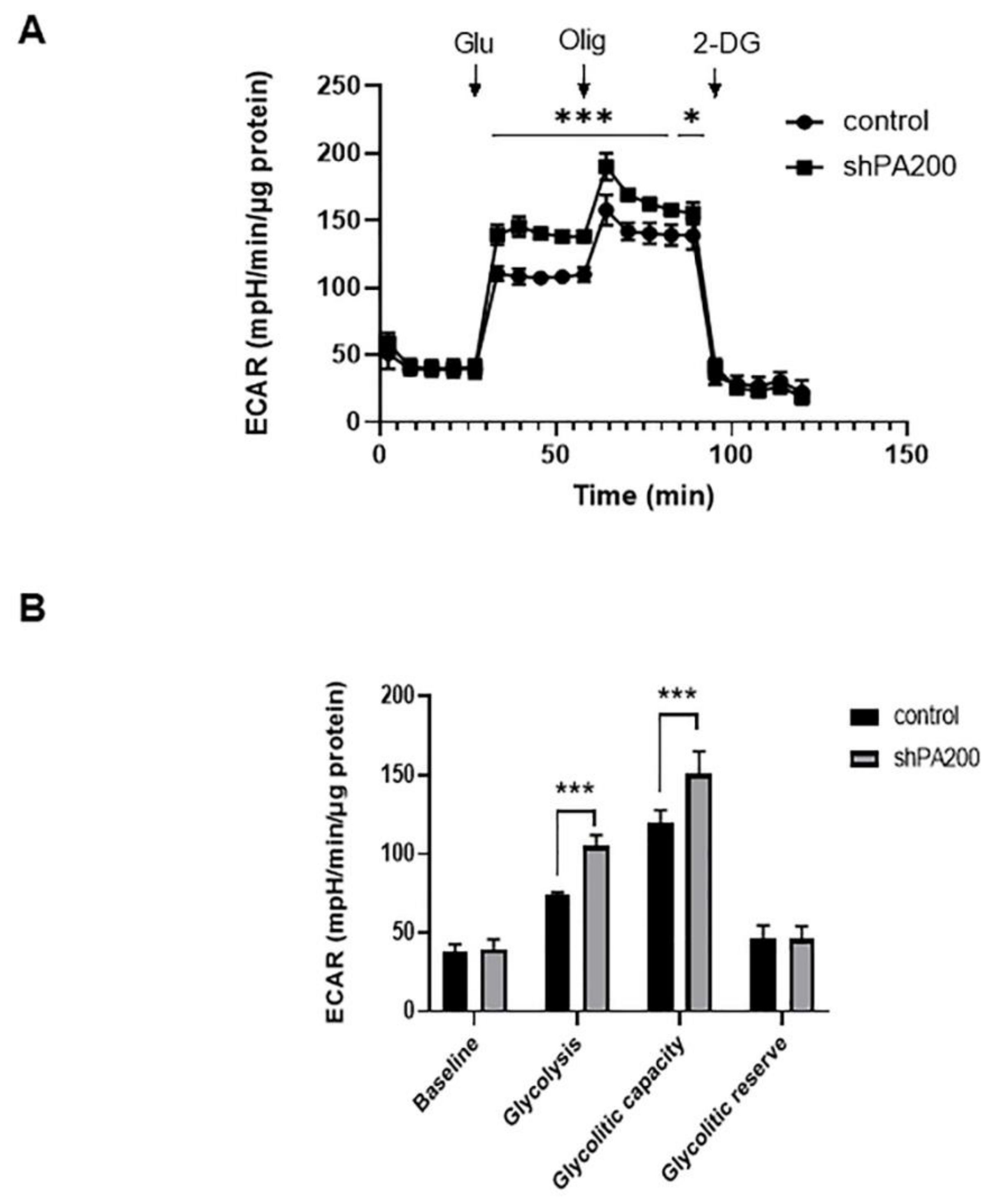
2.3. The Genetic Ablation of PA200 Results in Increased Glycolysis and Glycolytic Capacity
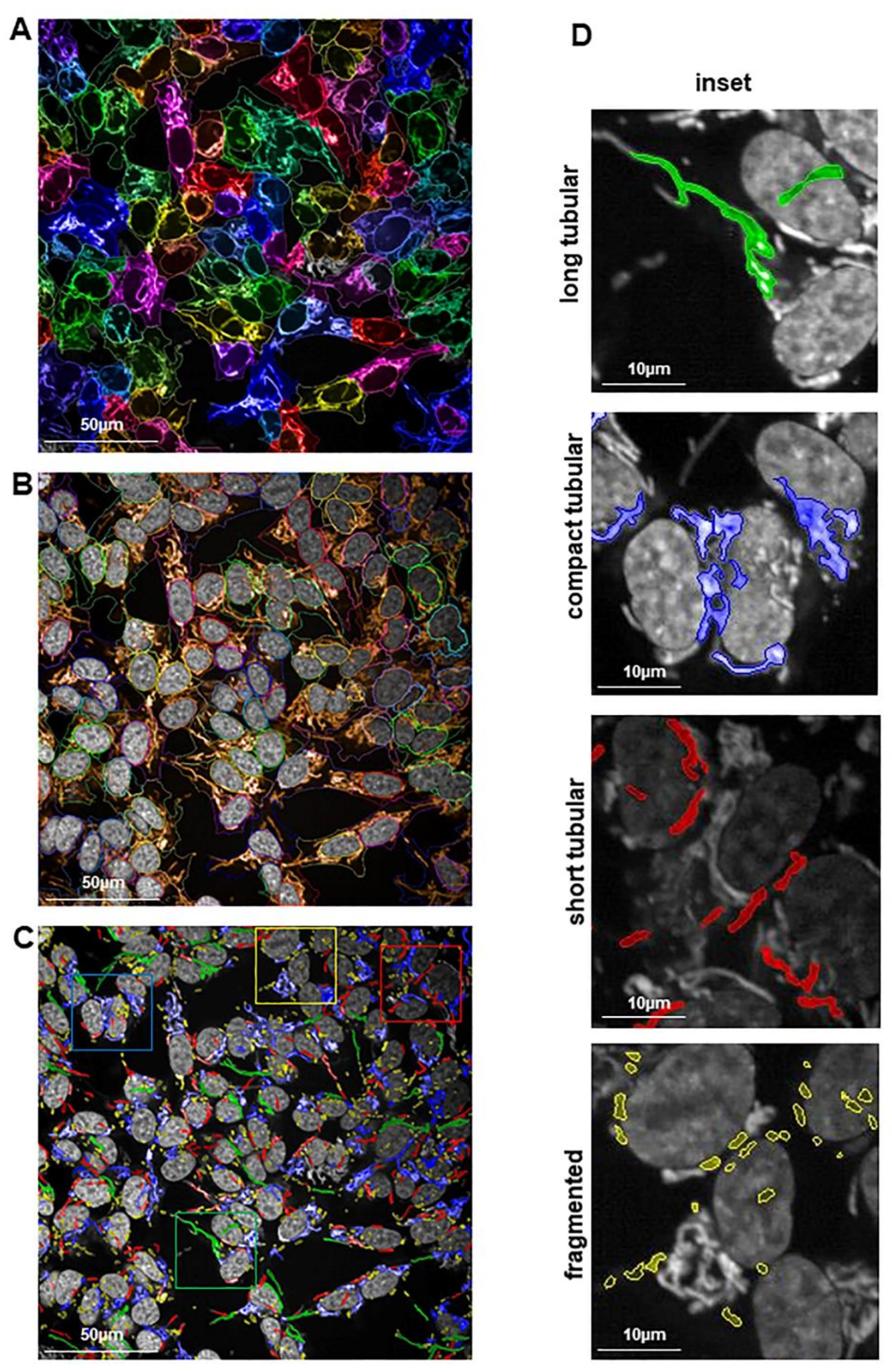
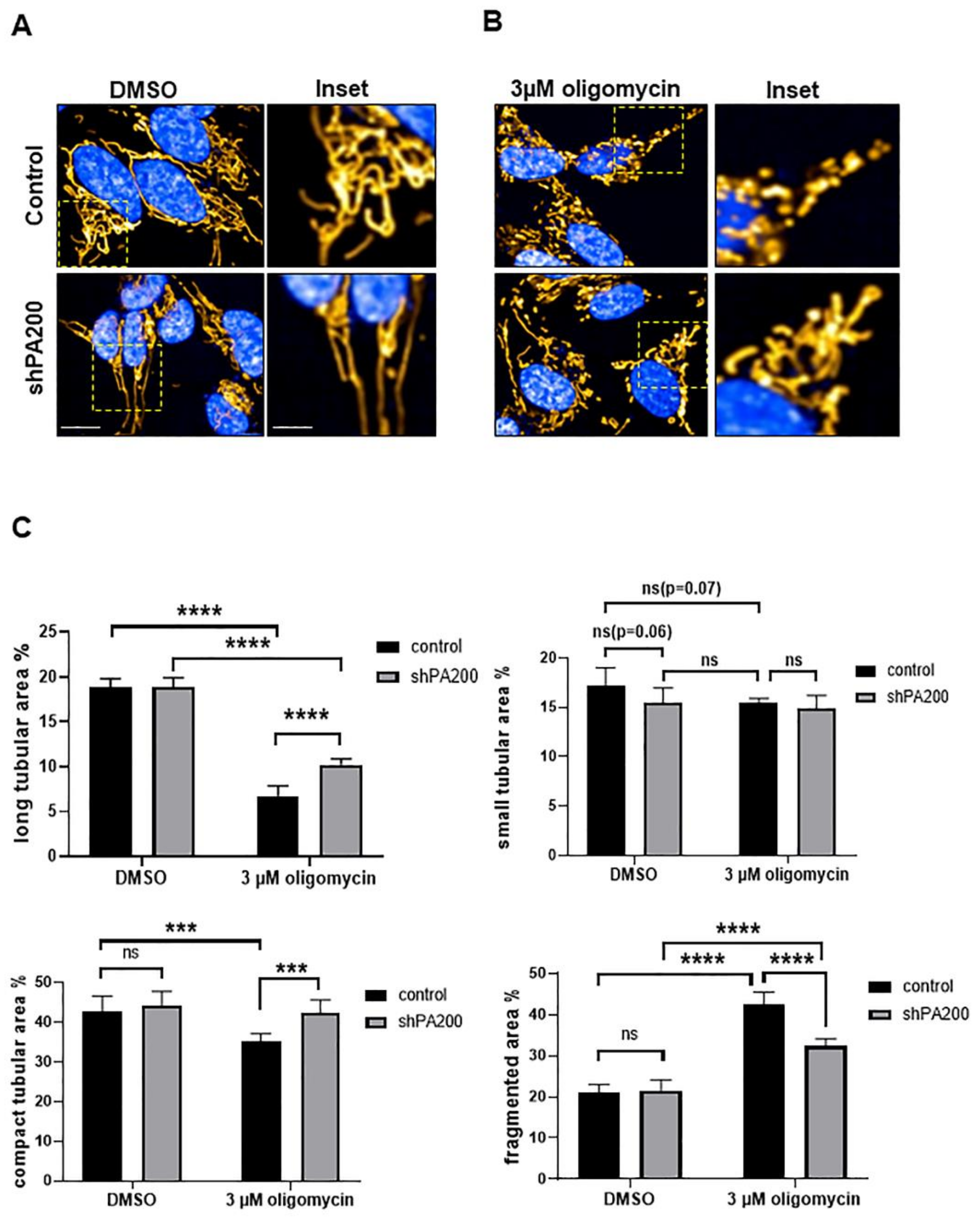
2.4. Preserved Long and Compact Tubular Mitochondrial Morphology in shPA200 Cells after Selective Mitochondrial Inhibitor Treatment
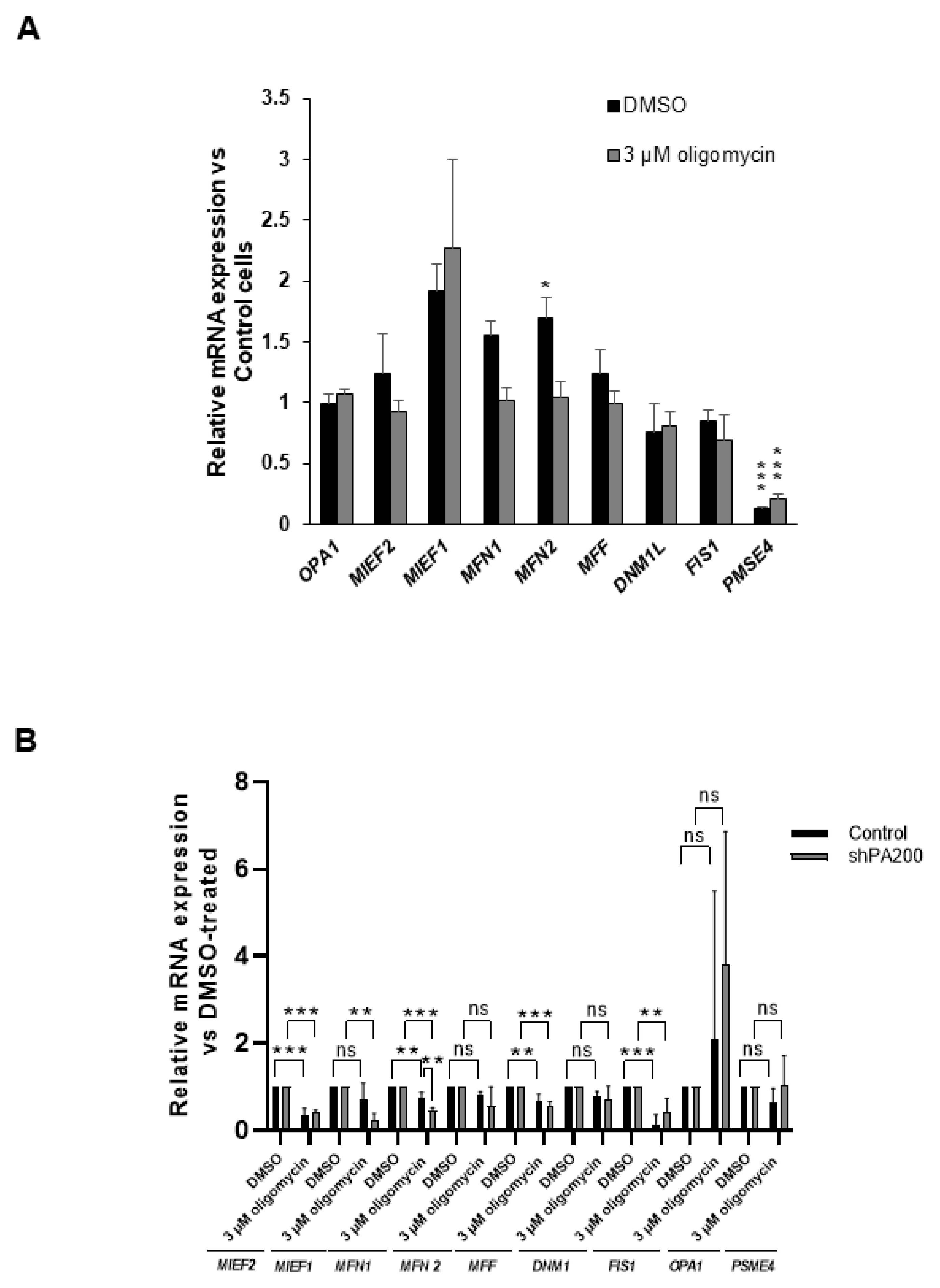
2.5. Effects of PA200 Knockdown on mRNA Expression of Genes Related to Mitochondrial Fusion and Fission
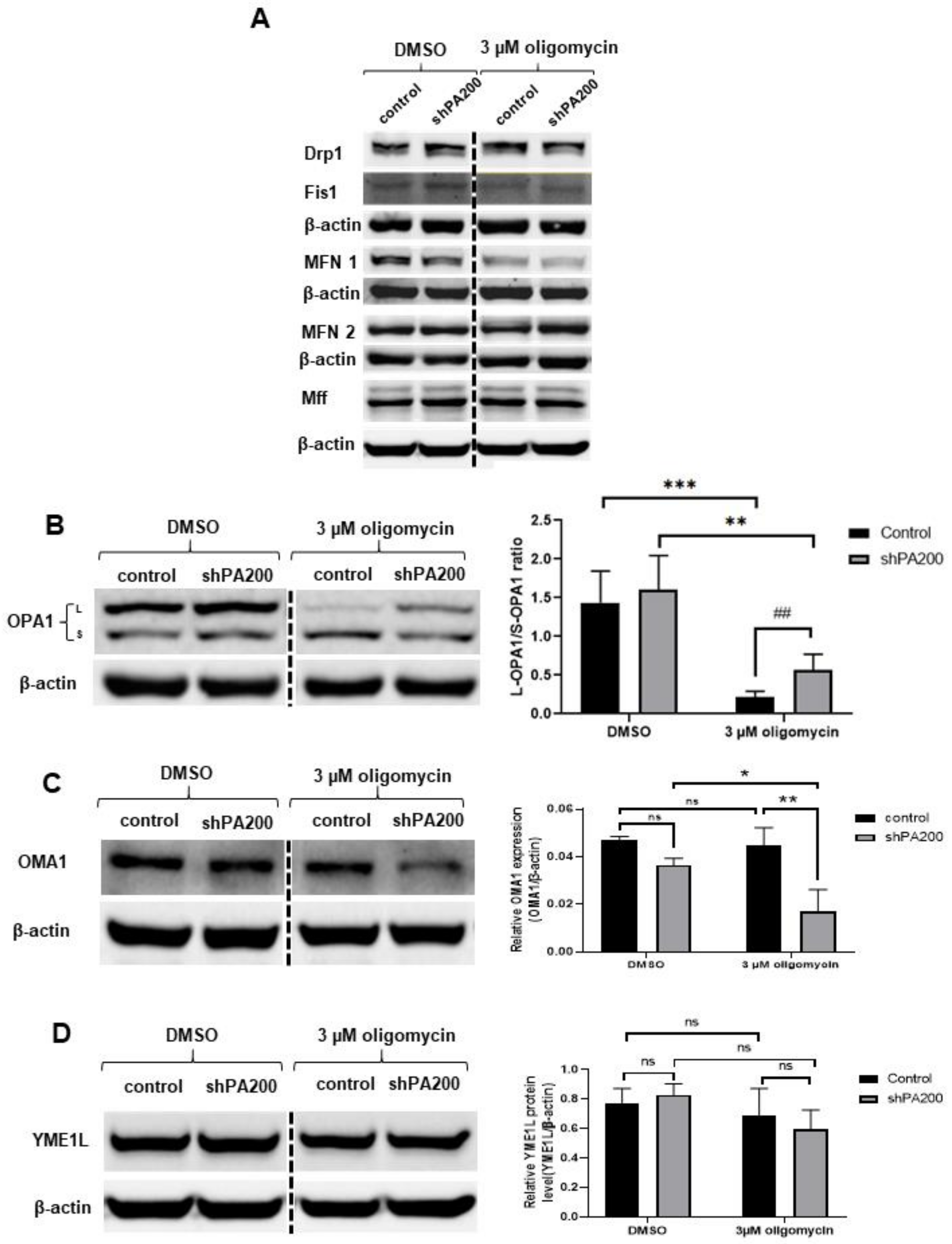
2.6. Genetic Ablation of PA200 Leads to Changes in Opa1 Processing in Cells Exposed to Selective Mitochondrial Insult
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Mitotracker Red CMXRos Staining for High-Content-Screening Confocal Microscopy (HCS)
4.3. Mitochondrial Morphology Analysis
4.4. RNA Extraction and Quantitative RT-PCR
4.5. Quantitative Real-Time PCR
4.6. Mitochondrial Fractionation
4.7. SDS-PAGE and Western Blot
4.8. Seahorse XF 96 Flux Analysis
4.8.1. Mitochondrial Stress Test Assay
4.8.2. Glycolysis Stress Test Assay
4.9. RNA-Seq
4.10. RNA-Seq Data Analysis
4.11. Functional Analysis of RNA-Seq Data
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haran, M.; Gross, A. Balancing glycolysis and mitochondrial OXPHOS: Lessons from the hematopoietic system and exercising muscles. Mitochondrion 2014, 19 Pt A, 3–7. [Google Scholar] [CrossRef]
- Rigoulet, M.; Bouchez, C.L.; Paumard, P.; Ransac, S.; Cuvellier, S.; Duvezin-Caubet, S.; Mazat, J.P.; Devin, A. Cell energy metabolism: An update. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148276. [Google Scholar] [CrossRef] [PubMed]
- Melser, S.; Lavie, J.; Bénard, G. Mitochondrial degradation and energy metabolism. Biochim. Biophys. Acta 2015, 1853, 2812–2821. [Google Scholar] [CrossRef]
- Desler, C.; Hansen, T.L.; Frederiksen, J.B.; Marcker, M.L.; Singh, K.K.; Juel Rasmussen, L. Is There a Link between Mitochondrial Reserve Respiratory Capacity and Aging? J. Aging Res. 2012, 2012, 192503. [Google Scholar] [CrossRef]
