
Genomic and Epigenomic Profile of Uterine Smooth Muscle Tumors of Uncertain Malignant Potential (STUMPs) Revealed Similarities and Differences with Leiomyomas and Leiomyosarcomas

 , , ,
, , , 
Abstract
1. Introduction
2. Results
2.1. Pathologic Features
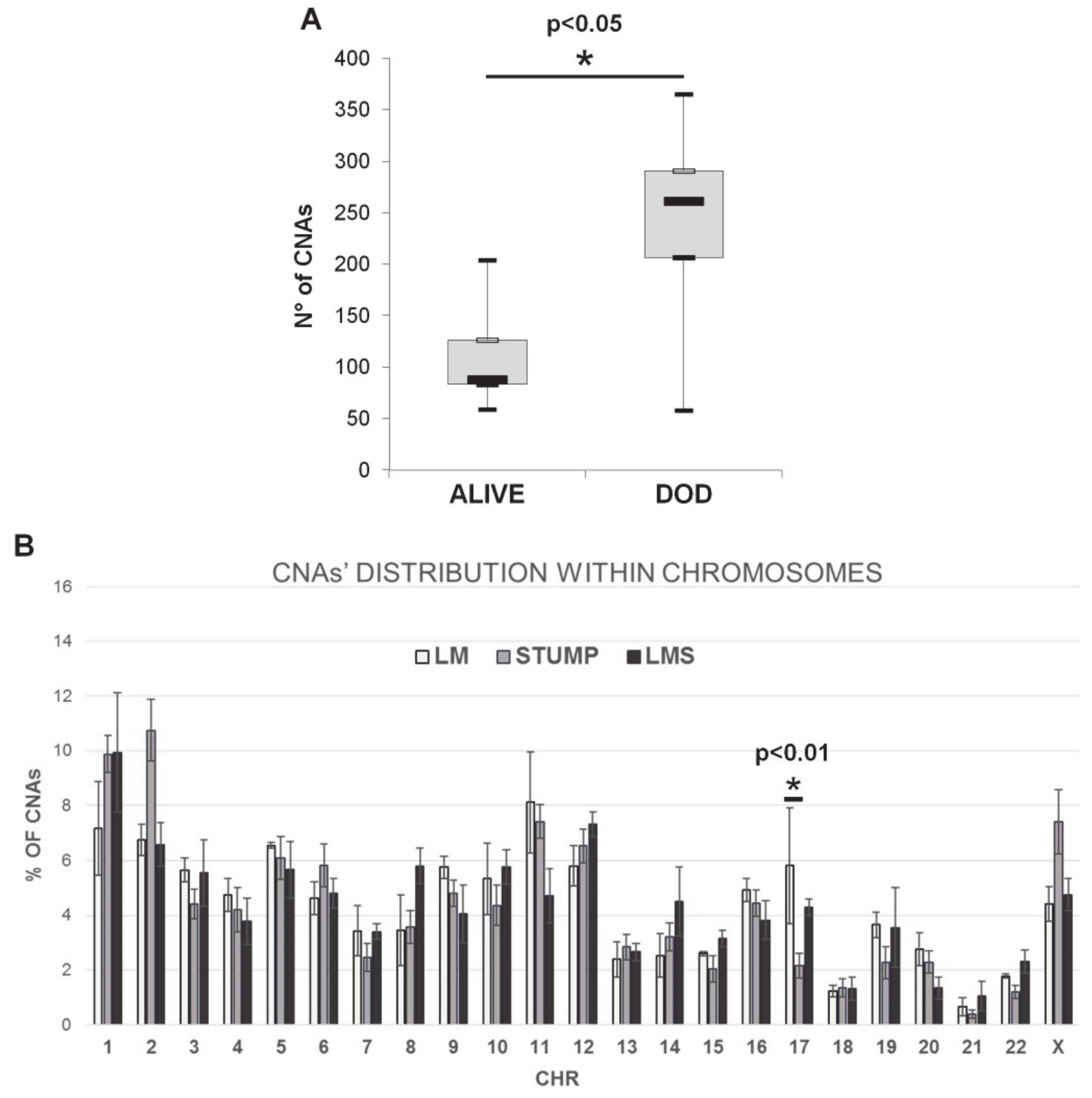
2.2. Array-CGH Results
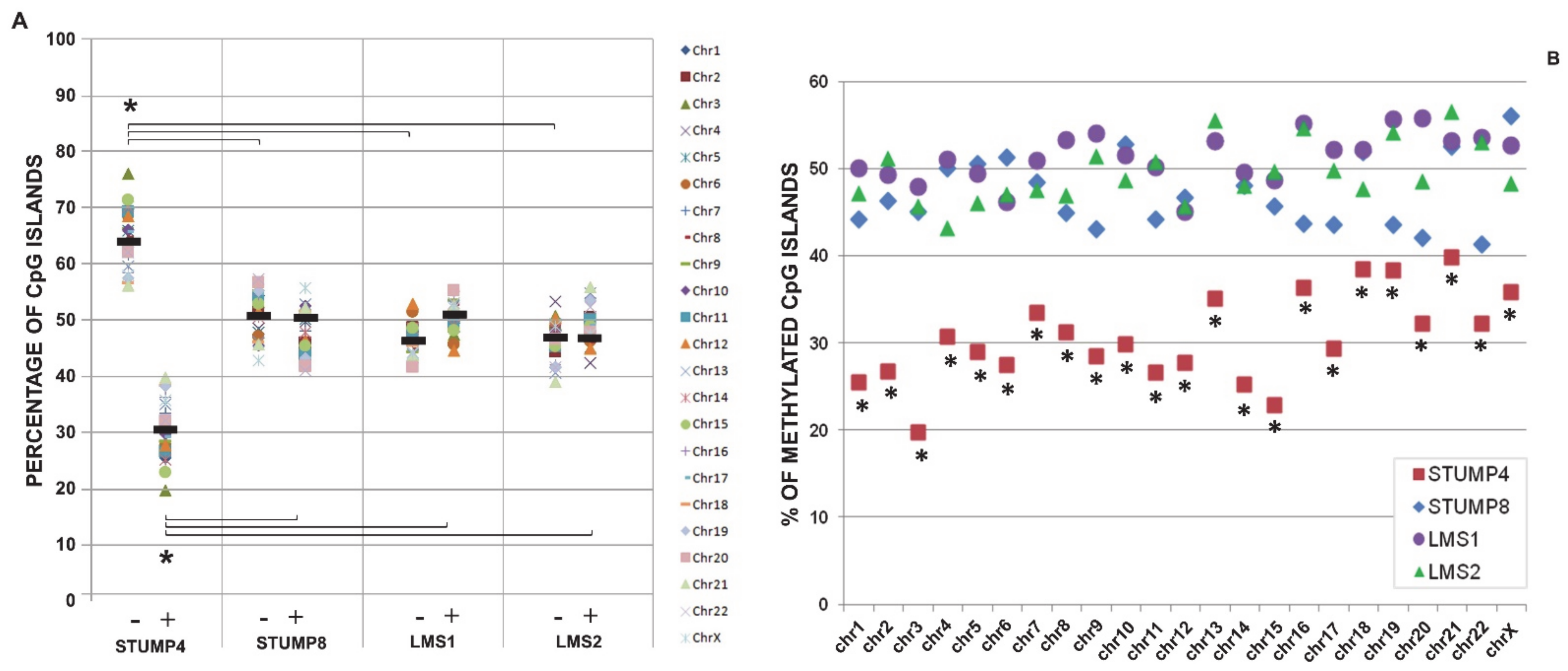
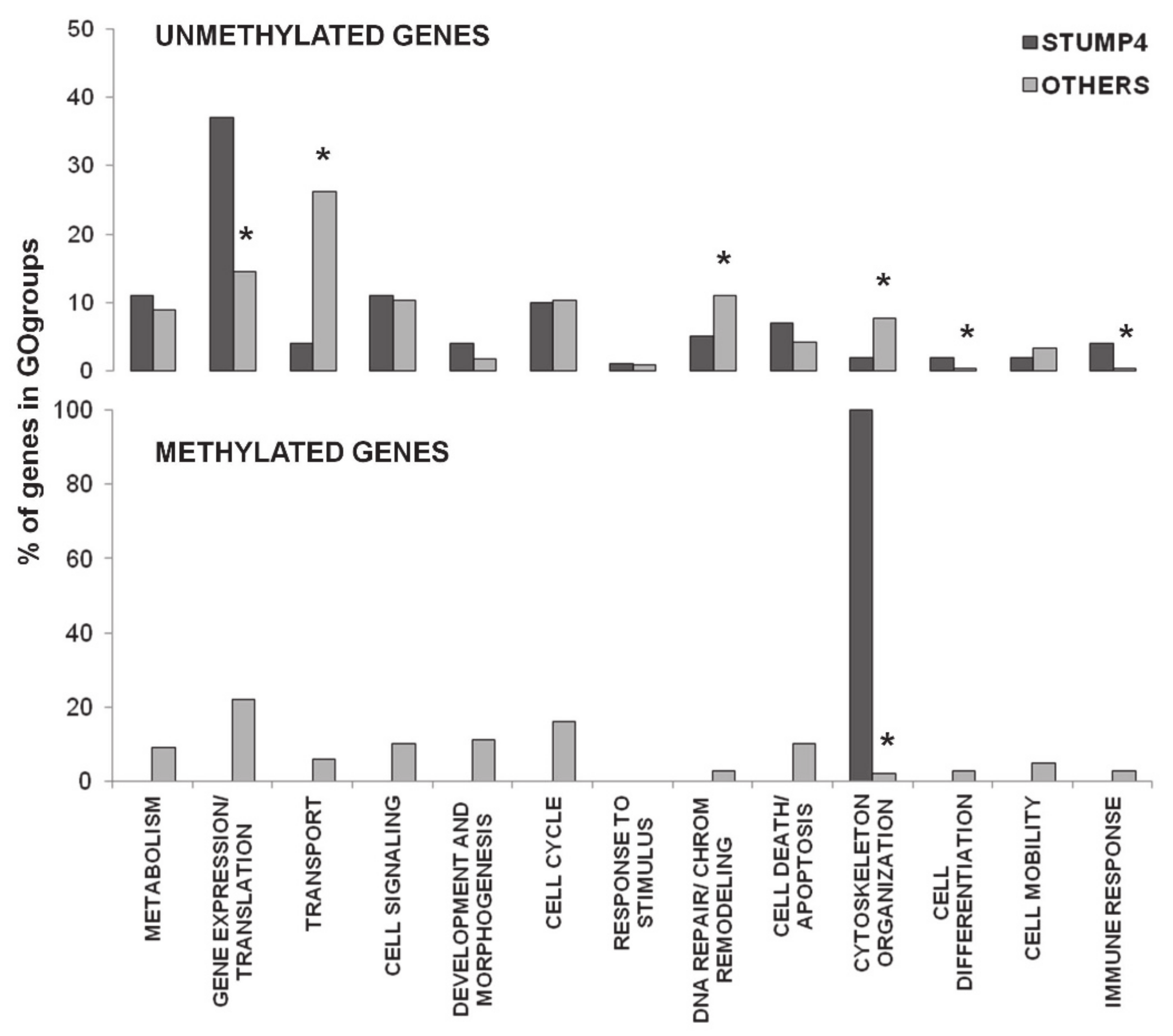
2.3. Promoter CpG Island Methylation
3. Discussion
4. Materials and Methods
4.1. Tumor Samples/Patients
4.2. Immunohistochemistry
4.3. DNA Extraction from FFPE Tissues
4.4. Array Comparative Genomic Hybridization (Array-CGH)
4.5. MeDIP-Chip
4.6. Bionformatic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CGH | Comparative Genomic Hybridization |
| CGIs | CpG Island |
| CNAs | Copy Number Alterations |
| GO | Gene Ontology |
| LM | Leiomyoma |
| LMS | Leiomyosarcoma |
| STUMPs | Uterine Smooth muscle Tumors of Uncertain Malignant Potential |
| USMTs | Uterine Smooth Muscle Tumors |
References
- Kurman, R.J.; Carcangiu, M.L.; Herrington, C.S.; Young, R.H. WHO Classification of Tumours of Female Reproductive Organs, 4th ed.; WHO Press, World Health Organization: Geneva, Switzerland, 2014; Volume 6. [Google Scholar]
- Ip, P.P.; Tse, K.Y.; Tam, K.F. Uterine Smooth Muscle Tumors Other Than the Ordinary Leiomyomas and Leiomyosarcomas: A Review of Selected Variants With Emphasis on Recent Advances and Unusual Morphology That May Cause Concern for Malignancy. Adv. Anat. Pathol. 2010, 17, 91–112. [Google Scholar] [CrossRef]
- Gadducci, A.; Zannoni, G.F. Uterine smooth muscle tumors of unknown malignant potential: A challenging question. Gynecol. Oncol. 2019, 154, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Croce, S.; Ducoulombier, A.; Ribeiro, A.; Lesluyes, T.; Noel, J.C.; Amant, F.; Guillou, L.; Stoeckle, E.; Devouassoux-Shisheboran, M.; Penel, N.; et al. Genome profiling is an efficient tool to avoid the STUMP classification of uterine smooth muscle lesions: A comprehensive array-genomic hybridization analysis of 77 tumors. Mod. Pathol. 2018, 31, 816–828. [Google Scholar] [CrossRef]
