
Fibrosis and Conduction Abnormalities as Basis for Overlap of Brugada Syndrome and Early Repolarization Syndrome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Abnormal Conduction in RVOT in Brugada Syndrome
3. Overlap in Mechanism of Brugada Syndrome and Early Repolarization Syndrome
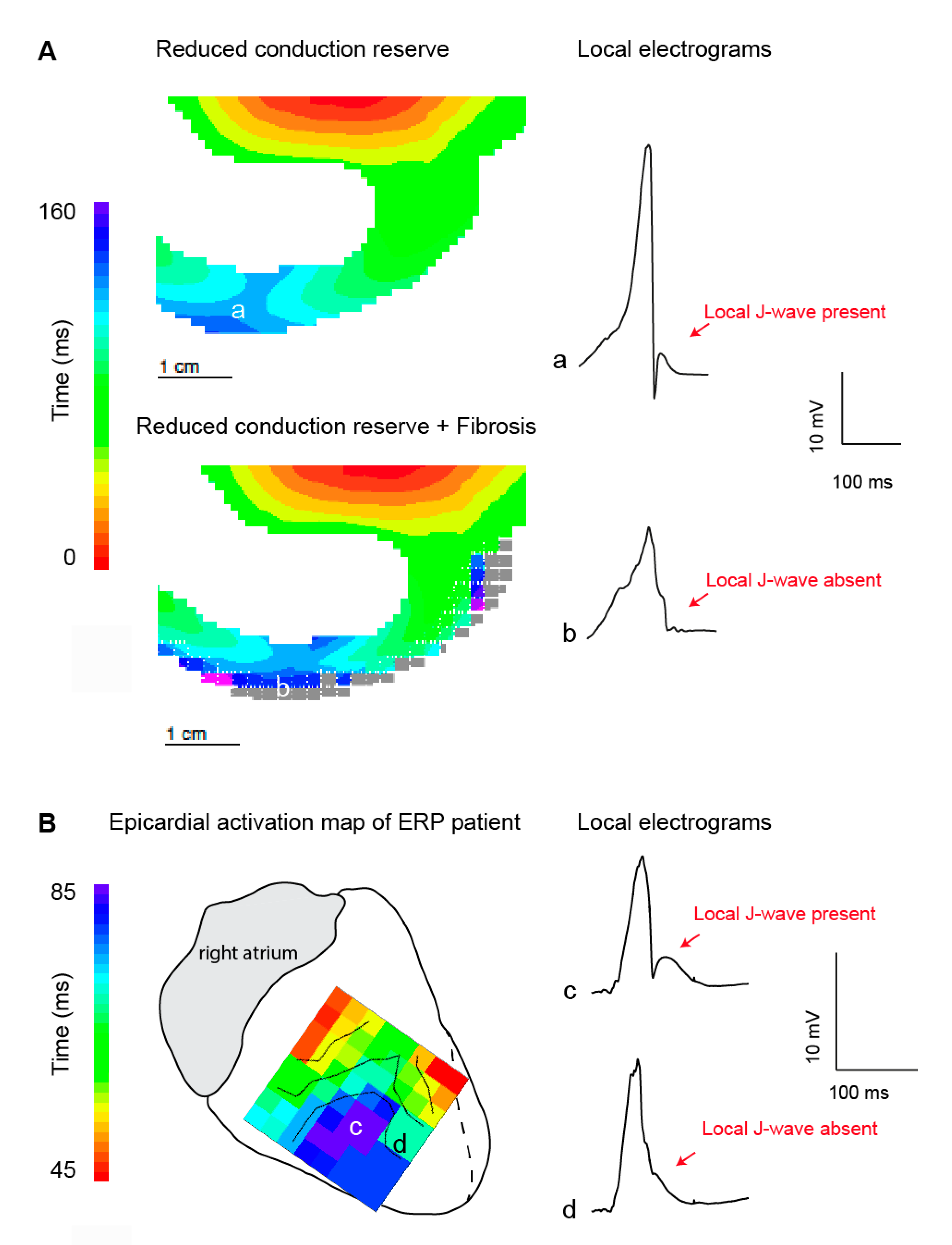
3.1. Structural Abnormalities and Fractionated Potentials
3.2. J-Waves in Local Electrograms and Excitation Failure
3.3. Generation of the J-Wave in the Electrocardiogram through Structural Abnormalities
3.4. Paradoxical Effect of Sodium Channel Blockade
4. Temporal Variability in Signs and Symptoms in Both Syndromes
5. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Antzelevitch, C.; Yan, G.X.; Ackerman, M.J.; Borggrefe, M.; Corrado, D.; Guo, J.; Gussak, I.; Hasdemir, C.; Horie, M.; Huikuri, H.; et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. Europace 2017, 19, 665–694. [Google Scholar]
- Nademanee, K.; Veerakul, G. Overlapping risks of early repolarization and brugada syndrome. J. Am. Coll. Cardiol. 2014, 63, 2139–2140. [Google Scholar] [CrossRef] [PubMed]
- Hoogendijk, M.G.; Coronel, R. J-wave syndrome(s). Trends Cardiovasc. Med. 2015, 25, 22–23. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.X.; Antzelevitch, C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation 1999, 100, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Lukas, A.; Antzelevitch, C. Differences in the electrophysiological response of canine ventricular epicardium and endocardium to ischemia. Role of the transient outward current. Circulation 1993, 88, 2903–2915. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.C.; Antzelevitch, C. Flecainide-induced arrhythmia in canine ventricular epicardium: Phase 2 reentry? Circulation 1993, 87, 562–572. [Google Scholar] [CrossRef]
- Postema, P.G.; van Dessel, P.F.; de Bakker, J.M.; Dekker, L.R.; Linnenbank, A.C.; Hoogendijk, M.G.; Coronel, R.; Tijssen, J.G.; Wilde, A.A.; Tan, H.L. Slow and discontinuous conduction conspire in Brugada syndrome: A right ventricular mapping and stimulation study. Circ. Arrhythmia Electrophysiol. 2008, 1, 379–386. [Google Scholar] [CrossRef]
- Coronel, R.; Casini, S.; Koopmann, T.T.; Wilms-Schopman, F.J.; Verkerk, A.O.; de Groot, J.R.; Bhuiyan, Z.; Bezzina, C.R.; Veldkamp, M.W.; Linnenbank, A.C.; et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: A combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005, 112, 2769–2777. [Google Scholar] [CrossRef]
- Nademanee, K.; Veerakul, G.; Chandanamattha, P.; Chaothawee, L.; Ariyachaipanich, A.; Jirasirirojanakorn, K.; Likittanasombat, K.; Bhuripanyo, K.; Ngarmukos, T. Prevention of ventricular fibrillation episodes in brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 2011, 123, 1270–1279. [Google Scholar] [CrossRef]
- Nademanee, K.; Raju, H.; de Noronha, S.V.; Papadakis, M.; Robinson, L.; Rothery, S.; Makita, N.; Kowase, S.; Boonmee, N.; Vitayakritsirikul, V.; et al. Fibrosis, Connexin-43, and Conduction Abnormalities in the Brugada Syndrome. J. Am. Coll. Cardiol. 2015, 66, 1976–1986. [Google Scholar] [CrossRef]
