
Alterations in Kynurenine and NAD+ Salvage Pathways during the Successful Treatment of Inflammatory Bowel Disease Suggest HCAR3 and NNMT as Potential Drug Targets




Abstract
:1. Introduction
2. Results
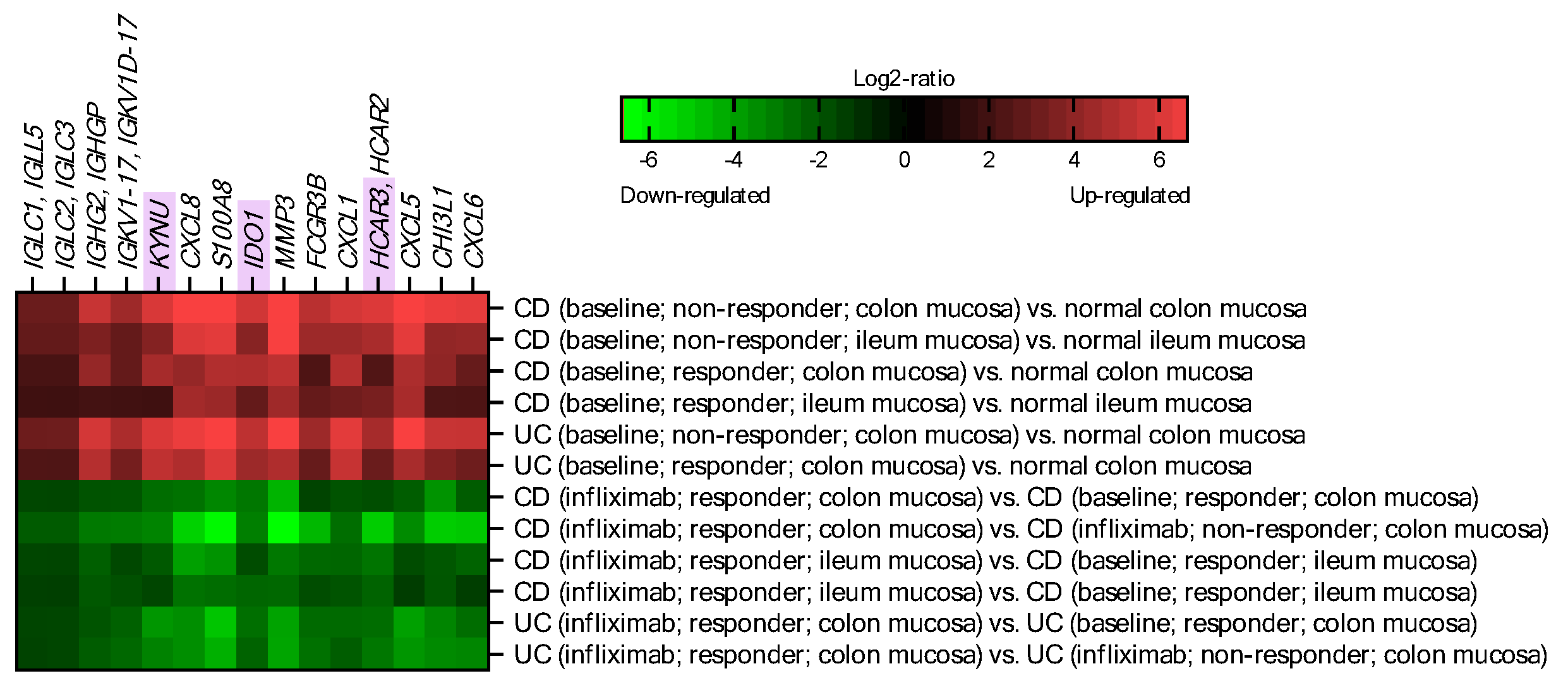
2.1. IDO1, KYNU and HCAR3 Are Upregulated in IBD and Suppressed upon Effective Infliximab Treatment
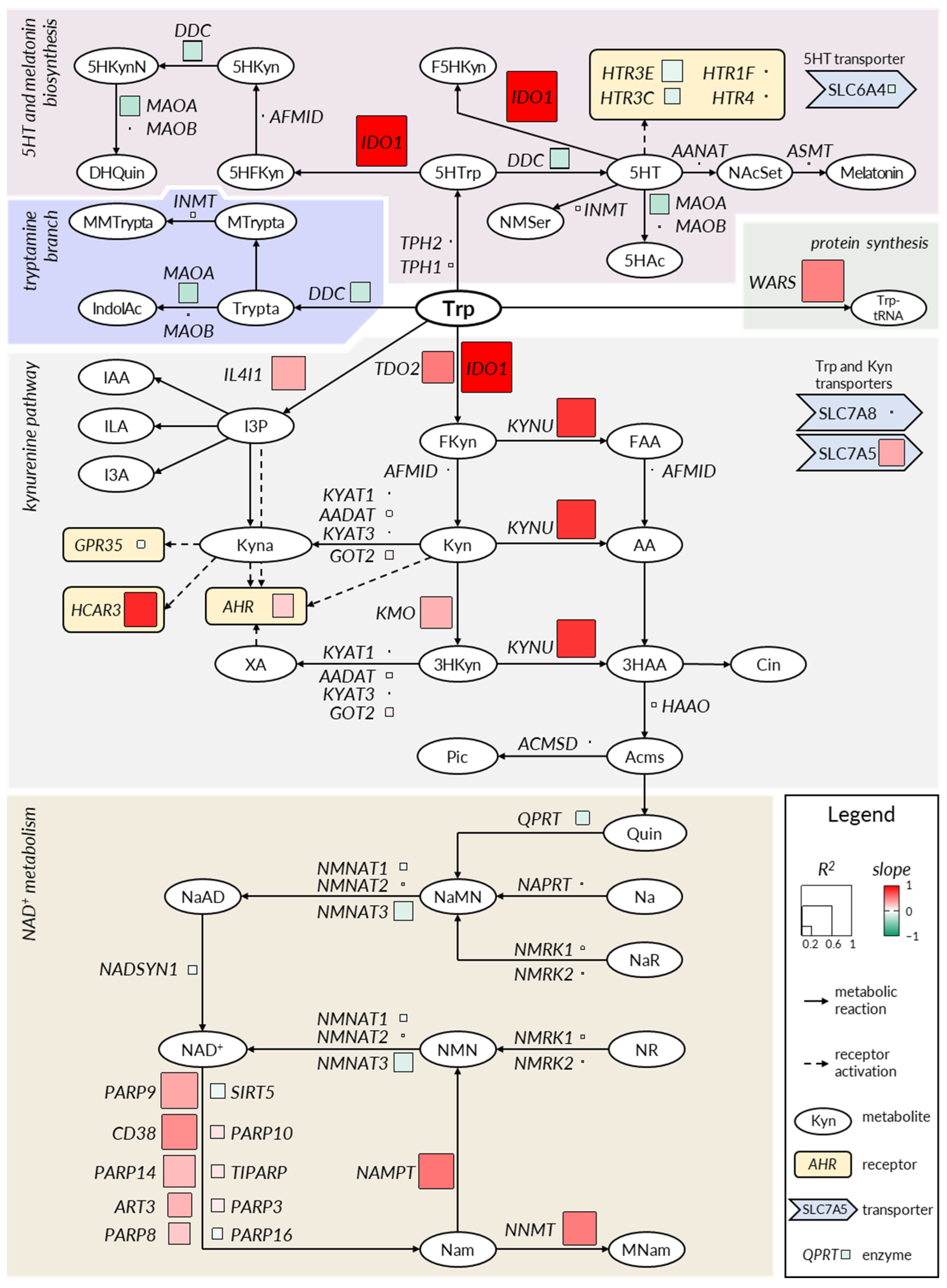
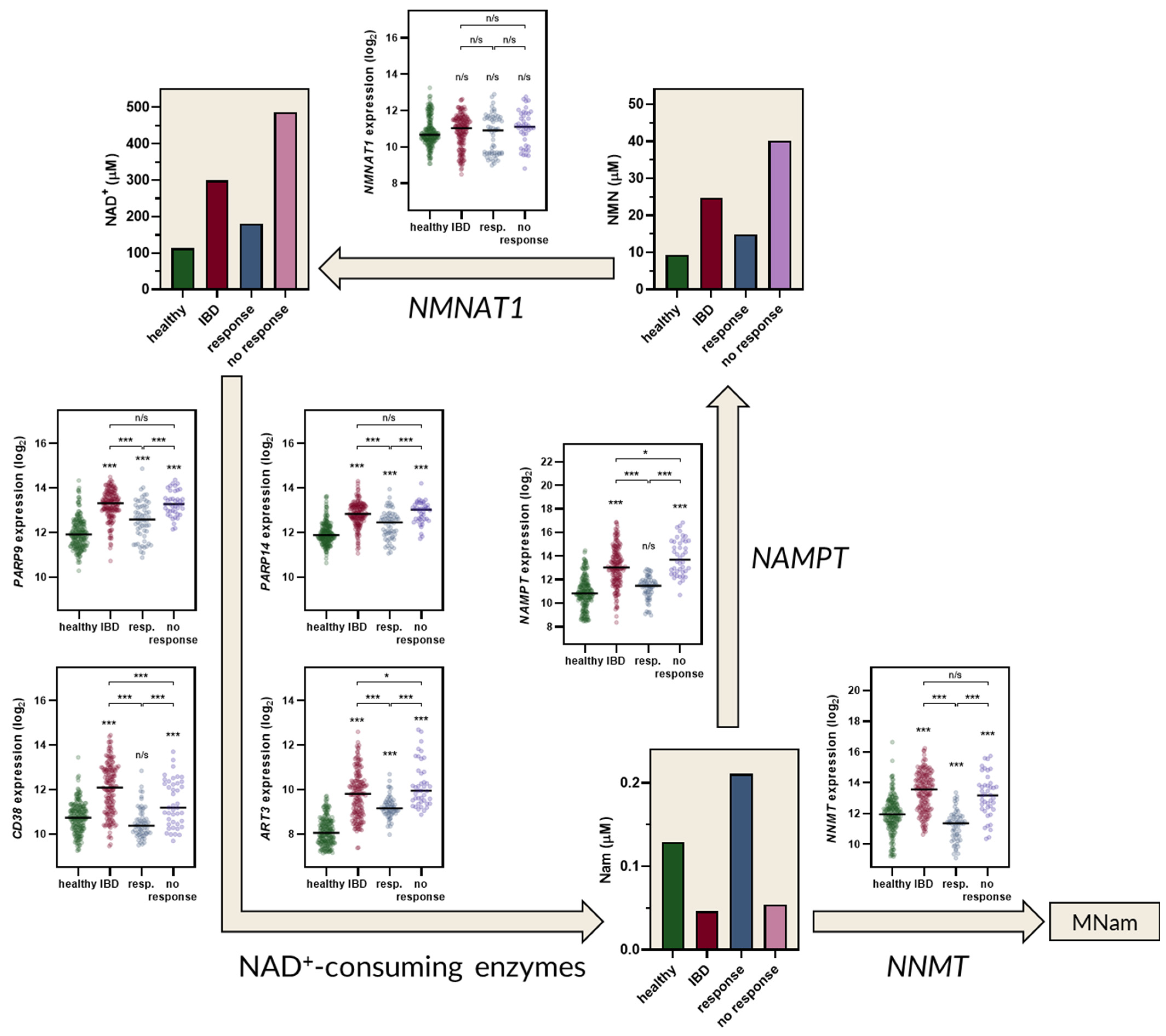
2.2. The Kynurenine and NAD+ Pathways Show a Consistent Imbalance in IBD
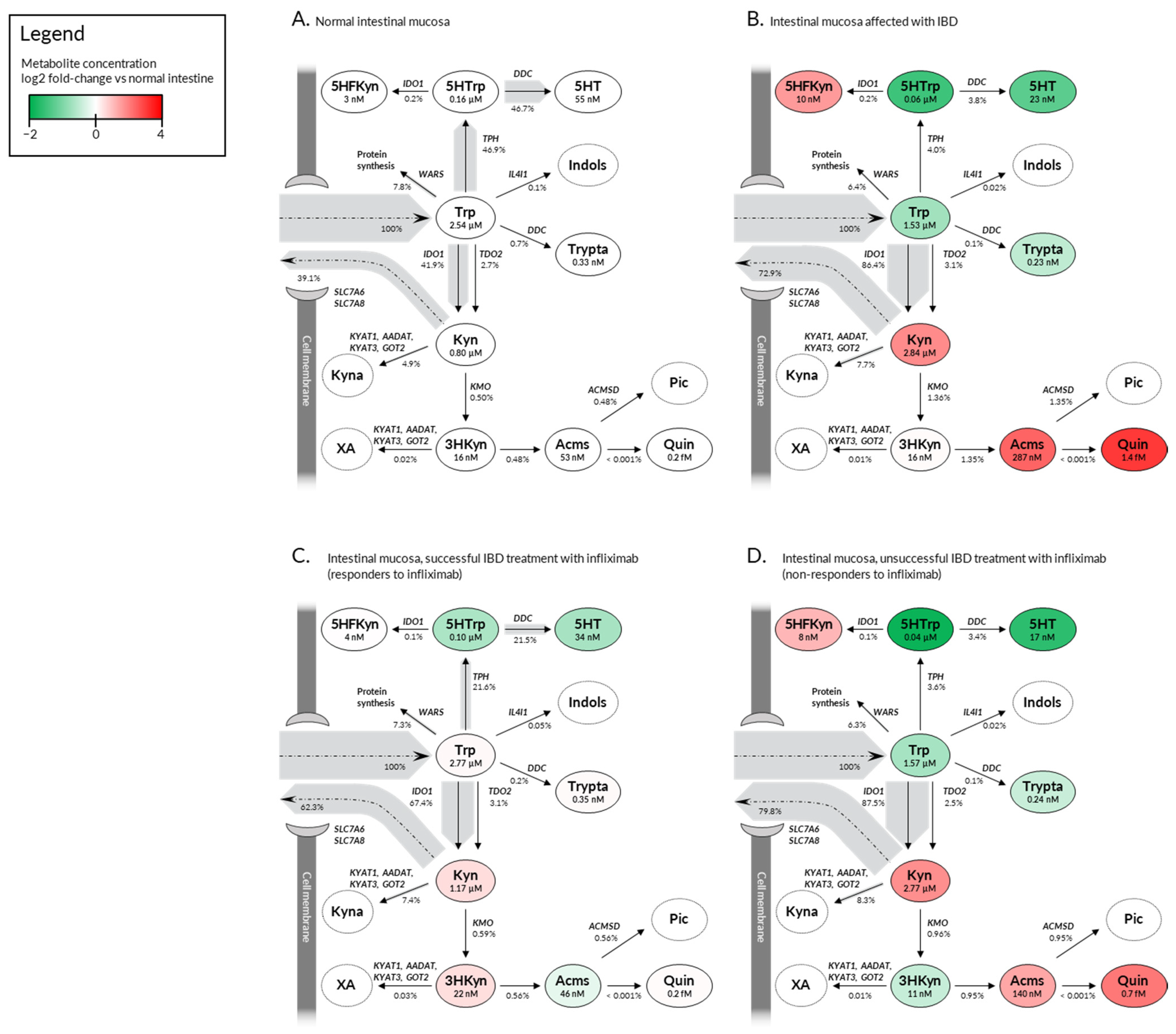
2.3. Deregulation in Gene Expression Shifts the Tryptophan Metabolite Landscape from Serotonin Production towards Kynurenine Pathway and NAD+ Synthesis
3. Discussion
4. Materials and Methods
4.1. Software
4.2. Identification of Genes Regulated during IBD Development and Treatment
4.3. Correlation Analysis
4.4. Tryptophan Metabolism Modeling
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaplan, G.G.; Ng, S.C. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology 2017, 152, 313–321.e312. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, O.H. New strategies for treatment of inflammatory bowel disease. Front. Med. 2014, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Nakase, H.; Uchino, M.; Shinzaki, S.; Matsuura, M.; Matsuoka, K.; Kobayashi, T.; Saruta, M.; Hirai, F.; Hata, K.; Hiraoka, S.; et al. Evidence-based clinical practice guidelines for inflammatory bowel disease 2020. J. Gastroenterol. 2021, 56, 489–526. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Y.M.; Yang, T.; An, L.Y.; Chen, X.Z.; Qi, Y.X.; He, H.Y.; Fan, H.B.; Sun, D.L. Evaluation of pharmacotherapy recommendations in guidelines for inflammatory bowel disease. J. Clin. Pharm. Ther. 2021, 46, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Kornbluth, A. Infliximab approved for use in Crohn’s disease: A report on the FDA GI Advisory Committee conference. Inflamm. Bowel Dis. 1998, 4, 328–329. [Google Scholar] [CrossRef]