- Conconi, D.; Chiappa, V.; Perego, P.; Redaelli, S.; Bovo, G.; Lavitrano, M.; Milani, R.; Dalprà, L.; Lissoni, A.A. Potential role of BCL2 in the recurrence of uterine smooth muscle tumors of uncertain malignant potential. Oncol. Rep. 2017, 37, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Baronchelli, S.; Bentivegna, A.; Redaelli, S.; Riva, G.; Butta, V.; Paoletta, L.; Isimbaldi, G.; Miozzo, M.; Tabano, S.; Daga, A.; et al. Delineating the cytogenomic and epigenomic landscapes of glioma stem cell lines. PLoS ONE 2013, 8, e57462. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.I.; Choi, M.C.; Heo, J.H.; Kim, K.A.; Jung, S.G.; Park, H.; Joo, W.D.; Song, S.H.; Kim, T.H.; Lee, C. A clinicopathologic review and obstetric outcome of uterine smooth muscle tumor of uncertain malignant potential (STUMP) in a single institution. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 228, 1–5. [Google Scholar] [CrossRef]
- Cornell, L.; Munck, J.M.; Alsinet, C.; Villanueva, A.; Ogle, L.; Willoughby, C.E.; Televantou, D.; Thomas, H.D.; Jackson, J.; Burt, A.D.; et al. DNA-PK-A candidate driver of hepatocarcinogenesis and tissue biomarker that predicts response to treatment and survival. Clin. Cancer Res. 2015, 21, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Cheng, S.; Zhu, Y.; Zhang, P.; Liu, N.; Xu, T.; Sun, C.; Lv, Y. Identification of PRKDC (Protein Kinase, DNA-Activated, Catalytic Polypeptide) as an essential gene for colorectal cancer (CRCs) cells. Gene 2016, 584, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wen, G.M.; Wu, C.A.; Jing, Z.L.; Li, D.Z.; Liu, G.L.; Wei, X.X.; Tang, M.S.; Li, Y.H.; Zhong, Y.; et al. PRKDC is a prognostic marker for poor survival in gastric cancer patients and regulates DNA damage response. Pathol. Res. Pract. 2019, 215, 152509. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, W.K.; Wen, G.M.; Tang, H.; Wu, C.A.; Wu, Y.X.; Jing, Z.L.; Tang, M.S.; Liu, G.L.; Li, D.Z.; et al. High expression of PRKDC promotes breast cancer cell growth via p38 MAPK signaling and is associated with poor survival. Mol. Genet. Genom. Med. 2019, 7, e908. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shilatifard, A. UTX Mutations in Human Cancer. Cancer Cell 2019, 35, 168–176. [Google Scholar] [CrossRef]
- Shevchenko, V.; Arnotskaya, N.; Zaitsev, S.; Sharma, A.; Sharma, H.S.; Bryukhovetskiy, A.; Pak, O.; Khotimchenko, Y.; Bryukhovetskiy, I. Proteins of Wnt signaling pathway in cancer stem cells of human glioblastoma. Int. Rev. Neurobiol. 2020, 151, 185–200. [Google Scholar] [PubMed]