- Ten Sande, J.N.; Coronel, R.; Conrath, C.E.; Driessen, A.H.G.; De Groot, J.R.; Tan, H.L.; Nademanee, K.; Wilde, A.A.M.; De Bakker, J.M.T.; Van Dessel, P.F.H.M. ST-Segment Elevation and Fractionated Electrograms in Brugada Syndrome Patients Arise from the Same Structurally Abnormal Subepicardial RVOT Area but Have a Different Mechanism. Circ. Arrhythmia Electrophysiol. 2015, 8, 1382–1392. [Google Scholar] [CrossRef]
- Hoogendijk, M.G.; Potse, M.; Linnenbank, A.C.; Verkerk, A.O.; Den Ruijter, H.M.; van Amersfoorth, S.C.; Klaver, E.C.; Beekman, L.; Bezzina, C.R.; Postema, P.G.; et al. Mechanism of right precordial ST-segment elevation in structural heart disease: Excitation failure by current-to-load mismatch. Heart Rhythm 2010, 7, 238–248. [Google Scholar] [CrossRef]
- Blok, M.; Boukens, B.J. Mechanisms of arrhythmias in the brugada syndrome. Int. J. Mol. Sci. 2020, 21, 7051. [Google Scholar] [CrossRef] [PubMed]
- Behr, E.R.; Ben-haim, Y.; Ackerman, M.J.; Krahn, A.D.; Wilde, A.A.M. OUP accepted manuscript. Eur. Heart J. 2021, 44, 1–12. [Google Scholar]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, e373–e407. [Google Scholar] [CrossRef] [PubMed]
- Boukens, B.J.D.; Benjacholamas, V.; van Amersfoort, S.; Meijborg, V.M.; Schumacher, C.; Jensen, B.; Haissaguerre, M.; Wilde, A.; Prechawat, S.; Huntrakul, A.; et al. Structurally Abnormal Myocardium Underlies Ventricular Fibrillation Storms in a Patient Diagnosed with the Early Repolarization Pattern. JACC Clin. Electrophysiol. 2020, 6, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Priori, S.; Pieroni, M.; Chimenti, C.; Napolitano, C.; Rivolta, I.; Sanna, T.; Bellocci, F.; Russo, M.A. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 2005, 112, 3680–3687. [Google Scholar] [CrossRef]
- Corrado, D.; Baso, C.; Buja, G.; Nava, A.; Rossi, L.; Thiene, G. Right bundle branch block, right precordial ST-segment elevation, and sudden death in young people. Circulation 2001, 103, 710–717. [Google Scholar] [CrossRef]
- Martini, B.; Nava, A.; Thiene, G.; Buja, G.F.; Canciani, B.; Scognamiglio, R.; Daliento, L.; Dalla Volta, S. Ventricular fibrillation without apparent heart disease: Description of six cases. Am. Heart J. 1989, 118, 1203–1209. [Google Scholar] [CrossRef]
- de Bakker, J.M.; van Capelle, F.J.; Janse, M.J.; Tasseron, S.; Vermeulen, J.T.; de Jonge, N.; Lahpor, J.R. Slow conduction in the infarcted human heart. “Zigzag” course of activation. Circulation 1993, 88, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Hoogendijk, M.G.; Potse, M.; Vinet, A.; de Bakker, J.M.; Coronel, R. ST segment elevation by current-to-load mismatch: An experimental and computational study. Heart Rhythm 2011, 8, 111–118. [Google Scholar] [CrossRef]
- Rohr, S.; Kucera, J.P.; Fast, V.G.; Kleber, A.G. Paradoxical improvement of impulse conduction in cardiac tissue by partial cellular uncoupling. Science 1997, 275, 841–844. [Google Scholar] [CrossRef]