- Dranka, B.P.; Benavides, G.A.; Diers, A.R.; Giordano, S.; Zelickson, B.R.; Reily, C.; Zou, L.; Chatham, J.C.; Hill, B.G.; Zhang, J.; et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 2011, 51, 1621–1635. [Google Scholar] [CrossRef]
- Schneider, L.; Giordano, S.; Zelickson, B.R.; S Johnson, M.; A Benavides, G.; Ouyang, X.; Fineberg, N.; Darley-Usmar, V.M.; Zhang, J. Differentiation of SH-SY5Y cells to a neuronal phenotype changes cellular bioenergetics and the response to oxidative stress. Free Radic. Biol. Med. 2011, 51, 2007–2017. [Google Scholar] [CrossRef]
- Patten, D.A.; McGuirk, S.; Anilkumar, U.; Antoun, G.; Gandhi, K.; Parmar, G.; Iqbal, M.A.; Wong, J.; Richardson, R.B.; St-Pierre, J.; et al. Altered mitochondrial fusion drives defensive glutathione synthesis in cells able to switch to glycolytic ATP production. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118854. [Google Scholar] [CrossRef]
- Shiraishi, T.; Verdone, J.E.; Huang, J.; Kahlert, U.D.; Hernandez, J.R.; Torga, G.; Zarif, J.C.; Epstein, T.; Gatenby, R.; McCartney, A.; et al. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 2015, 6, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biophys. Acta 2011, 1807, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Chen, L.; Winger, A.J.; Knowlton, A.A. Mitochondrial dynamic changes in health and genetic diseases. Mol. Biol. Rep. 2014, 41, 7053–7062. [Google Scholar] [CrossRef]
- Cid-Castro, C.; Hernández-Espinosa, D.R.; Morán, J. ROS as Regulators of Mitochondrial Dynamics in Neurons. Cell Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Shirendeb, U.P. Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington’s disease. Biochim. Biophys. Acta 2012, 1822, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Otsu, K. Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem. J. 2018, 475, 839–852. [Google Scholar] [CrossRef]
- Márton, J.; Fodor, T.; Nagy, L.; Vida, A.; Kis, G.; Brunyánszki, A.; Antal, M.; Lüscher, B.; Bai, P. PARP10 (ARTD10) modulates mitochondrial function. PLoS ONE 2018, 13, e0187789. [Google Scholar] [CrossRef]