- Li, M.; Ke, J.; Wang, Q.; Qian, H.; Yang, L.; Zhang, X.; Xiao, J.; Ding, H.; Shan, X.; Liu, Q.; et al. Upregulation of ROCK2 in gastric cancer cell promotes tumor cell proliferation, metastasis and invasion. Clin. Exp. Med. 2017, 17, 519–529. [Google Scholar] [CrossRef]
- Hu, J.; Wang, L.; Zhao, W.; Huang, Y.; Wang, Z.; Shen, H. mi-R4435-2HG promotes proliferation and inhibits apoptosis of cancer cells in ovarian carcinoma by upregulating ROCK2. Oncol. Lett. 2020, 19, 1305–1309. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Yi, X.; Deng, J.; Zou, Y.; Wang, S.; Shan, W.; Liu, P.; Zhang, Z.; Chen, L.; Hao, L. ROCK2 promotes osteosarcoma growth and metastasis by modifying PFKFB3 ubiquitination and degradation. Exp. Cell Res. 2019, 385, 111689. [Google Scholar] [CrossRef] [PubMed]
- Mijanović, O.; Branković, A.; Panin, A.N.; Savchuk, S.; Timashev, P.; Ulasov, I.; Lesniak, M.S. Cathepsin B: A sellsword of cancer progression. Cancer Lett. 2019, 449, 207–214. [Google Scholar] [CrossRef]
- Ho, K.H.; Cheng, C.H.; Chou, C.M.; Chen, P.H.; Liu, A.J.; Lin, C.W.; Shih, C.M.; Chen, K.C. miR-140 targeting CTSB signaling suppresses the mesenchymal transition and enhances temozolomide cytotoxicity in glioblastoma multiforme. Pharm. Res. 2019, 147, 104390. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 2020, 6, 41–56. [Google Scholar] [CrossRef]
- Yang, M.; Chen, H.; Zhou, L.; Chen, K.; Su, F. Expression profile and prognostic values of STAT family members in non-small cell lung cancer. Am. J. Transl. Res. 2019, 11, 4866–4880. [Google Scholar]
- Wang, Y.; Sun, W.; Yang, J.; Yang, L.; Li, C.; Liu, H.; Liu, X.; Jiao, B. PUM2 Promotes Glioblastoma Cell Proliferation and Migration via Repressing BTG1 Expression. Cell Struct. Funct. 2019, 44, 29–39. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Y.; Li, C.; Liu, J.; Ren, H.; Li, L.; Zheng, X.; Wang, H.; Han, Z. RNA binding protein PUM2 promotes the stemness of breast cancer cells via competitively binding to neuropilin-1 (NRP-1) mRNA with miR-376a. Biomed. Pharm. 2019, 114, 108772. [Google Scholar] [CrossRef]