- Papamichael, K.; Lin, S.; Moore, M.; Papaioannou, G.; Sattler, L.; Cheifetz, A.S. Infliximab in inflammatory bowel disease. Ther. Adv. Chronic Dis. 2019, 10, 2040622319838443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vulliemoz, M.; Brand, S.; Juillerat, P.; Mottet, C.; Ben-Horin, S.; Michetti, P. TNF-Alpha Blockers in Inflammatory Bowel Diseases: Practical Recommendations and a User’s Guide: An Update. Digestion 2020, 101 (Suppl. S1), 16–26. [Google Scholar] [CrossRef]
- Nielsen, O.H.; Ainsworth, M.A. Tumor necrosis factor inhibitors for inflammatory bowel disease. N. Engl. J. Med. 2013, 369, 754–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, 1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapolka, N.J.; Taghon, G.J.; Rowe, J.B.; Morgan, W.M.; Enten, J.F.; Lambert, N.A.; Isom, D.G. DCyFIR: A high-throughput CRISPR platform for multiplexed G protein-coupled receptor profiling and ligand discovery. Proc. Natl. Acad. Sci. USA 2020, 117, 13117–13126. [Google Scholar] [CrossRef]
- Zhai, L.; Ladomersky, E.; Lenzen, A.; Nguyen, B.; Patel, R.; Lauing, K.L.; Wu, M.; Wainwright, D.A. IDO1 in cancer: A Gemini of immune checkpoints. Cell. Mol. Immunol. 2018, 15, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Stavrum, A.K.; Heiland, I.; Schuster, S.; Puntervoll, P.; Ziegler, M. Model of tryptophan metabolism, readily scalable using tissue-specific gene expression data. J. Biol. Chem. 2013, 288, 34555–34566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockwoldt, M.; Houry, D.; Niere, M.; Gossmann, T.I.; Reinartz, I.; Schug, A.; Ziegler, M.; Heiland, I. Identification of evolutionary and kinetic drivers of NAD-dependent signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 15957–15966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadik, A.; Somarribas Patterson, L.F.; Ozturk, S.; Mohapatra, S.R.; Panitz, V.; Secker, P.F.; Pfander, P.; Loth, S.; Salem, H.; Prentzell, M.T.; et al. IL4I1 Is a Metabolic Immune Checkpoint that Activates the AHR and Promotes Tumor Progression. Cell 2020, 182, 1252–1270.e34. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Gostner, J.M.; Geisler, S.; Stonig, M.; Mair, L.; Sperner-Unterweger, B.; Fuchs, D. Tryptophan Metabolism and Related Pathways in Psychoneuroimmunology: The Impact of Nutrition and Lifestyle. Neuropsychobiology 2020, 79, 89–99. [Google Scholar] [CrossRef]
