
Programmed Death-Ligand 2 Deficiency Exacerbates Experimental Autoimmune Myocarditis in Mice

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
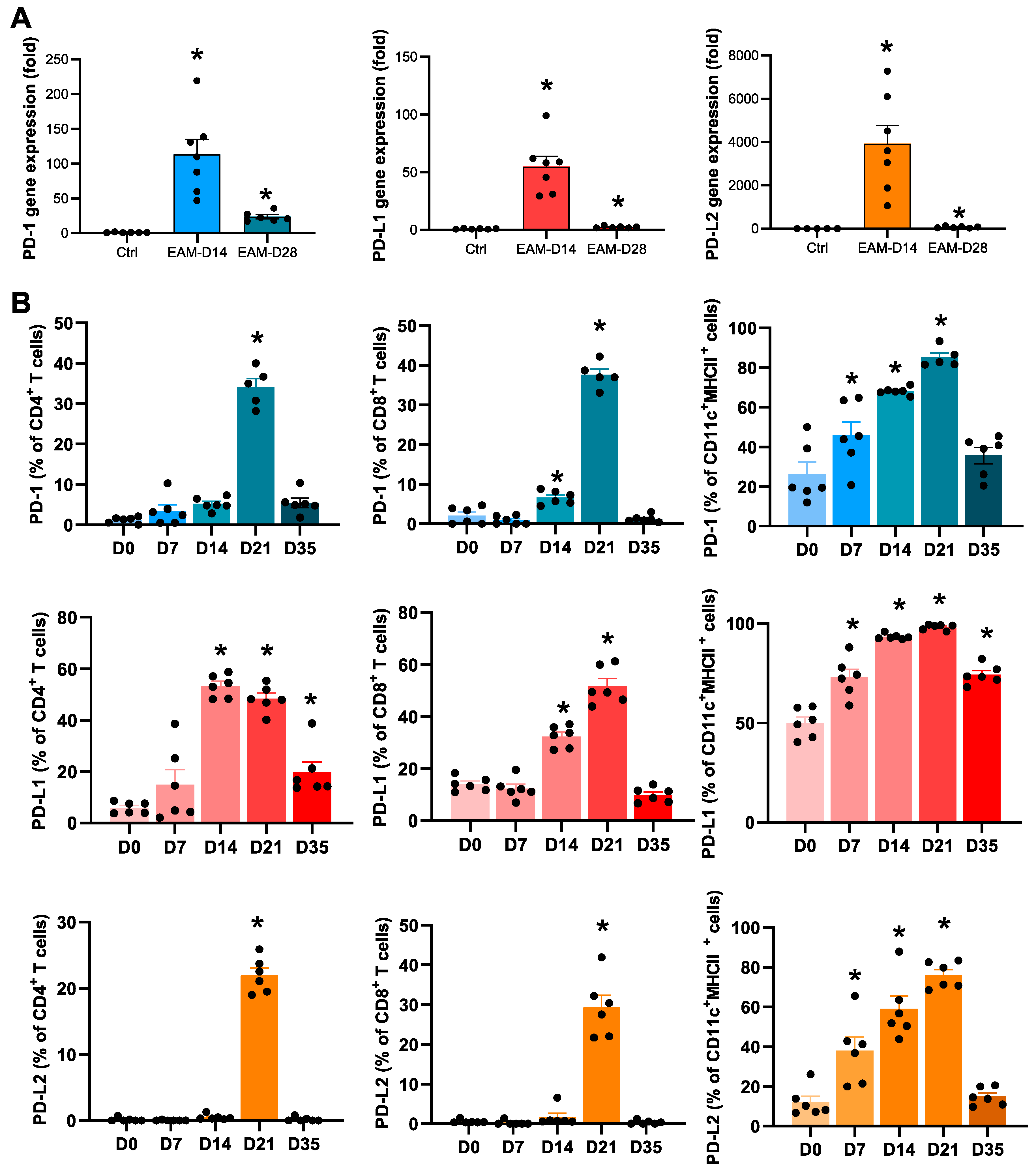
2.1. Expression Levels of PD-1 and PD-Ls in the EAM Hearts
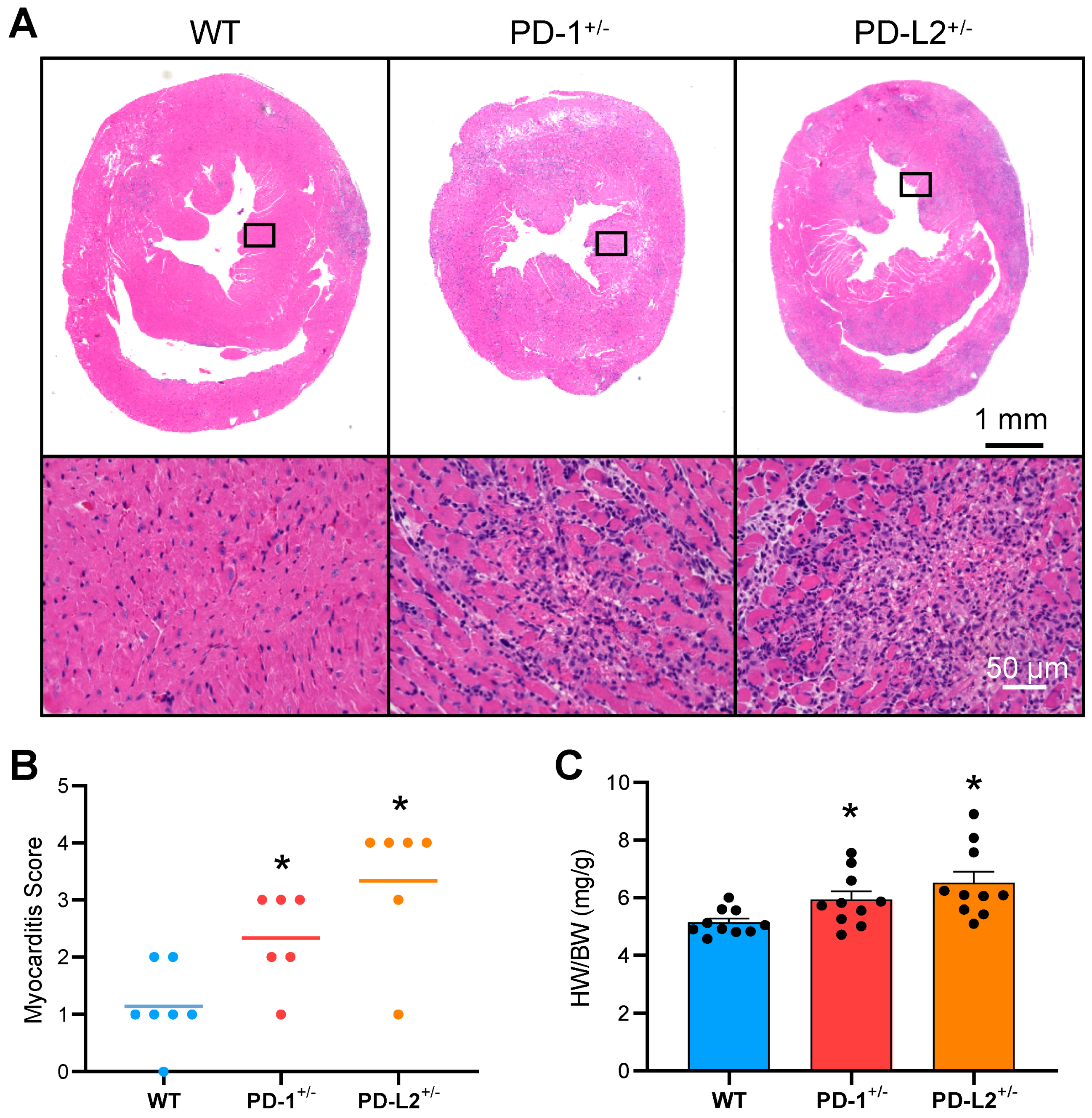
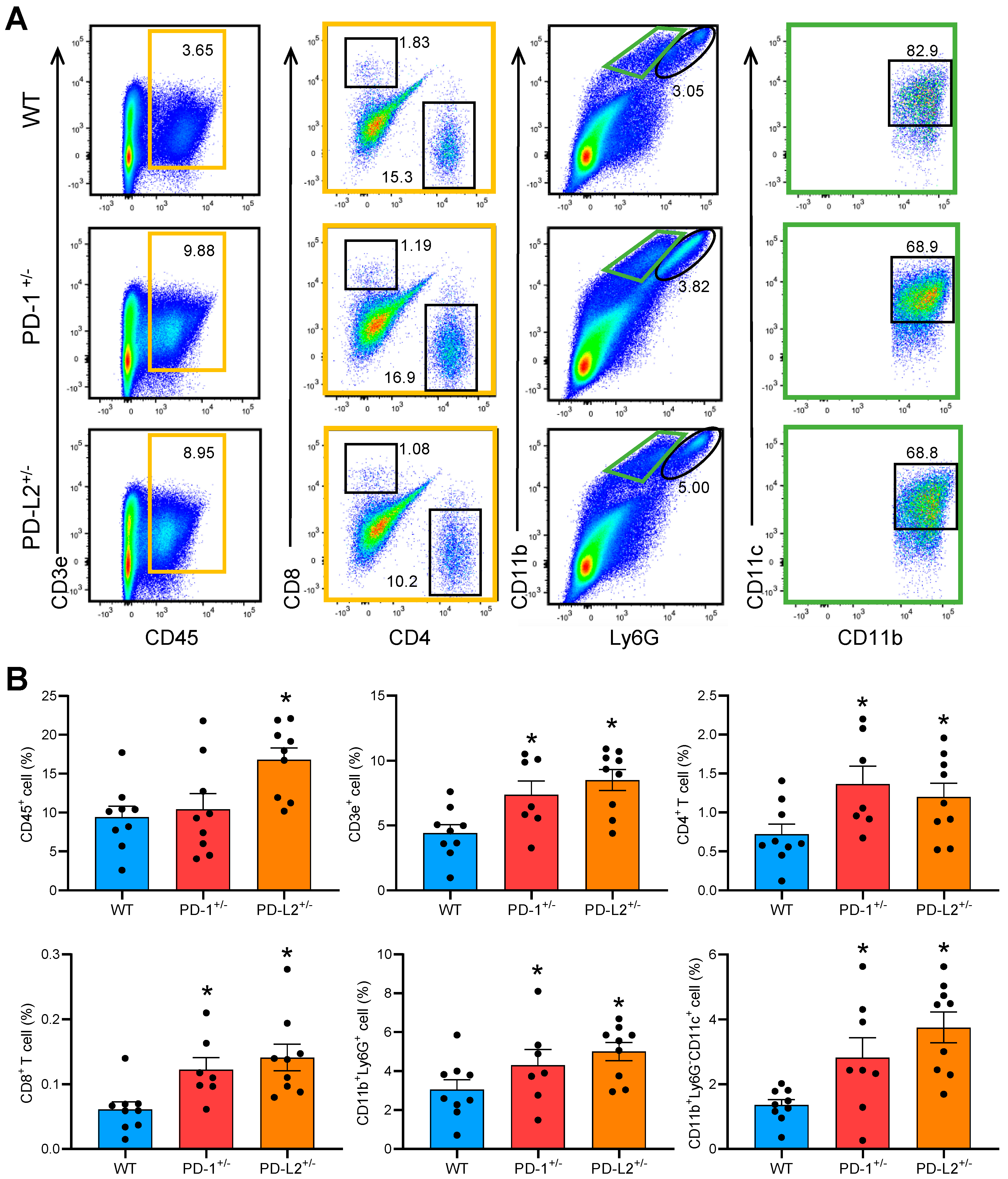
2.2. PD-L2 Deficiency Exacerbates Cardiac Inflammation in EAM
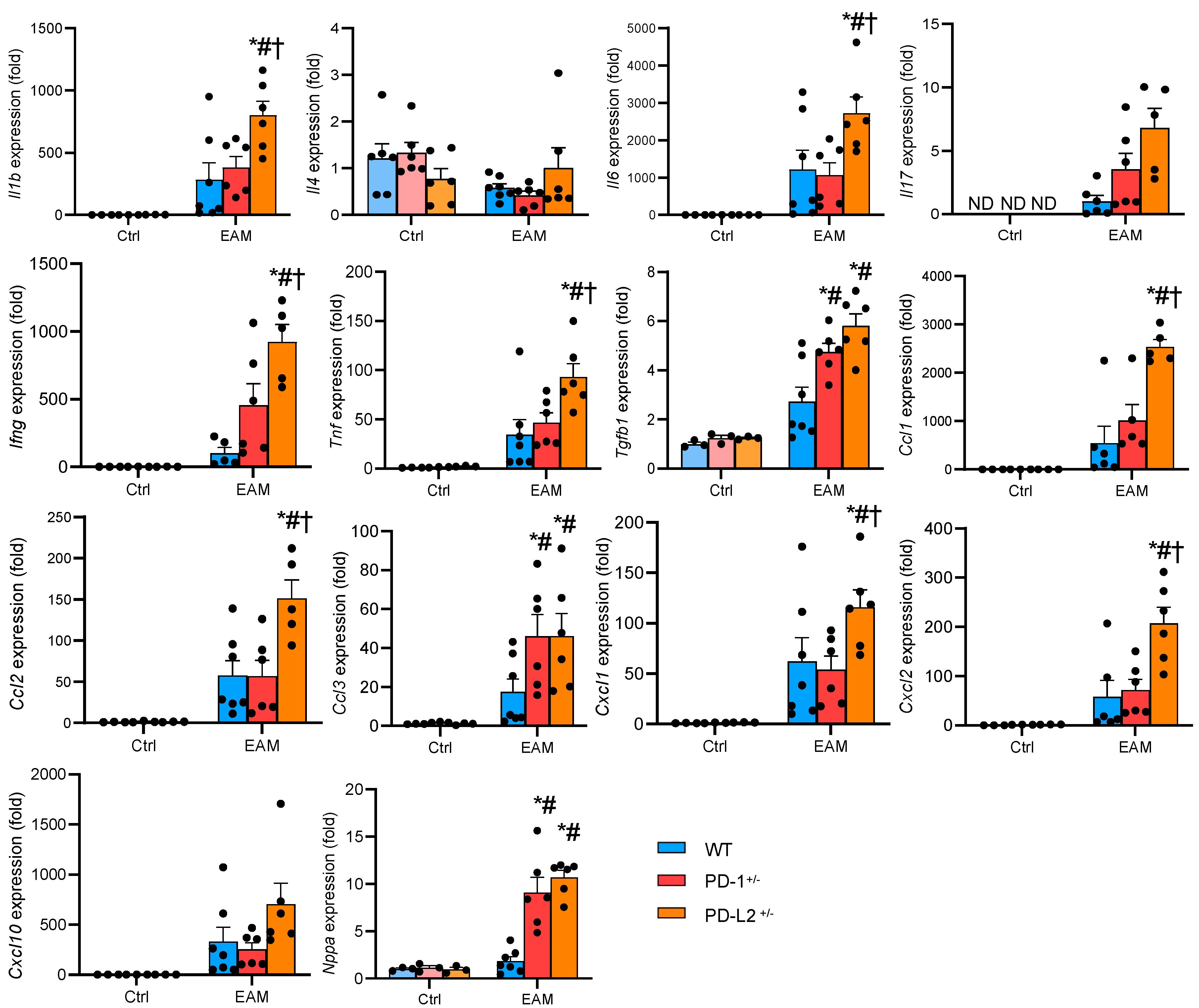
2.3. PD-L2 Deficiency Promotes Proinflammatory Cytokine Expression in the EAM Heart
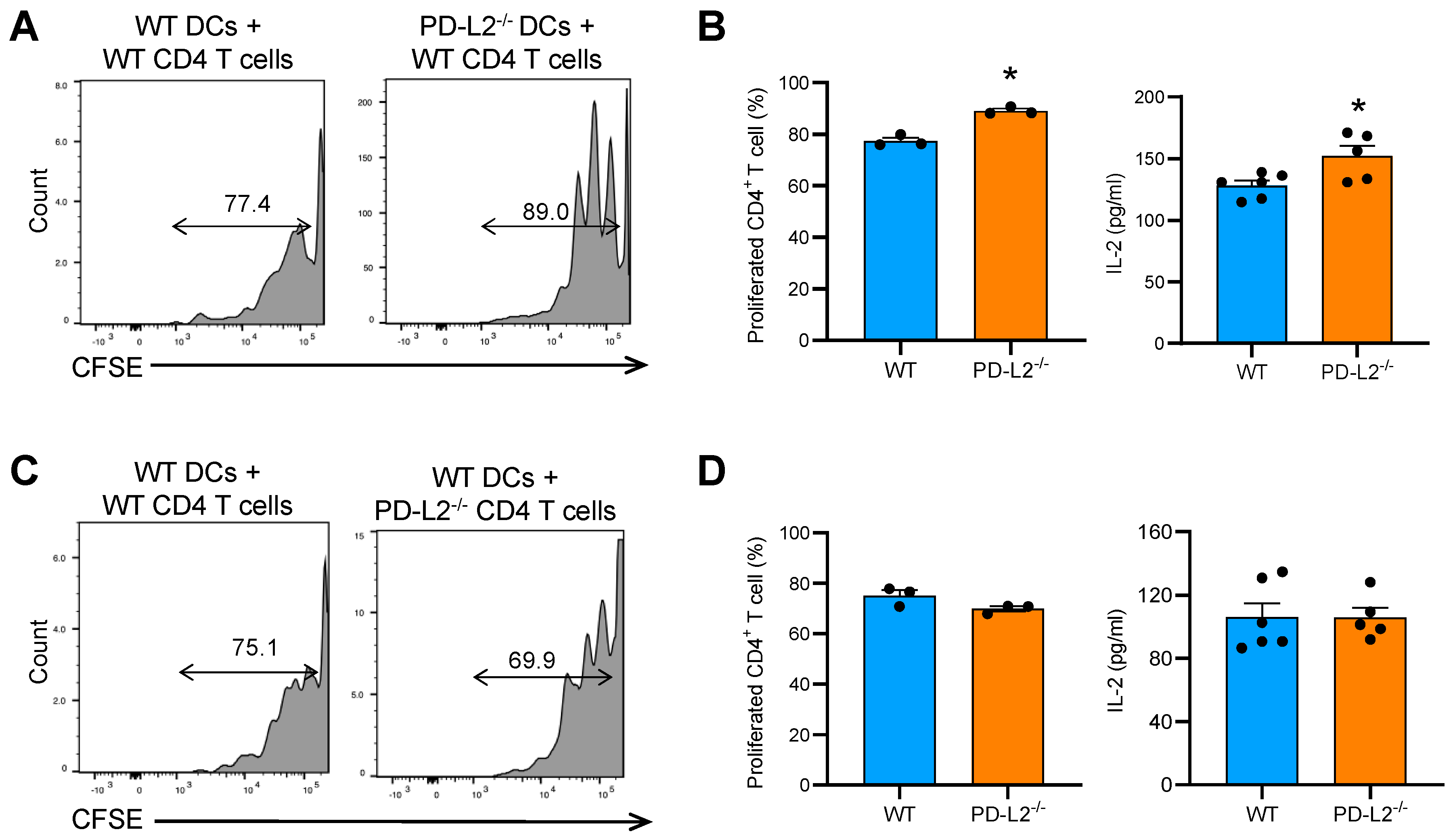
2.4. PD-L2 Deficiency in Dendritic Cells Promotes CD4+ T Cell Proliferation
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. EAM Induction
4.3. Histopathological Examination
4.4. Flow Cytometric Analyses
4.5. RNA Extraction and Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction
4.6. T Cell Proliferation Assay
4.7. ELISA
4.8. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | antigen-presenting cell |
| BMDC | bone marrow-derived dendritic cell |
| CCL | chemokine (C-C motif) ligand |
| CFSE | carboxyfluorescein succinimidyl ester |
| CXCL | chemokine (C-X-C motif) ligand |
| DC | dendritic cell |
| EAM | experimental autoimmune myocarditis |
| H&E | hematoxylin and eosin |
| IFN-γ | interferon-γ |