- Van Rijen, H.V.M.; De Bakker, J.M.T.; Van Veen, T.A.B. Hypoxia, electrical uncoupling, and conduction slowing: Role of conduction reserve. Cardiovasc. Res. 2005, 66, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Kléber, A.G.; Rudy, Y.; Kleber, A.G.; Rudy, Y.; Kléber, A.G.; Rudy, Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol. Rev. 2004, 84, 431–488. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Brugada, P.; Borggrefe, M.; Brugada, J.; Brugada, R.; Corrado, D.; Gussak, I.; LeMarec, H.; Nademanee, K.; Perez Riera, A.R.; et al. Brugada syndrome: Report of the second consensus conference. Heart Rhythm. 2005, 2, 429–440. [Google Scholar] [CrossRef]
- De Bakker, J.M.T.; Wittkampf, F.H.M. The pathophysiologic basis of fractionated and complex electrograms and the impact of recording techniques on their detection and interpretation. Circ. Arrhythmia Electrophysiol. 2010, 3, 204–213. [Google Scholar] [CrossRef]
- Haissaguerre, M.; Shoda, M.; Jais, P.; Nogami, A.; Shah, D.C.; Kautzner, J.; Arentz, T.; Kalushe, D.; Lamaison, D.; Griffith, M.; et al. Mapping and ablation of idiopathic ventricular fibrillation. Circulation 2002, 106, 962–967. [Google Scholar] [CrossRef]
- Boukens, B.J.J.B.J.; Sylva, M.; De Gier-De Vries, C.; Remme, C.A.C.A.; Bezzina, C.R.C.R.; Christoffels, V.M.V.M.; Coronel, R.; de Gier-de, V.C.; Remme, C.A.C.A.; Bezzina, C.R.C.R.; et al. Reduced sodium channel function unmasks residual embryonic slow conduction in the adult right ventricular outflow tract. Circ. Res. 2013, 113, 137–141. [Google Scholar] [CrossRef]
- Boukens, B.J.D.; Christoffels, V.M.; Coronel, R.; Moorman, A.F.M. Developmental Basis for Electrophysiological Heterogeneity in the Ventricular and Outflow Tract Myocardium as a Substrate for Life-Threatening Ventricular Arrhythmias. SDAA 2009, 104, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, K.; Watanabe, I.; Okumura, Y.; Takagi, Y.; Ashino, S.; Kofune, M.; Sugimura, H.; Nakai, T.; Kasamaki, Y.; Hirayama, A.; et al. Right ventricular histological substrate and conduction delay in patients with Brugada syndrome. Int. Heart J. 2010, 51, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, P. Brugada syndrome and myocardial histology: Where may the truth lie? Arch. Cardiovasc. Dis. 2019, 112, 367–369. [Google Scholar] [CrossRef]
- Nakasuka, K.; Noda, T.; Miyamoto, K.; Kusano, K. QRS alternans due to localized intraventricular block during ventricular tachycardia in Uhl’s anomaly: A case report. Eur. Hear. J. Case Rep. 2019, 3, 1–5. [Google Scholar] [CrossRef]
- MacFarlane, P.W.; Antzelevitch, C.; Haissaguerre, M.; Huikuri, H.V.; Potse, M.; Rosso, R.; Sacher, F.; Tikkanen, J.T.; Wellens, H.; Yan, G.X. The early repolarization pattern: A consensus paper. J. Am. Coll. Cardiol. 2015, 66, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Nagase, S.; Tanaka, M.; Morita, H.; Nakagawa, K.; Wada, T.; Murakami, M.; Nishii, N.; Nakamura, K.; Ito, H.; Ohe, T.; et al. Local Left Ventricular Epicardial J Waves and Late Potentials in Brugada Syndrome Patients with Inferolateral Early Repolarization Pattern. Front. Physiol. 