- Dudzinska, E.; Szymona, K.; Kloc, R.; Gil-Kulik, P.; Kocki, T.; Swistowska, M.; Bogucki, J.; Kocki, J.; Urbanska, E.M. Increased expression of kynurenine aminotransferases mRNA in lymphocytes of patients with inflammatory bowel disease. Therap. Adv. Gastroenterol. 2019, 12, 1756284819881304. [Google Scholar] [CrossRef]
- Gerner, R.R.; Klepsch, V.; Macheiner, S.; Arnhard, K.; Adolph, T.E.; Grander, C.; Wieser, V.; Pfister, A.; Moser, P.; Hermann-Kleiter, N.; et al. NAD metabolism fuels human and mouse intestinal inflammation. Gut 2018, 67, 1813–1823. [Google Scholar] [CrossRef]
- Joisten, N.; Ruas, J.L.; Braidy, N.; Guillemin, G.J.; Zimmer, P. The kynurenine pathway in chronic diseases: A compensatory mechanism or a driving force? Trends Mol. Med. 2021, 27, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Hasler, R.; et al. Increased Tryptophan Metabolism Is Associated With Activity of Inflammatory Bowel Diseases. Gastroenterology 2017, 153, 1504–1516.e1502. [Google Scholar] [CrossRef] [Green Version]
- Forrest, C.M.; Youd, P.; Kennedy, A.; Gould, S.R.; Darlington, L.G.; Stone, T.W. Purine, kynurenine, neopterin and lipid peroxidation levels in inflammatory bowel disease. J. Biomed. Sci. 2002, 9, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Coates, M.D.; Tekin, I.; Vrana, K.E.; Mawe, G.M. Review article: The many potential roles of intestinal serotonin (5-hydroxytryptamine, 5-HT) signalling in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2017, 46, 569–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magro, F.; Vieira-Coelho, M.A.; Fraga, S.; Serrao, M.P.; Veloso, F.T.; Ribeiro, T.; Soares-da-Silva, P. Impaired synthesis or cellular storage of norepinephrine, dopamine, and 5-hydroxytryptamine in human inflammatory bowel disease. Dig. Dis. Sci. 2002, 47, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Capurso, L.; Friedmann, C.A. Distribution of 5-OH tryptamine (serotonin) in ulcerative colitis. Proc. R. Soc. Med. 1970, 63 (Suppl. S1), 20–21. [Google Scholar]
- Coates, M.D.; Mahoney, C.R.; Linden, D.R.; Sampson, J.E.; Chen, J.; Blaszyk, H.; Crowell, M.D.; Sharkey, K.A.; Gershon, M.D.; Mawe, G.M.; et al. Molecular defects in mucosal serotonin content and decreased serotonin reuptake transporter in ulcerative colitis and irritable bowel syndrome. Gastroenterology 2004, 126, 1657–1664. [Google Scholar] [CrossRef]