- Antico Arciuch, V.G.; Elguero, M.E.; Poderoso, J.J.; Carreras, M.C. Mitochondrial regulation of cell cycle and proliferation. Antioxid. Redox Signal. 2012, 16, 1150–1180. [Google Scholar] [CrossRef]
- Tanwar, D.K.; Parker, D.J.; Gupta, P.; Spurlock, B.; Alvarez, R.D.; Basu, M.K.; Mitra, K. Crosstalk between the mitochondrial fission protein, Drp1, and the cell cycle is identified across various cancer types and can impact survival of epithelial ovarian cancer patients. Oncotarget 2016, 7, 60021–60037. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Bleazard, W.; McCaffery, J.M.; King, E.J.; Bale, S.; Mozdy, A.; Tieu, Q.; Nunnari, J.; Shaw, J.M. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1999, 1, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Sesaki, H.; Jensen, R.E. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J. Cell Biol. 1999, 147, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Tieu, Q.; Okreglak, V.; Naylor, K.; Nunnari, J. The WD repeat protein, Mdv1p, functions as a molecular adaptor by interacting with Dnm1p and Fis1p during mitochondrial fission. J. Cell Biol. 2002, 158, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Koirala, S.; Guo, Q.; Kalia, R.; Bui, H.T.; Eckert, D.M.; Frost, A.; Shaw, J.M. Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc. Natl. Acad. Sci. USA 2013, 110, E1342–E1351. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.H.; Brisch, E.; Keegan, B.R.; Bleazard, W.; Shaw, J.M. The GTPase effector domain sequence of the Dnm1p GTPase regulates self-assembly and controls a rate-limiting step in mitochondrial fission. Mol. Biol. Cell 2001, 12, 2756–2766. [Google Scholar] [CrossRef]
- Del Dotto, V.; Mishra, P.; Vidoni, S.; Fogazza, M.; Maresca, A.; Caporali, L.; McCaffery, J.M.; Cappelletti, M.; Baruffini, E.; Lenaers, G.; et al. OPA1 Isoforms in the Hierarchical Organization of Mitochondrial Functions. Cell Rep. 2017, 19, 2557–2571. [Google Scholar] [CrossRef]
