
Genetic Background Influences Severity of Colonic Aganglionosis and Response to GDNF Enemas in the Holstein Mouse Model of Hirschsprung Disease

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
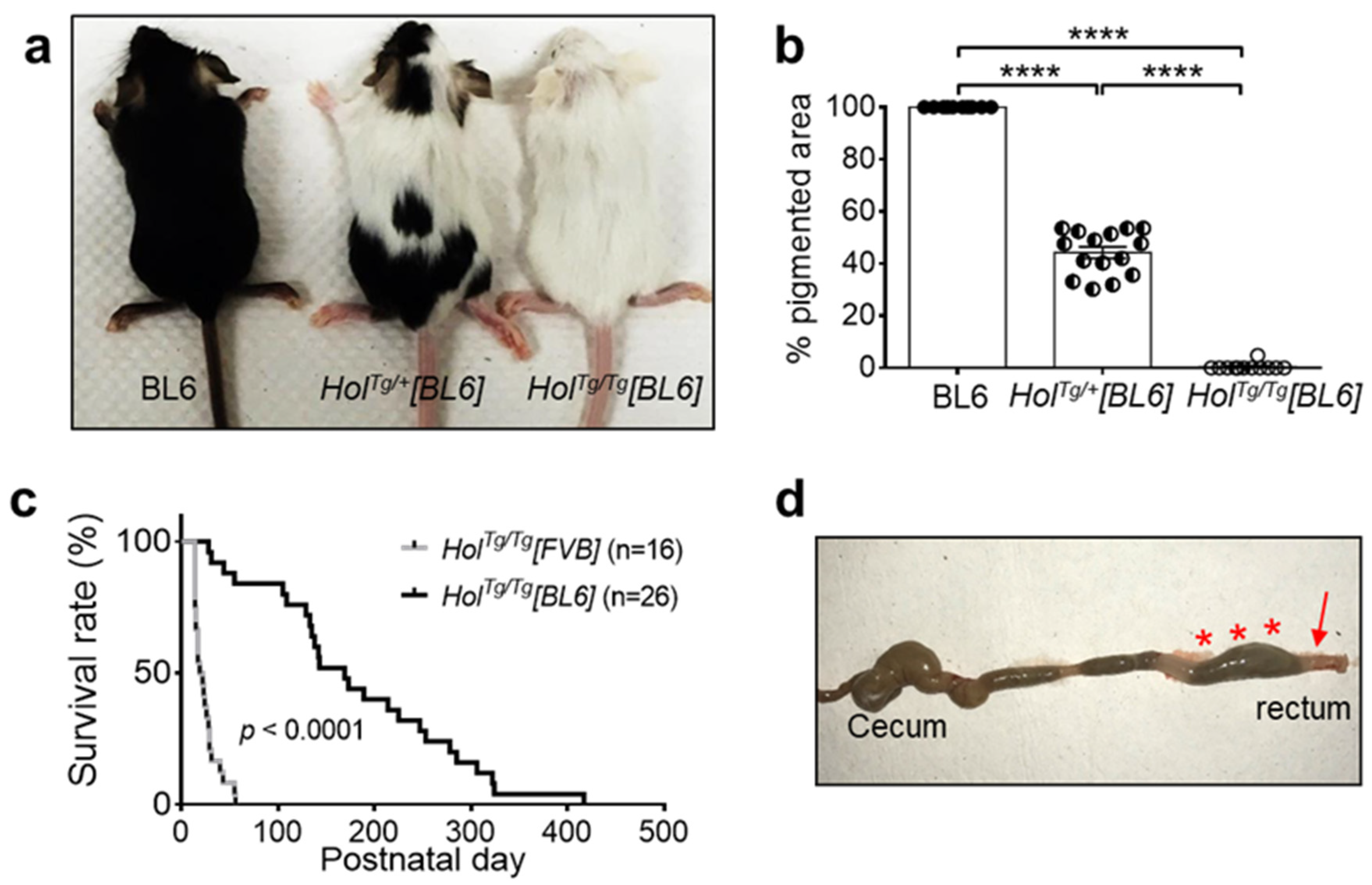