| IL | interleukin |
| MyHC-α | myosin heavy chain-α |
| PD-1 | programmed death 1 |
| PD-L1 | programmed death ligand 1 |
| PD-L2 | programmed death ligand 2 |
| TCR | T cell receptor |
| TGF-β | transforming growth factor beta |
| TNF | tumor necrosis factor |
| WT | wild-type |
References
- Li, S.; Tajiri, K. A Lack of Biomarkers for Cardiac Complications of Immune Checkpoint Inhibitor Therapy. Intern. Med. 2021, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, K.; Ieda, M. Cardiac Complications in Immune Checkpoint Inhibition Therapy. Front. Cardiovasc. Med. 2019, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, K.; Aonuma, K.; Sekine, I. Immune checkpoint inhibitor-related myocarditis. Jpn. J. Clin. Oncol. 2018, 48, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Tanaka, Y.; Nishio, R.; Mitsuiye, T.; Mizoguchi, A.; Wang, J.; Ishida, M.; Hiai, H.; Matsumori, A.; Minato, N.; et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat. Med. 2003, 9, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Okazaki, I.-M.; Yoshida, T.; Chikuma, S.; Kato, Y.; Nakaki, F.; Hiai, H.; Honjo, T.; Okazaki, T. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int. Immunol. 2010, 22, 443–452. [Google Scholar] [CrossRef]