- Del Dotto, V.; Fogazza, M.; Carelli, V.; Rugolo, M.; Zanna, C. Eight human OPA1 isoforms, long and short: What are they for? Biochim. Biophys. Acta 2018, 1859, 263–269. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease—The long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef] [PubMed]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef]
- Del Dotto, V.; Fogazza, M.; Lenaers, G.; Rugolo, M.; Carelli, V.; Zanna, C. OPA1: How much do we know to approach therapy? Pharmacol. Res. 2018, 131, 199–210. [Google Scholar] [CrossRef]
- Landes, T.; Leroy, I.; Bertholet, A.; Diot, A.; Khosrobakhsh, F.; Daloyau, M.; Davezac, N.; Miquel, M.C.; Courilleau, D.; Guillou, E.; et al. OPA1 (dys)functions. Semin. Cell Dev. Biol. 2010, 21, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Douida, A.; Batista, F.; Robaszkiewicz, A.; Boto, P.; Aladdin, A.; Szenykiv, M.; Czinege, R.; Virág, L.; Tar, K. The proteasome activator PA200 regulates expression of genes involved in cell survival upon selective mitochondrial inhibition in neuroblastoma cells. J. Cell Mol. Med. 2020, 24, 6716–6730. [Google Scholar] [CrossRef]
- Ustrell, V.; Hoffman, L.; Pratt, G.; Rechsteiner, M. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 2002, 21, 3516–3525. [Google Scholar] [CrossRef]
- Sadre-Bazzaz, K.; Whitby, F.G.; Robinson, H.; Formosa, T.; Hill, C.P. Structure of a Blm10 complex reveals common mechanisms for proteasome binding and gate opening. Mol. Cell 2010, 37, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.X.; Pang, Y.; Liu, C.H.; Haratake, K.; Du, B.Y.; Ji, D.Y.; Wang, G.F.; Zhu, Q.Q.; Song, W.; Yu, Y.; et al. Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell 2013, 153, 1012–1024. [Google Scholar] [CrossRef]
- Toste Rêgo, A.; da Fonseca, P.C.A. Characterization of Fully Recombinant Human 20S and 20S-PA200 Proteasome Complexes. Mol. Cell 2019, 76, 138–147.e135. [Google Scholar] [CrossRef]
- Guan, H.; Wang, Y.; Yu, T.; Huang, Y.; Li, M.; Saeed, A.F.U.H.; Perčulija, V.; Li, D.; Xiao, J.; Wang, D.; et al. Cryo-EM structures of the human PA200 and PA200-20S complex reveal regulation of proteasome gate opening and two PA200 apertures. PLoS Biol. 2020, 18, e3000654. [Google Scholar] [CrossRef] [PubMed]