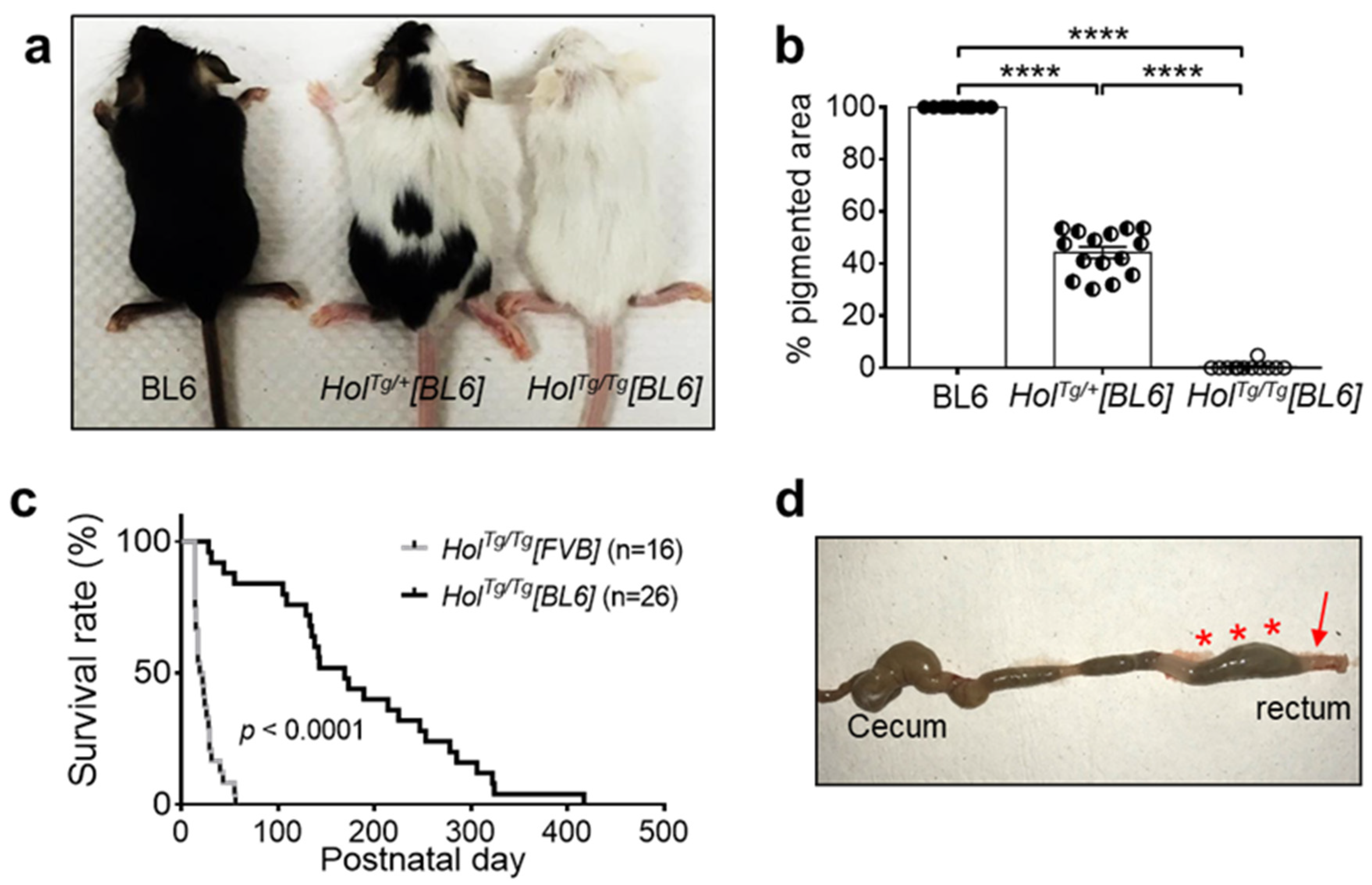
2.1. Increased Survival of HolTg/Tg[BL6] Mice Compared