2017, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Takigawa, M.; Relan, J.; Martin, R.; Kim, S.; Kitamura, T.; Cheniti, G.; Vlachos, K.; Pillois, X.; Frontera, A.; Massoullié, G.; et al. Detailed Analysis of the Relation Between Bipolar Electrode Spacing and Far- and Near-Field Electrograms. JACC Clin. Electrophysiol. 2019, 5, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Avella, A.; D’Amati, G.; Pappalardo, A.; Re, F.; Silenzi, P.F.; Laurenzi, F.; De Girolamo, P.; Pelargonio, G.; Russo, A. Dello; Baratta, P.; et al. Diagnostic value of endomyocardial biopsy guided by electroanatomic voltage mapping in arrhythmogenic right ventricular cardiomyopathy/dysplasia. J. Cardiovasc. Electrophysiol. 2008, 19, 1127–1134. [Google Scholar] [CrossRef]
- Nademanee, K.; Haissaguerre, M.; Hocini, M.; Nogami, A.; Cheniti, G.; Duchateau, J.; Behr, E.R.; Saba, M.; Bokan, R.; Lou, Q.; et al. Mapping and Ablation of Ventricular Fibrillation Associated with Early Repolarization Syndrome. Circulation 2019, 140, 1477–1490. [Google Scholar] [CrossRef] [PubMed]
- Boukens, B.J.; Opthof, T.; Coronel, R. J-Waves in Epicardial Electrograms Can Guide Ablation of Arrhythmogenic Substrates. SDAA 2019, 124. [Google Scholar] [CrossRef] [PubMed]
- Vigmond, E.; Efimov, I.R.; Rentschler, S.; Coronel, R.; Boukens, B.J. Fractionated electrograms with ST-segment elevation recorded from the human RVOT. HeartRhythm Case Rep. 2017, 3, 546–550. [Google Scholar] [CrossRef][Green Version]
- Potse, M.; Dubé, B.; Richer, J.; Vinet, A.; Gulrajani, R.M. A comparison of monodomain and bidomain reaction-diffusion models for action potential propagation in the human heart. IEEE Trans. Biomed. Eng. 2006, 53, 2425–2435. [Google Scholar] [CrossRef] [PubMed]
- ten Tusscher, K.H.W.J.; Noble, D.; Noble, P.J.; Panfilov, A.V. A model for human ventricular tissue. AJP Hear. Circ. Physiol. 2004, 286, H1573–H1589. [Google Scholar] [CrossRef] [PubMed]
- Vigmond, E.J.; Weber dos Santos, R.; Prassl, A.J.; Deo, M.; Plank, G. Solvers for the cardiac bidomain equations. Prog. Biophys. Mol. Biol. 2008, 96, 3–18. [Google Scholar] [CrossRef]
- Vigmond, E.J. The electrophysiological basis of MAP recordings. Cardiovasc. Res. 2005, 68, 502–503. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meijborg, V.M.F.; Potse, M.; Conrath, C.E.; Belterman, C.N.W.; De Bakker, J.M.T.; Coronel, R. Reduced sodium current in the lateral ventricular wall induces inferolateral J-waves. Front. Physiol. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Ikeda, T.; Abe, A.; Yusu, S.; Nakamura, K.; Ishiguro, H.; Mera, H.; Yotsukura, M.; Yoshino, H. The full stomach test as a novel diagnostic technique for identifying patients at risk of Brugada syndrome. J. Cardiovasc. Electrophysiol. 2006, 17, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Nademanee, K.; Veerakul, G.; Nimmannit, S.; Chaowakul, V.; Bhuripanyo, K.; Likittanasombat, K.; Tunsanga, K.; Kuasirikul, S.; Malasit, P.; Tansupasawadikul, S.; et al. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation 1997, 96, 2595–2600. [Google Scholar] [CrossRef]