- El-Salhy, M.; Danielsson, A.; Stenling, R.; Grimelius, L. Colonic endocrine cells in inflammatory bowel disease. J. Intern. Med. 1997, 242, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Sikander, A.; Sinha, S.K.; Prasad, K.K.; Rana, S.V. Association of Serotonin Transporter Promoter Polymorphism (5-HTTLPR) with Microscopic Colitis and Ulcerative Colitis. Dig. Dis. Sci. 2015, 60, 887–894. [Google Scholar] [CrossRef]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood-brain barrier transport of kynurenines: Implications for brain synthesis and metabolism. J. Neurochem. 1991, 56, 2007–2017. [Google Scholar] [CrossRef]
- Fujigaki, S.; Saito, K.; Takemura, M.; Fujii, H.; Wada, H.; Noma, A.; Seishima, M. Species differences in L-tryptophan-kynurenine pathway metabolism: Quantification of anthranilic acid and its related enzymes. Arch. Biochem. Biophys. 1998, 358, 329–335. [Google Scholar] [CrossRef]
- Uwai, Y.; Honjo, H.; Iwamoto, K. Interaction and transport of kynurenic acid via human organic anion transporters hOAT1 and hOAT3. Pharmacol. Res. 2012, 65, 254–260. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Ala, M. Tryptophan metabolites modulate inflammatory bowel disease and colorectal cancer by affecting immune system. Int. Rev. Immunol. 2021, 1–20. [Google Scholar] [CrossRef]
- Gasaly, N.; Hermoso, M.A.; Gotteland, M. Butyrate and the Fine-Tuning of Colonic Homeostasis: Implication for Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 3061. [Google Scholar] [CrossRef]
- Ngui, I.Q.H.; Perera, A.P.; Eri, R. Does NLRP3 Inflammasome and Aryl Hydrocarbon Receptor Play an Interlinked Role in Bowel Inflammation and Colitis-Associated Colorectal Cancer? Molecules 2020, 25, 2427. [Google Scholar] [CrossRef] [PubMed]
- Pernomian, L.; Duarte-Silva, M.; de Barros Cardoso, C.R. The Aryl Hydrocarbon Receptor (AHR) as a Potential Target for the Control of Intestinal Inflammation: Insights from an Immune and Bacteria Sensor Receptor. Clin. Rev. Allergy Immunol. 2020, 59, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Ma, N.; He, T.; Johnston, L.J.; Ma, X. Tryptophan (Trp) modulates gut homeostasis via aryl hydrocarbon receptor (AhR). Crit. Rev. Food Sci. Nutr. 2020, 60, 1760–1768. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [Green Version]