to HolTg/Tg[FVB] Mice
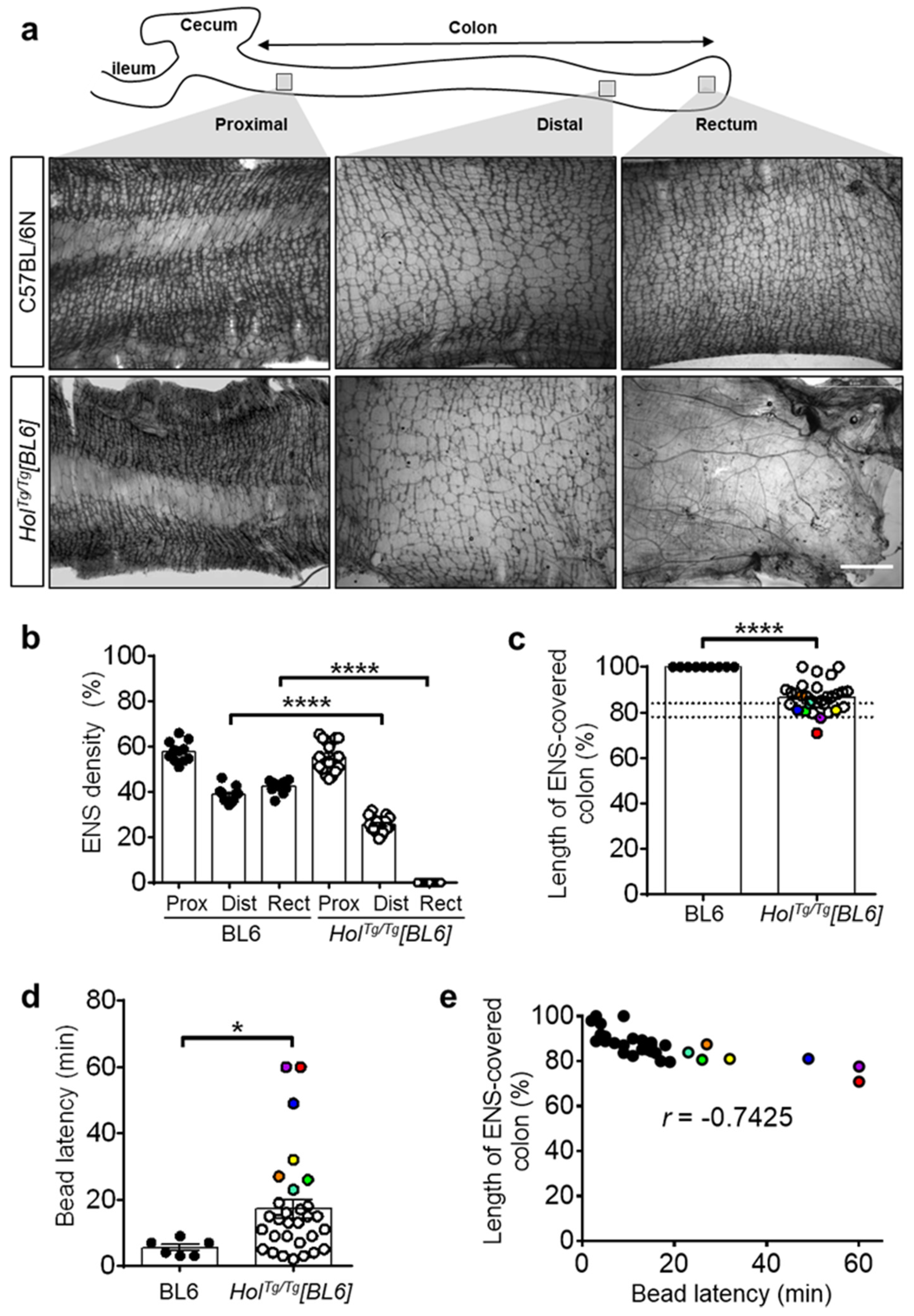
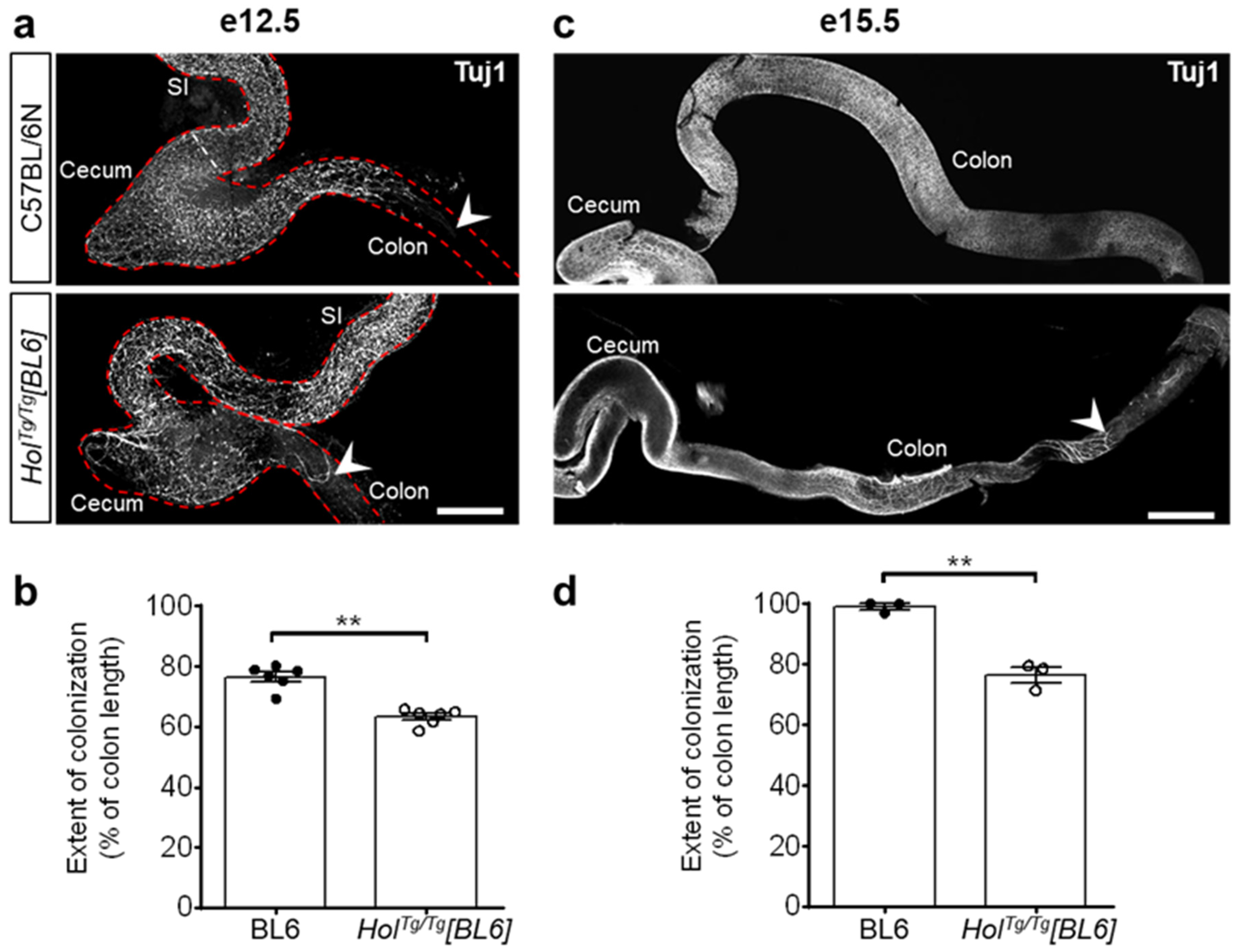