- Lucas, J.A.; Menke, J.; Rabacal, W.A.; Schoen, F.J.; Sharpe, A.H.; Kelley, V.R. Programmed death ligand 1 regulates a critical checkpoint for autoimmune myocarditis and pneumonitis in MRL mice. J. Immunol. 2008, 181, 2513–2521. [Google Scholar] [CrossRef] [PubMed]
- Tarrio, M.L.; Grabie, N.; Bu, D.; Sharpe, A.H.; Lichtman, A.H. PD-1 protects against inflammation and myocyte damage in T cell-mediated myocarditis. J. Immunol. 2012, 188, 4876–4884. [Google Scholar] [CrossRef]
- Tsuruoka, K.; Wakabayashi, S.; Morihara, H.; Matsunaga, N.; Fujisaka, Y.; Goto, I.; Imagawa, A.; Asahi, M. Exacerbation of autoimmune myocarditis by an immune checkpoint inhibitor is dependent on its time of administration in mice. Int. J. Cardiol. 2020, 313, 67–75. [Google Scholar] [CrossRef]
- Pummerer, C.L.; Luze, K.; Grässl, G.; Bachmaier, K.; Offner, F.; Burrell, S.K.; Lenz, D.M.; Zamborelli, T.J.; Penninger, J.M.; Neu, N. Identification of cardiac myosin peptides capable of inducing autoimmune myocarditis in BALB/c mice. J. Clin. Investig. 1996, 97, 2057–2062. [Google Scholar] [CrossRef]
- Neu, N.; Rose, N.R.; Beisel, K.W.; Herskowitz, A.; Gurri-Glass, G.; Craig, S.W. Cardiac myosin induces myocarditis in genetically predisposed mice. J. Immunol. 