- Abron, J.D.; Singh, N.P.; Mishra, M.K.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S.; Singh, U.P. An endogenous aryl hydrocarbon receptor ligand, ITE, induces regulatory T cells and ameliorates experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G220–G230. [Google Scholar] [CrossRef]
- Gasaly, N.; de Vos, P.; Hermoso, M.A. Impact of Bacterial Metabolites on Gut Barrier Function and Host Immunity: A Focus on Bacterial Metabolism and Its Relevance for Intestinal Inflammation. Front. Immunol. 2021, 12, 658354. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, D.; Zhang, Y.; Chen, J.; Zhang, Y.; Liao, C.; Qin, S.; Tian, Y.; Zhang, Z.; Xu, F. Functional metabolomics reveal the role of AHR/GPR35 mediated kynurenic acid gradient sensing in chemotherapy-induced intestinal damage. Acta Pharm. Sin. B 2021, 11, 763–780. [Google Scholar] [CrossRef]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metghalchi, S.; Ponnuswamy, P.; Simon, T.; Haddad, Y.; Laurans, L.; Clement, M.; Dalloz, M.; Romain, M.; Esposito, B.; Koropoulis, V.; et al. Indoleamine 2,3-Dioxygenase Fine-Tunes Immune Homeostasis in Atherosclerosis and Colitis through Repression of Interleukin-10 Production. Cell Metab. 2015, 22, 460–471. [Google Scholar] [CrossRef] [Green Version]
- Tiszlavicz, Z.; Nemeth, B.; Fulop, F.; Vecsei, L.; Tapai, K.; Ocsovszky, I.; Mandi, Y. Different inhibitory effects of kynurenic acid and a novel kynurenic acid analogue on tumour necrosis factor-α (TNF-α) production by mononuclear cells, HMGB1 production by monocytes and HNP1-3 secretion by neutrophils. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 383, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef] [Green Version]
- Kapolka, N.J.; Isom, D.G. HCAR3: An underexplored metabolite sensor. Nat. Rev. Drug Discov. 2020, 19, 745. [Google Scholar] [CrossRef]