- Aladdin, A.; Yao, Y.; Yang, C.; Kahlert, G.; Ghani, M.; Király, N.; Boratkó, A.; Uray, K.; Dittmar, G.; Tar, K. The Proteasome Activators Blm10/PA200 Enhance the Proteasomal Degradation of N-Terminal Huntingtin. Biomolecules 2020, 10, 1581. [Google Scholar] [CrossRef]
- Tar, K.; Dange, T.; Yang, C.; Yao, Y.; Bulteau, A.L.; Salcedo, E.F.; Braigen, S.; Bouillaud, F.; Finley, D.; Schmidt, M. Proteasomes associated with the Blm10 activator protein antagonize mitochondrial fission through degradation of the fission protein Dnm1. J. Biol. Chem. 2014, 289, 12145–12156. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Fovez, Q.; Germain, N.; Khamari, R.; Kluza, J. Mitochondrial spare respiratory capacity: Mechanisms, regulation, and significance in non-transformed and cancer cells. FASEB J. 2020, 34, 13106–13124. [Google Scholar] [CrossRef]
- Stroud, D.A.; Surgenor, E.E.; Formosa, L.E.; Reljic, B.; Frazier, A.E.; Dibley, M.G.; Osellame, L.D.; Stait, T.; Beilharz, T.H.; Thorburn, D.R.; et al. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 2016, 538, 123–126. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.P.; Cameron, R.B.; Speiser, J.L.; Wolf, B.J.; Peterson, Y.K.; Schnellmann, R.G.; Beeson, C.C.; Rohrer, B. Quantitative analysis of mitochondrial morphology and membrane potential in living cells using high-content imaging, machine learning, and morphological binning. Biochim. Biophys. Acta 2015, 1853, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Han, S.Y.; Jeong, Y.J.; Choi, Y.; Hwang, S.K.; Bae, Y.S.; Chang, Y.C. Mitochondrial dysfunction induces the invasive phenotype, and cell migration and invasion, through the induction of AKT and AMPK pathways in lung cancer cells. Int. J. Mol. Med. 2018, 42, 1644–1652. [Google Scholar] [CrossRef]
- Hao, W.; Chang, C.P.; Tsao, C.C.; Xu, J. Oligomycin-induced bioenergetic adaptation in cancer cells with heterogeneous bioenergetic organization. J. Biol. Chem. 2010, 285, 12647–12654. [Google Scholar] [CrossRef]