1987, 139, 3630–3636. [Google Scholar] [PubMed]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Dorfman, D.M.; Ma, F.-R.; Sullivan, E.L.; Munoz, O.; Wood, C.R.; Greenfield, E.A.; Freeman, G.J. Blockade of Programmed Death-1 Ligands on Dendritic Cells Enhances T Cell Activation and Cytokine Production. J. Immunol. 2003, 170, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, O.; Montes-Servín, E.; Hernandez-Martinez, J.M.; Cardona, A.F.; Casas-Ruiz, E.; Crispín, J.C.; Motola, D.; Flores-Estrada, D.; Barrera, L. Expression of PD-1/PD-L1 and PD-L2 in peripheral T-cells from non-small cell lung cancer patients. Oncotarget 2017, 8, 101994–102005. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.C.; Hsu, H.S.; Li, A.F.Y.; Chen, Y.J. Clinical relevance of PD-L1 and PD-L2 overexpression in patients with esophageal squamous cell carcinoma. J. Thorac. Dis. 2018, 10, 4433–4444. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Yearley, J.H.; Annamalai, L.; Pryzbycin, C.; Rini, B. Association of PD-L1, PD-L2, and immune response markers in matched renal clear cell carcinoma primary and metastatic tissue specimens. Am. J. Clin. Pathol. 2019, 151, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Tumang, J.R.; Gao, W.; Bai, C.; Rothstein, T.L.; Rothstein, T.L. PD-L2 expression extends beyond dendritic cells/macrophages to B1 cells enriched for VH11/VH12 and phosphatidylcholine binding. Eur. J. Immunol. 2007, 37, 2405–2410. [Google Scholar] [CrossRef]