- Boyle, P.M.; Franceschi, W.H.; Constantin, M.; Hawks, C.; Desplantez, T.; Trayanova, N.A.; Vigmond, E.J. New insights on the cardiac safety factor: Unraveling the relationship between conduction velocity and robustness of propagation. J. Mol. Cell. Cardiol. 2019, 128, 117–128. [Google Scholar] [CrossRef]
- Xiao, Y.F.; Kang, J.X.; Morgan, J.P.; Leaf, A. Blocking effects of polyunsaturated fatty acids on Na+ channels of neonatal rat ventricular myocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 11000–11004. [Google Scholar] [CrossRef]
- Den Ruijter, H.M.; Berecki, G.; Opthof, T.; Verkerk, A.O.; Zock, P.L.; Coronel, R. Pro- and antiarrhythmic properties of a diet rich in fish oil. Cardiovasc. Res. 2007, 73, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Meijborg, V.M.F.; Boukens, B.J.D.; Janse, M.J.; Salavatian, S.; Dacey, M.J.; Yoshie, K.; Opthof, T.; Swid, M.A.; Hoang, J.D.; Hanna, P.; et al. Stellate ganglion stimulation causes spatiotemporal changes in ventricular repolarization in pig. Heart Rhythm 2020, 17, 795–803. [Google Scholar] [CrossRef]
- Joyner, R.W.; Sugiura, H.; Tan, R.C. Unidirectional block between isolated rabbit ventricular cells coupled by a variable resistance. Biophys. J. 1991, 60, 1038–1045. [Google Scholar] [CrossRef][Green Version]
- Nabauer, M.; Beuckelmann, D.J.; Uberfuhr, P.; Steinbeck, G. Regional differences in current density and rate-dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation 1996, 93, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Scrocco, C.; Behr, E.R. J-Wave Syndromes: Where’s the Scar? JACC Clin. Electrophysiol. 2020, 6, 1862–1863. [Google Scholar] [CrossRef] [PubMed]
- Boukens, B.J.; Nademanee, K.; Coronel, R. Reply: J-Wave Syndromes: Where’s the Scar? JACC Clin. Electrophysiol. 2020, 6, 1863–1864. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boukens, B.J.; Potse, M.; Coronel, R. Fibrosis and Conduction Abnormalities as Basis for Overlap of Brugada Syndrome and Early Repolarization Syndrome. Int. J. Mol. Sci. 2021, 22, 1570. https://doi.org/10.3390/ijms22041570
Boukens BJ, Potse M, Coronel R. Fibrosis and Conduction Abnormalities as Basis for Overlap of Brugada Syndrome and Early Repolarization Syndrome. International Journal of Molecular Sciences. 2021; 22(4):1570. https://doi.org/10.3390/ijms22041570
Chicago/Turabian StyleBoukens, Bastiaan J., Mark Potse, and Ruben Coronel. 2021. "Fibrosis and Conduction Abnormalities as Basis for Overlap of Brugada Syndrome and Early Repolarization Syndrome" International Journal of Molecular Sciences 22, no. 4: 1570. https://doi.org/10.3390/ijms22041570
APA StyleBoukens, B. J., Potse, M., & Coronel, R. (2021). Fibrosis and Conduction Abnormalities as Basis for Overlap of Brugada Syndrome and Early Repolarization Syndrome. International Journal of Molecular Sciences, 22(4), 1570. https://doi.org/10.3390/ijms22041570