- Giannini, G.; Ambrosini, M.I.; Di Marcotullio, L.; Cerignoli, F.; Zani, M.; MacKay, A.R.; Screpanti, I.; Frati, L.; Gulino, A. EGF- and cell-cycle-regulated STAG1/PMEPA1/ERG1.2 belongs to a conserved gene family and is overexpressed and amplified in breast and ovarian cancer. Mol. Carcinog. 2003, 38, 188–200. [Google Scholar] [CrossRef]
- Xiang, J.; Fang, L.; Luo, Y.; Yang, Z.; Liao, Y.; Cui, J.; Huang, M.; Yang, Z.; Huang, Y.; Fan, X.; et al. Levels of human replication factor C4, a clamp loader, correlate with tumor progression and predict the prognosis for colorectal cancer. J. Transl. Med. 2014, 12, 320. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.X.; Pan, Y.Y.; You, C.G. CDK1, CCNB1, CDC20, BUB1, MAD2L1, MCM3, BUB1B, MCM2, and RFC4 May Be Potential Therapeutic Targets for Hepatocellular Carcinoma Using Integrated Bioinformatic Analysis. Biomed. Res. Int. 2019, 2019, 1245072. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.H.; Liu, H.; Lin, Y.H.; Chi, H.C.; Huang, Y.H.; Yang, C.C.; Yeh, C.T.; Tan, B.C.; Lin, K.H. ChIP-on-chip analysis of thyroid hormone-regulated genes and their physiological significance. Oncotarget 2016, 7, 22448–22459. [Google Scholar] [CrossRef]
- Liu, Y.; Tao, Z.; Qu, J.; Zhou, X.; Zhang, C. Long non-coding RNA PCAT7 regulates ELF2 signaling through inhibition of miR-134-5p in nasopharyngeal carcinoma. Biochem. Biophys. Res. Commun. 2017, 491, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu. Rev. Pharm. Toxicol. 2005, 45, 629–656. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef]
- Hoffmann, M.J.; Schulz, W.A. Causes and consequences of DNA hypomethylation in human cancer. Biochem. Cell Biol. 2005, 83, 296–321. [Google Scholar] [CrossRef]
- Cui, H.; Onyango, P.; Brandenburg, S.; Wu, Y.; Hsieh, C.L.; Feinberg, A.P. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 2002, 62, 6442–6446. [Google Scholar]
- Laner, T.; Schulz, W.A.; Engers, R.; Müller, M.; Florl, A.R. Hypomethylation of the XIST gene promoter in prostate cancer. Oncol. Res. 2005, 15, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.W.; Kempson, R.L.; Hendrickson, M.R. Problematic uterine smooth muscle neoplasms. A clinicopathologic study of 213 cases. Am. J. Surg. Pathol. 1994, 18, 535–558. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Laury, A.L.; Nucci, M.R.; Quade, B.J. Predictors of adverse outcome in uterine smooth muscle tumours of uncertain malignant potential (STUMP): A clinicopathological analysis of 22 cases with a proposal for the inclusion of additional histological parameters. Histopathology 2018, 73, 284–298. [Google Scholar] [CrossRef]