- Peters, A.; Krumbholz, P.; Jager, E.; Heintz-Buschart, A.; Cakir, M.V.; Rothemund, S.; Gaudl, A.; Ceglarek, U.; Schoneberg, T.; Staubert, C. Metabolites of lactic acid bacteria present in fermented foods are highly potent agonists of human hydroxycarboxylic acid receptor 3. PLoS Genet. 2019, 15, e1008145. [Google Scholar] [CrossRef] [Green Version]
- Mandrika, I.; Tilgase, A.; Petrovska, R.; Klovins, J. Hydroxycarboxylic Acid Receptor Ligands Modulate Proinflammatory Cytokine Expression in Human Macrophages and Adipocytes without Affecting Adipose Differentiation. Biol. Pharm. Bull. 2018, 41, 1574–1580. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Mohammed, A.; Wong, H.R.; Palaniyar, N.; Kamaleswaran, R. Machine Learning Identifies Complicated Sepsis Course and Subsequent Mortality Based on 20 Genes in Peripheral Blood Immune Cells at 24 H Post-ICU Admission. Front. Immunol. 2021, 12, 592303. [Google Scholar] [CrossRef]
- Li, Y.; Lin, M.; Wang, K.; Zhan, Y.; Gu, W.; Gao, G.; Huang, Y.; Chen, Y.; Huang, T.; Wang, J. A module of multifactor-mediated dysfunction guides the molecular typing of coronary heart disease. Mol. Genet. Genom. Med. 2020, 8, e1415. [Google Scholar] [CrossRef]
- Yang, X.; Wei, W.; Tan, S.; Guo, L.; Qiao, S.; Yao, B.; Wang, Z. Identification and verification of HCAR3 and INSL5 as new potential therapeutic targets of colorectal cancer. World J. Surg. Oncol. 2021, 19, 248. [Google Scholar] [CrossRef]
- Offermanns, S. Hydroxy-Carboxylic Acid Receptor Actions in Metabolism. Trends Endocrinol. Metab. 2017, 28, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Raker, V.K.; Becker, C.; Steinbrink, K. The cAMP Pathway as Therapeutic Target in Autoimmune and Inflammatory Diseases. Front. Immunol. 2016, 7, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colletti, S.; Ijzerman, A.; Lovenberg, T.; Offermanns, S.; Semple, G.; Waters, M.; Wise, A. Hydroxycarboxylic acid receptors (version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS Guide Pharmacol. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Glaria, E.; Valledor, A.F. Roles of CD38 in the Immune Response to Infection. Cells 2020, 9, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Zhang, A.; Du, Y.; Fang, M.; Minze, L.J.; Liu, Y.J.; Li, X.C.; Zhang, Z. Identification of poly(ADP-ribose) polymerase 9 (PARP9) as a noncanonical sensor for RNA virus in dendritic cells. Nat. Commun. 2021, 12, 2681. [Google Scholar] [CrossRef]