- Messal, N.; Serriari, N.E.; Pastor, S.; Nunès, J.A.; Olive, D. PD-L2 is expressed on activated human T cells and regulates their function. Mol. Immunol. 2011, 48, 2214–2219. [Google Scholar] [CrossRef]
- Jubel, J.M.; Barbati, Z.R.; Burger, C.; Wirtz, D.C.; Schildberg, F.A. The Role of PD-1 in Acute and Chronic Infection. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Zhang, Y.; Chung, Y.; Bishop, C.; Daugherty, B.; Chute, H.; Holst, P.; Kurahara, C.; Lott, F.; Sun, N.; Welcher, A.A.; et al. Regulation of T cell activation and tolerance by PDL2. Proc. Natl. Acad. Sci. USA 2006, 103, 11695–11700. [Google Scholar] [CrossRef]
- Cuppari, C.; Amatruda, M.; Ceravolo, G.; Ceravolo, M.D.; Oreto, L.; Colavita, L.; Barbalace, A.; Calabrò, M.P.; Salpietro, C. Myocarditis in children—From infection to autoimmunity. J. Biol. Regul. Homeost. Agents 2020, 34, 37–41. [Google Scholar] [PubMed]
- Kania, G.; Blyszczuk, P.; Müller-Edenborn, B.; Eriksson, U. Novel therapeutic options in inflammatory cardiomyopathy. Swiss Med. Wkly. 2013, 143, w13841. [Google Scholar] [CrossRef] [PubMed]