- Ip, P.P.; Cheung, A.N.; Clement, P.B. Uterine smooth muscle tumors of uncertain malignant potential (STUMP): A clinicopathologic analysis of 16 cases. Am. J. Surg. Pathol. 2009, 33, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.W.; Shaw, C.A.; Scott, D.A. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am. J. Med. Genet. A 2007, 143, 1679–1686. [Google Scholar] [CrossRef]
- Straussman, R.; Nejman, D.; Roberts, D.; Steinfeld, I.; Blum, B.; Benvenisty, N.; Simon, I.; Yakhini, Z.; Cedar, H. Developmental programming of CpG island methylation profiles in the human genome. Nat. Struct. Mol. Biol. 2009, 16, 564–571. [Google Scholar] [CrossRef]
- Beissbarth, T.; Speed, T.P. GOstat: Find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics 2004, 20, 1464–1465. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Patient | Sample | Histotype | Age at Diagnosis | Surgery | Mitosis | KI-67 | Markers | ER | PgR | p16 | p53 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | LM1 | Leiomyoma | 28 | n.a. | 0 | 5% | n.a. | n.a. | n.a. | n.a. | n.a. |
| 2 | LM2 | Leiomyoma | 52 | vaginal histerectomy, bilateral salpingo oohorectomy | <1 × 10 HPF | 0 | n.a. | n.a. | n.a. | n.a. | n.a. |
| 3 | LM3 | Leiomyoma | 40 | n.a. | 0 | <5% | n.a. | n.a. | n.a. | n.a. | n.a. |
| 4 | STUMP1 | STUMP | 56 | n.a. | 2 × 10 HPF | 10% | Actin 3+ | neg. | neg. | neg. | 2+ |
| STUMP2 | STUMP | 59 | n.a. | 5 × 10 HPF | 10% | Actin 3+ | neg. | neg. | neg. | 3+ | |
| 5 | STUMP3 | STUMP | 47 | n.a. | 4–5 × 10 HPF | <1% | Actin 2+ | neg. | neg. | neg. | neg. |
| US1 | Undiff. sarcoma | 48 | total abdominal histerectomy, bilateral salpingo oophorectomy | 14 × 10 HPF | 20% | Actin r.e. | neg. | neg. | neg. | 3+ | |
| 6 | STUMP4 | STUMP | 41 | Myomectomy | 3 × 10 HPF | <5% | Actin 1+, Desmin 3+, CD10 1+ | 30% | 50% | neg. | 2+ |
| LMS1 | Leiomyosarcoma | 45 | n.a. | 15 × 10 HPF | 40% | Actin 1+, Desmin 3+, CD10 1+ | neg. | 60% | neg. | neg. | |
| 7 | STUMP5 | STUMP | 26 | n.a. | 6-7 × 10 HPF | 30% | Actin 1+ | neg. | neg. | neg. | neg. |