- Gao, Y.; Martin, N.I.; van Haren, M.J. Nicotinamide N-methyl transferase (NNMT): An emerging therapeutic target. Drug Discov. Today 2021, 26, 2699–2706. [Google Scholar] [CrossRef]
- Hruz, T.; Laule, O.; Szabo, G.; Wessendorp, F.; Bleuler, S.; Oertle, L.; Widmayer, P.; Gruissem, W.; Zimmermann, P. Genevestigator v3: A reference expression database for the meta-analysis of transcriptomes. Adv. Bioinform. 2008, 2008, 420747. [Google Scholar] [CrossRef] [PubMed]
- Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhal, M.; Xu, L.; Mendes, P.; Kummer, U. COPASI–A COmplex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar] [CrossRef] [Green Version]
- Arijs, I.; De Hertogh, G.; Lemaire, K.; Quintens, R.; Van Lommel, L.; Van Steen, K.; Leemans, P.; Cleynen, I.; Van Assche, G.; Vermeire, S.; et al. Mucosal gene expression of antimicrobial peptides in inflammatory bowel disease before and after first infliximab treatment. PLoS ONE 2009, 4, e7984. [Google Scholar] [CrossRef]
- Li, C.; Donizelli, M.; Rodriguez, N.; Dharuri, H.; Endler, L.; Chelliah, V.; Li, L.; He, E.; Henry, A.; Stefan, M.I.; et al. BioModels Database: An enhanced, curated and annotated resource for published quantitative kinetic models. BMC Syst. Biol. 2010, 4, 92. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Hara, N.; Shibata, T.; Osago, H.; Tsuchiya, M. The simultaneous measurement of nicotinamide adenine dinucleotide and related compounds by liquid chromatography/electrospray ionization tandem mass spectrometry. Anal. Biochem. 2006, 352, 282–285. [Google Scholar] [CrossRef]
- Sallin, O.; Reymond, L.; Gondrand, C.; Raith, F.; Koch, B.; Johnsson, K. Semisynthetic biosensors for mapping cellular concentrations of nicotinamide adenine dinucleotides. eLife 2018, 7, e32638. [Google Scholar] [CrossRef] [PubMed]