- Elgass, K.; Pakay, J.; Ryan, M.T.; Palmer, C.S. Recent advances into the understanding of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Henriques, M.; Joaquim, M. Mitofusins: Disease Gatekeepers and Hubs in Mitochondrial Quality Control by E3 Ligases. Front. Physiol. 2019, 10, 517. [Google Scholar] [CrossRef]
- Pagliuso, A.; Cossart, P.; Stavru, F. The ever-growing complexity of the mitochondrial fission machinery. Cell Mol. Life Sci. 2018, 75, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Dasgupta, A.; Lin, J.; Potus, F.; Bonnet, S.; Iremonger, J.; Fu, J.; Mewburn, J.; Wu, D.; Dunham-Snary, K.; et al. Epigenetic Dysregulation of the Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increases Mitotic Mitochondrial Fission and Promotes Pulmonary Arterial Hypertension: Mechanistic and Therapeutic Implications. Circulation 2018, 138, 287–304. [Google Scholar] [CrossRef] [PubMed]
- Samangouei, P.; Crespo-Avilan, G.E.; Cabrera-Fuentes, H.; Hernández-Reséndiz, S.; Ismail, N.I.; Katwadi, K.B.; Boisvert, W.A.; Hausenloy, D.J. MiD49 and MiD51: New mediators of mitochondrial fission and novel targets for cardioprotection. Cond. Med. 2018, 1, 239–246. [Google Scholar] [PubMed]
- Yang, F.; Li, B.; Yang, Y.; Huang, M.; Liu, X.; Zhang, Y.; Liu, H.; Zhang, L.; Pan, Y.; Tian, S.; et al. Leptin enhances glycolysis via OPA1-mediated mitochondrial fusion to promote mesenchymal stem cell survival. Int. J. Mol. Med. 2019, 44, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Amini, P.; Stojkov, D.; Felser, A.; Jackson, C.B.; Courage, C.; Schaller, A.; Gelman, L.; Soriano, M.E.; Nuoffer, J.M.; Scorrano, L.; et al. Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat. Commun. 2018, 9, 2958. [Google Scholar] [CrossRef] [PubMed]
- Duvezin-Caubet, S.; Jagasia, R.; Wagener, J.; Hofmann, S.; Trifunovic, A.; Hansson, A.; Chomyn, A.; Bauer, M.F.; Attardi, G.; Larsson, N.G.; et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J. Biol. Chem. 2006, 281, 37972–37979. [Google Scholar] [CrossRef]
- Baricault, L.; Ségui, B.; Guégand, L.; Olichon, A.; Valette, A.; Larminat, F.; Lenaers, G. OPA1 cleavage depends on decreased mitochondrial ATP level and bivalent metals. Exp. Cell Res. 2007, 313, 3800–3808. [Google Scholar] [CrossRef]