| n.a. | Leiomyosarcoma | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | |
| 8 | n.a. | STUMP | 47 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
| STUMP6 | STUMP | 54 | total abdominal histerectomy, bilateral salpingo oophorectomy | 2 × 10 HPF | 3% | Actin 3+, Desmin 3+, CD10 neg. | 90% | 70% | neg. | 2+ | |
| 9 | n.a. | STUMP | 60 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | |
| STUMP7 | STUMP | 65 | total abdominal histerectomy, bilateral salpingo oophorectomy | 5–6 × 10 HPF | n.a. | Actin 3+, Desmin 1+, CD10 neg. | 50% | neg. | neg. | neg. | |
| 10 | STUMP8 | STUMP | 40 | Myomectomy | 9 × 10 HPF | 30% | Actin 1+ | neg. | 20% | neg. | 3+ |
| 11 | STUMP9 | STUMP | 31 | n.a. | 1 × 50 HPF | <1% | Actin neg., Desmin 2+ | neg. | 70% | neg. | neg. |
| 12 | STUMP10 | STUMP | 30 | Myomectomy | 4–6 × 10 HPF | <1% | Actin 2+ | 10% | 70% | neg. | neg. |
| 13 | STUMP11 | STUMP | 42 | total abdominal histetectomy, left salpingo oophorectomy | 1–2 × 10 HPF | <1% | Actin neg. | neg. | 40% | neg. | r.e. |
| 14 | STUMP12 | STUMP | 47 | total abdominal histerectomy, bilateral salpingo oophorectomy | 4–5 × 10 HPF | <5% | Actin 1+ | neg. | neg. | neg. | neg. |
| 15 | STUMP13 | STUMP | 58 | total abdominal histerectomy, bilateral salpingo oophorectomy | 1–2 × 10 HPF | <1% | Actin 1+ | neg. | 50% | neg. | neg. |
| 16 | STUMP14 | STUMP | 34 | laparothomy | 0–1 × 10 HPF | 1% | Actin 3+, Desmin 3+ | 30% | 80% | neg. | neg. |
| 17 | LMS2 | Leiomyosarcoma | 49 | total abdominal histerectomy, bilateral salpingo oophorectomy | 9 × 10 HPF | 30% | Actin 2+, Desmin 2+ | 20% | 90% | neg. | neg. |
| 18 | LMS3 | Leiomyosarcoma | 63 | n.a. | 15 × 10 HPF | 10% | Actin 1+ | neg. | neg. | neg. | 3+ |
| 19 | LMS4 | Leiomyosarcoma | 50 | laparoscopic histerectomy, bilateral adnexectomy | 3–4 × 10 HPF | 10% | Actin 1+, Desmin 1+ | neg. | 30% | neg. | r.e. |
| n.a. | Leiomyosarcoma | 54 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | |
| 20 | n.a. | Leiomyosarcoma | 68 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
| LMS5 | Leiomyosarcoma | 73 | n.a. | 1 × 10 HPF | <1% | Actin 1+ | 40% | 60% | r.e. | r.e. | |
| n.a. | Leiomyosarcoma | 77 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
| Patient | Sample | Follow Up | OS | CNAs | ||