- Elamm, C.; Fairweather, D.; Cooper, L.T. Pathogenesis and diagnosis of myocarditis. Heart 2012, 98, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Kindermann, I.; Barth, C.; Mahfoud, F.; Ukena, C.; Lenski, M.; Yilmaz, A.; Klingel, K.; Kandolf, R.; Sechtem, U.; Cooper, L.T.; et al. Update on myocarditis. J. Am. Coll. Cardiol. 2012, 59, 779–792. [Google Scholar] [CrossRef]
- Abenavoli, F.M.; Inchingolo, A.D.; Inchingolo, A.M.; Dipalma, G.; Inchingolo, F. Periodontal neoformations and myocarditis onset: Is it more than a simple coincidence? J. Biol. Regul. Homeost. Agents 2019, 33, 987–989. [Google Scholar]
- An, H.; Wang, Z.; Zheng, J. Inhibitory effects of apolipoprotein m on expressions of nf-κb, il-6, il-1β and other related factors in mice with myocarditis. J. Biol. Regul. Homeost. Agents 2019, 33, 1155–1160. [Google Scholar]
- Tajiri, K.; Imanaka-Yoshida, K.; Matsubara, A.; Tsujimura, Y.; Hiroe, M.; Naka, T.; Shimojo, N.; Sakai, S.; Aonuma, K.; Yasutomi, Y. Suppressor of Cytokine Signaling 1 DNA Administration Inhibits Inflammatory and Pathogenic Responses in Autoimmune Myocarditis. J. Immunol. 2012, 189, 2043–2053. [Google Scholar] [CrossRef]