- MacVicar, T.D.; Lane, J.D. Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. J. Cell Sci. 2014, 127, 2313–2325. [Google Scholar] [CrossRef]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Stiburek, L.; Cesnekova, J.; Kostkova, O.; Fornuskova, D.; Vinsova, K.; Wenchich, L.; Houstek, J.; Zeman, J. YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol. Biol. Cell 2012, 23, 1010–1023. [Google Scholar] [CrossRef]
- Jiang, T.-X.; Ma, S.; Han, X.; Luo, Z.-Y.; Zhu, Q.-Q.; Chiba, T.; Xie, W.; Lin, K.; Qiu, X.-B. Proteasome activator PA200 maintains stability of histone marks during transcription and aging. Theranostics 2021, 11, 1458–1472. [Google Scholar] [CrossRef]
- Lopez, A.D.; Tar, K.; Krügel, U.; Dange, T.; Ros, I.G.; Schmidt, M. Proteasomal degradation of Sfp1 contributes to the repression of ribosome biogenesis during starvation and is mediated by the proteasome activator Blm10. Mol. Biol. Cell 2011, 22, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Morino, K.; Mengistu, L.; Ishibashi, T.; Kiriyama, K.; Ikami, T.; Maegawa, H. Amla Enhances Mitochondrial Spare Respiratory Capacity by Increasing Mitochondrial Biogenesis and Antioxidant Systems in a Murine Skeletal Muscle Cell Line. Oxid. Med. Cell Longev. 2016, 2016, 1735841. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Schmitt, K.; Grimm, A.; Dallmann, R.; Oettinghaus, B.; Restelli, L.M.; Witzig, M.; Ishihara, N.; Mihara, K.; Ripperger, J.A.; Albrecht, U.; et al. Circadian Control of DRP1 Activity Regulates Mitochondrial Dynamics and Bioenergetics. Cell Metab. 2018, 27, 657–666.e655. [Google Scholar] [CrossRef]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
|---|---|---|
| hPSME4 | ATGGAGAGTGCCTGAACTATTG | GTAGGTCAGCACACTTCCTATTC |
| hFIS1 | AGCTGGTGTCTGTGGAGGAC | ACGATGCCTTTACGGATGTC |
| hMFN1 | CGGAACTTGATCGAATAGCC | AGAGCTCTTCCCACTGCTTG |
| hMFN2 | ATGCATCCCCACTTAAGCAC | AGCACCTCACTGATGCCTCT |
| hDNM1L | AGATCTCATCCCGCTGGTC | CAGATCCTCGAGGCAAGAAG |
| hMIEF2 | GCAGAGTTCTCCCAGAAACG | GTCTGCCTTGGTGTCATCCT |
| hMIEF1 | GCAAAGGCAAGAAGGATGAC | CTTCATGTCCCTGTTCAGCA |