- Llamas-Velasco, M.; Bonay, P.; Jose Concha-Garzon, M.; Corvo-Villen, L.; Vara, A.; Cibrian, D.; Sanguino-Pascual, A.; Sanchez-Madrid, F.; de la Fuente, H.; Dauden, E. Immune cells from patients with psoriasis are defective in inducing indoleamine 2,3-dioxygenase expression in response to inflammatory stimuli. Br. J. Dermatol. 2017, 176, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Nollen, E.A.A.; Rohrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Auwerx, J. Modulating NAD+ metabolism, from bench to bedside. EMBO J. 2017, 36, 2670–2683. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calculated Intestine Level 1 | Calculated Brain Level [12] | Calculated Liver Level [12] | Calculated Serum Level 2 | Measured Serum Level [18] | ||
|---|---|---|---|---|---|---|
| Trp | normal | 2.5 µM | 3.9 µM | 0.1 µM | 46.4 µM | 48.1 ± 13.1 µM |
| IBD | 1.5 µM | 39.3 µM | 35.6 ± 11.1 µM (UC) 37.8 ± 9.4 µM (CD) | |||
| Kyn | normal | 0.8 µM | 0.5 µM | 2.6 µM | 3.9 µM | 2.9 ± 1.2 µM |
| IBD | 2.8 µM | 5.9 µM | 5.5 ± 5.2 µM (UC) 8.4 ± 7.4 µM (CD) | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wnorowski, A.; Wnorowska, S.; Kurzepa, J.; Parada-Turska, J. Alterations in Kynurenine and NAD+ Salvage Pathways during the Successful Treatment of Inflammatory Bowel Disease Suggest HCAR3 and NNMT as Potential Drug Targets. Int. J. Mol. Sci. 2021, 22, 13497. https://doi.org/10.3390/ijms222413497
Wnorowski A, Wnorowska S, Kurzepa J, Parada-Turska J. Alterations in Kynurenine and NAD+ Salvage Pathways during the Successful Treatment of Inflammatory Bowel Disease Suggest HCAR3 and NNMT as Potential Drug Targets. International Journal of Molecular Sciences. 2021; 22(24):13497. https://doi.org/10.3390/ijms222413497
Chicago/Turabian StyleWnorowski, Artur, Sylwia Wnorowska, Jacek Kurzepa, and Jolanta Parada-Turska. 2021. "Alterations in Kynurenine and NAD+ Salvage Pathways during the Successful Treatment of Inflammatory Bowel Disease Suggest HCAR3 and NNMT as Potential Drug Targets" International Journal of Molecular Sciences 22, no. 24: 13497. https://doi.org/10.3390/ijms222413497
APA StyleWnorowski, A., Wnorowska, S., Kurzepa, J., & Parada-Turska, J. (2021). Alterations in Kynurenine and NAD+ Salvage Pathways during the Successful Treatment of Inflammatory Bowel Disease Suggest HCAR3 and NNMT as Potential Drug Targets. International Journal of Molecular Sciences, 22(24), 13497. https://doi.org/10.3390/ijms222413497