- Tajiri, K.; Shimojo, N.; Sakai, S.; Machino-Ohtsuka, T.; Imanaka-Yoshida, K.; Hiroe, M.; Tsujimura, Y.; Kimura, T.; Sato, A.; Yasutomi, Y.; et al. Pitavastatin regulates helper T-Cell differentiation and ameliorates autoimmune myocarditis in mice. Cardiovasc. Drugs Ther. 2013, 27, 413–424. [Google Scholar] [CrossRef]
- Tajiri, K.; Sakai, S.; Kimura, T.; Machino-Ohtsuka, T.; Murakoshi, N.; Xu, D.; Wang, Z.; Sato, A.; Miyauchi, T.; Aonuma, K. Endothelin receptor antagonist exacerbates autoimmune myocarditis in mice. Life Sci. 2014, 118, 288–296. [Google Scholar] [CrossRef]
- Eriksson, U.; Ricci, R.; Hunziker, L.; Kurrer, M.O.; Oudit, G.Y.; Watts, T.H.; Sonderegger, I.; Bachmaier, K.; Kopf, M.; Penninger, J.M. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat. Med. 2003, 9, 1484–1490. [Google Scholar] [CrossRef]
- Valaperti, A.; Marty, R.R.; Kania, G.; Germano, D.; Mauermann, N.; Dirnhofer, S.; Leimenstoll, B.; Blyszczuk, P.; Dong, C.; Mueller, C.; et al. CD11b+ monocytes abrogate Th17 CD4+ T cell-mediated experimental autoimmune myocarditis. J. Immunol. 2008, 180, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.B.; Kukutsch, N.; Ogilvie, A.L.; Rössner, S.; Koch, F.; Romani, N.; Schuler, G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 1999, 223, 77–92. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Tajiri, K.; Murakoshi, N.; Xu, D.; Yonebayashi, S.; Okabe, Y.; Yuan, Z.; Feng, D.; Inoue, K.; Aonuma, K.; et al. Programmed Death-Ligand 2 Deficiency Exacerbates Experimental Autoimmune Myocarditis in Mice. Int. J. Mol. Sci. 2021, 22, 1426. https://doi.org/10.3390/ijms22031426
Li S, Tajiri K, Murakoshi N, Xu D, Yonebayashi S, Okabe Y, Yuan Z, Feng D, Inoue K, Aonuma K, et al. Programmed Death-Ligand 2 Deficiency Exacerbates Experimental Autoimmune Myocarditis in Mice. International Journal of Molecular Sciences. 2021; 22(3):1426. https://doi.org/10.3390/ijms22031426
Chicago/Turabian StyleLi, Siqi, Kazuko Tajiri, Nobuyuki Murakoshi, DongZhu Xu, Saori Yonebayashi, Yuta Okabe, Zixun Yuan, Duo Feng, Keiko Inoue, Kazuhiro Aonuma, and et al. 2021. "Programmed Death-Ligand 2 Deficiency Exacerbates Experimental Autoimmune Myocarditis in Mice" International Journal of Molecular Sciences 22, no. 3: 1426. https://doi.org/10.3390/ijms22031426
APA StyleLi, S., Tajiri, K., Murakoshi, N., Xu, D., Yonebayashi, S., Okabe, Y., Yuan, Z., Feng, D., Inoue, K., Aonuma, K., Shimoda, Y., Song, Z., Mori, H., Huang, H., Aonuma, K., & Ieda, M. (2021). Programmed Death-Ligand 2 Deficiency Exacerbates Experimental Autoimmune Myocarditis in Mice. International Journal of Molecular Sciences, 22(3), 1426. https://doi.org/10.3390/ijms22031426