|---|---|---|---|---|---|---|
| Total | Gain | Loss | ||||
| 1 | LM1 | NED | 6 | 110 | 106 | 4 |
| 2 | LM2 | NED | 12 | 123 | 114 | 9 |
| 3 | LM3 | n.a. | 2 | 268 | 239 | 29 |
| 4 | STUMP1 | n.a. | 5 | 25 | 23 | 2 |
| STUMP2 | 17 | 17 | 0 | |||
| 5 | STUMP3 | DOD | 2 | 365 | 342 | 23 |
| US1 | 28 | 28 | 0 | |||
| 6 | STUMP4 | DOD | 7 | 256 | 238 | 18 |
| LMS1 | 126 | 121 | 5 | |||
| 7 | STUMP5 | DOD | 1 | 58 | 58 | 0 |
| n.a. | ||||||
| 8 | n.a. | ALIVE | 13 | |||
| STUMP6 | 116 | 108 | 8 | |||
| 9 | n.a. | NED | 8 | |||
| STUMP7 | 94 | 88 | 6 | |||
| 10 | STUMP8 | NED | 14 | 88 | 80 | 8 |
| 11 | STUMP9 | ALIVE | 9 | 82 | 81 | 1 |
| 12 | STUMP10 | n.a. | 10 | 40 | 38 | 2 |
| 13 | STUMP11 | NED | 18 | 97 | 97 | 0 |
| 14 | STUMP12 | NED | 10 | 156 | 155 | 1 |
| 15 | STUMP13 | NED | 16 | 85 | 81 | 4 |
| 16 | STUMP14 | NED | 7 | 59 | 58 | 1 |
| 17 | LMS2 | ALIVE | 8 | 204 | 194 | 10 |
| 18 | LMS3 | n.a. | n.a | 199 | 189 | 10 |
| 19 | LMS4 | DOD | 7 | 262 | 234 | 28 |
| n.a. | ||||||
| 20 | n.a. | AWD | ||||
| LMS5 | 23 | 166 | 148 | 18 | ||
| n.a. | ||||||
| PRKDC | MLL3 | ROCK2 | CTSB | STAT2 | STAG1 | RFC4 | CCRN4L, ELF2 | PUM2 | BCL2 | |
|---|---|---|---|---|---|---|---|---|---|---|
| STUMP2 | + | |||||||||
| STUMP3 | + | + | + | + | + | + | ||||
| STUMP4 | + | + | + | + | + | + | + | |||
| STUMP5 | + | |||||||||
| STUMP6 | + | + | ||||||||
| STUMP7 | + | + | + | |||||||
| STUMP9 | + | |||||||||
| STUMP10 | + | |||||||||
| STUMP11 | + | + | + | |||||||
| STUMP12 | + | + | + | |||||||
| STUMP13 | + | + | ||||||||
| LMS1 | + | + | + | + | ||||||
| LMS2 | + | + | + | + | + | |||||
| LMS3 | + | + | + | + | + | + | + | |||
| LMS4 | + | + | + | + | + | |||||
| LMS5 | + | + | + | + | + | + | + |
| Cell Signaling Pathway | Genes with a Significant GOterm | Unmethylated Genes in STUMP4 (Methylated in LMS1) | Methylated Genes in STUMP4 (Unmethylated in LMS1) |
|---|---|---|---|
| NFKβ cascade | 22 | NDFIP1, MAP3K7IP2, PLK2, NDFIP2, NLRC3, PPM1A, SECTM1, TRIM13, IRAK1, TSPAN6, VAPA, TNFAIP3, BIRK2, C9orf89, EDG2, GOLT1B, LTBR, PRDX3, TNFRSF1A, RPS6KB2 | ZDHHC13, TFG |
| WNT receptor signaling pathway | 16 | CSNK2B, LRRFIP2, PYGO2, PORCN, DKK1, FZD3, FZD6, FZD8, HBP1, WNT10A, WNT2B, WNT6, WNT7A, DIXDC1, TLE4, WNT11 | / |
| Smoothened signaling pathway (Hedgehog signaling pathway) | 1 | CTNNA1 | / |