| hOPA1 | CACTTCCTGGGTCATTCCTG | TGCTTCGTGAAACCAGATGT |
| hMFF | AAACGCTGACCTGGAACAAG | TTTTCAGTGCCAGGGGTTTA |
| hβ-actin | GACCCAGATCATGTTTGAGACC | CATCACGATGCCAGTGGTAC |
| Antibody | Source | Catalog Number | Host | Dilution |
|---|---|---|---|---|
| Drp1 | BD Biosciences | # 611112 | Mouse | 1:1000 |
| Mfn1 | Abnova | # H00055669-M04 | Mouse | 1:1000 |
| Mfn2 | Sigma Aldrich | # WH0009927M3 | Mouse | 1:800 |
| Opa1 | Novus Biologicals | # NB110-55290 | Rabbit | 1:1000 |
| OXPHOS | Abcam | # ab110413 | Mouse | 1:250 |
| OMA1 | SantaCruz Biotechnology | # sc-515788 | Mouse | 1:500 |
| YME1L | Proteintech | # 11510-1-AP | Rabbit | 1:1000 |
| Fis1 | Invitrogen, Thermo Fisher | # PA1-41082 | Rabbit | 1:1000 |
| Hsp60 | Invitrogen, Thermo Fisher | # MA3-012 | Mouse | 1:1000 |
| β-actin-HRP | SantaCruz Biotechnology | # sc-1616 | Goat | 1:5000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Douida, A.; Batista, F.; Boto, P.; Regdon, Z.; Robaszkiewicz, A.; Tar, K. Cells Lacking PA200 Adapt to Mitochondrial Dysfunction by Enhancing Glycolysis via Distinct Opa1 Processing. Int. J. Mol. Sci. 2021, 22, 1629. https://doi.org/10.3390/ijms22041629
Douida A, Batista F, Boto P, Regdon Z, Robaszkiewicz A, Tar K. Cells Lacking PA200 Adapt to Mitochondrial Dysfunction by Enhancing Glycolysis via Distinct Opa1 Processing. International Journal of Molecular Sciences. 2021; 22(4):1629. https://doi.org/10.3390/ijms22041629
Chicago/Turabian StyleDouida, Abdennour, Frank Batista, Pal Boto, Zsolt Regdon, Agnieszka Robaszkiewicz, and Krisztina Tar. 2021. "Cells Lacking PA200 Adapt to Mitochondrial Dysfunction by Enhancing Glycolysis via Distinct Opa1 Processing" International Journal of Molecular Sciences 22, no. 4: 1629. https://doi.org/10.3390/ijms22041629
APA StyleDouida, A., Batista, F., Boto, P., Regdon, Z., Robaszkiewicz, A., & Tar, K. (2021). Cells Lacking PA200 Adapt to Mitochondrial Dysfunction by Enhancing Glycolysis via Distinct Opa1 Processing. International Journal of Molecular Sciences, 22(4), 1629. https://doi.org/10.3390/ijms22041629