| Negative regulation of WNT pathway | 1 | FRZB | / |
| Regulation ARF GTPase activity (Ras protein transduction) | 24 | ARGEF2, PSD3, CENTB2, CENTD1, GNB1, DOK3, EVI5, PSD4, RASSF1, RASSF6, GBF1, DDEF1, DOK2, SMAP1, PSCD1, PSCD4, ARFGEF1, CTGLF4 | ARFGAP1, FBXO8, IQSEC1, TBC1D22A, TBC1D15, TBC1D16 |
| Ras GTPase activity | 15 | TBC1D5, TBC1D8, TBC1D9, TBC1D9B, TBC1D2B, TBC1D4, EVI5L, SGSM1, TBC1D10A, TBC1D20, TBC1D25, TBC1D10C, TBC1D12, CENTG1, RABGAP1 | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conconi, D.; Redaelli, S.; Lissoni, A.A.; Cilibrasi, C.; Perego, P.; Gautiero, E.; Sala, E.; Paderno, M.; Dalprà, L.; Landoni, F.; et al. Genomic and Epigenomic Profile of Uterine Smooth Muscle Tumors of Uncertain Malignant Potential (STUMPs) Revealed Similarities and Differences with Leiomyomas and Leiomyosarcomas. Int. J. Mol. Sci. 2021, 22, 1580. https://doi.org/10.3390/ijms22041580
Conconi D, Redaelli S, Lissoni AA, Cilibrasi C, Perego P, Gautiero E, Sala E, Paderno M, Dalprà L, Landoni F, et al. Genomic and Epigenomic Profile of Uterine Smooth Muscle Tumors of Uncertain Malignant Potential (STUMPs) Revealed Similarities and Differences with Leiomyomas and Leiomyosarcomas. International Journal of Molecular Sciences. 2021; 22(4):1580. https://doi.org/10.3390/ijms22041580
Chicago/Turabian StyleConconi, Donatella, Serena Redaelli, Andrea Alberto Lissoni, Chiara Cilibrasi, Patrizia Perego, Eugenio Gautiero, Elena Sala, Mariachiara Paderno, Leda Dalprà, Fabio Landoni, and et al. 2021. "Genomic and Epigenomic Profile of Uterine Smooth Muscle Tumors of Uncertain Malignant Potential (STUMPs) Revealed Similarities and Differences with Leiomyomas and Leiomyosarcomas" International Journal of Molecular Sciences 22, no. 4: 1580. https://doi.org/10.3390/ijms22041580
APA StyleConconi, D., Redaelli, S., Lissoni, A. A., Cilibrasi, C., Perego, P., Gautiero, E., Sala, E., Paderno, M., Dalprà, L., Landoni, F., Lavitrano, M., Roversi, G., & Bentivegna, A. (2021). Genomic and Epigenomic Profile of Uterine Smooth Muscle Tumors of Uncertain Malignant Potential (STUMPs) Revealed Similarities and Differences with Leiomyomas and Leiomyosarcomas. International Journal of Molecular Sciences, 22(4), 1580. https://doi.org/10.3390/ijms22041580