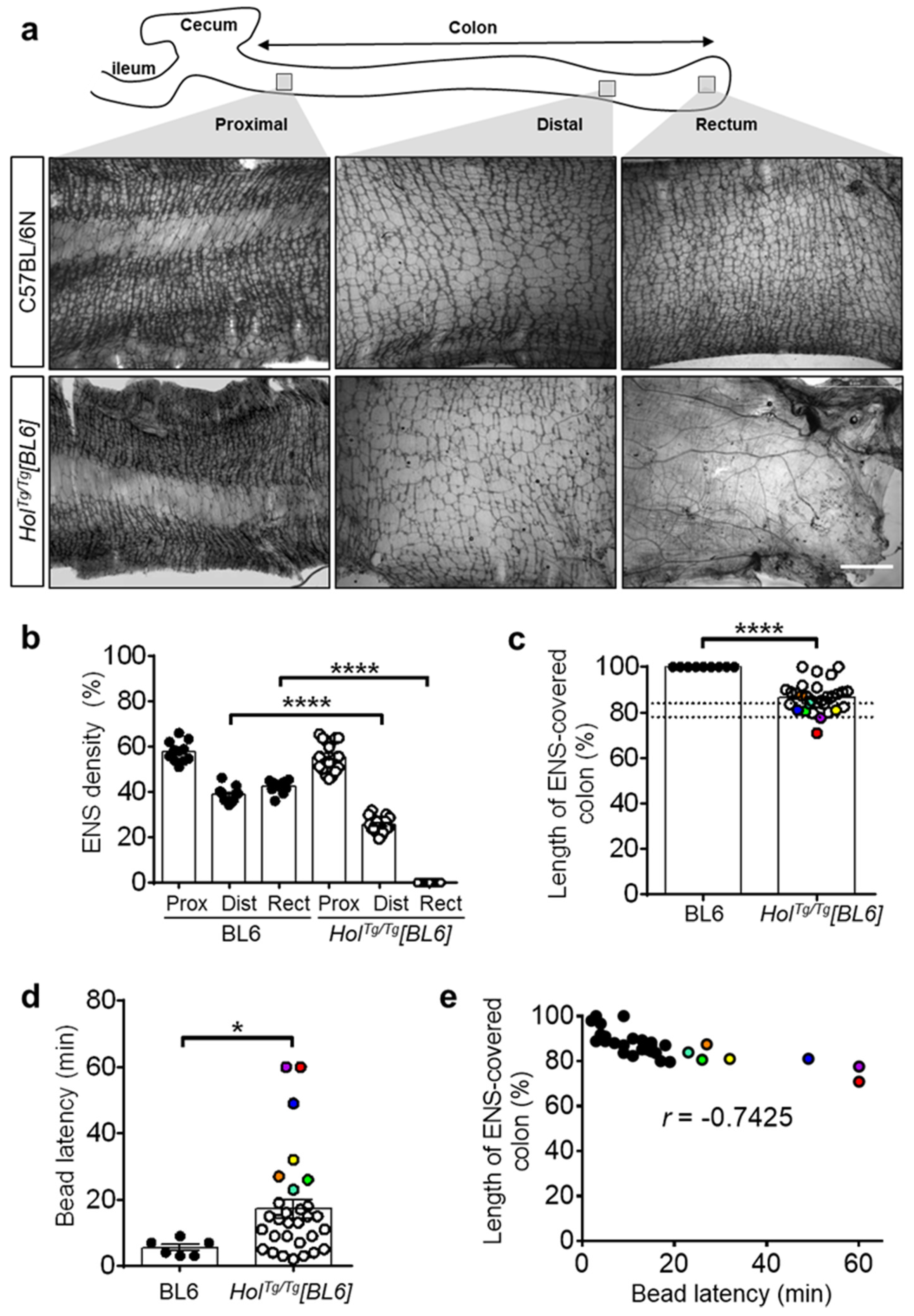
2.2. Short-Segment Aganglionosis and Colonic Dysmotility in HolTg/Tg[BL6] Mice
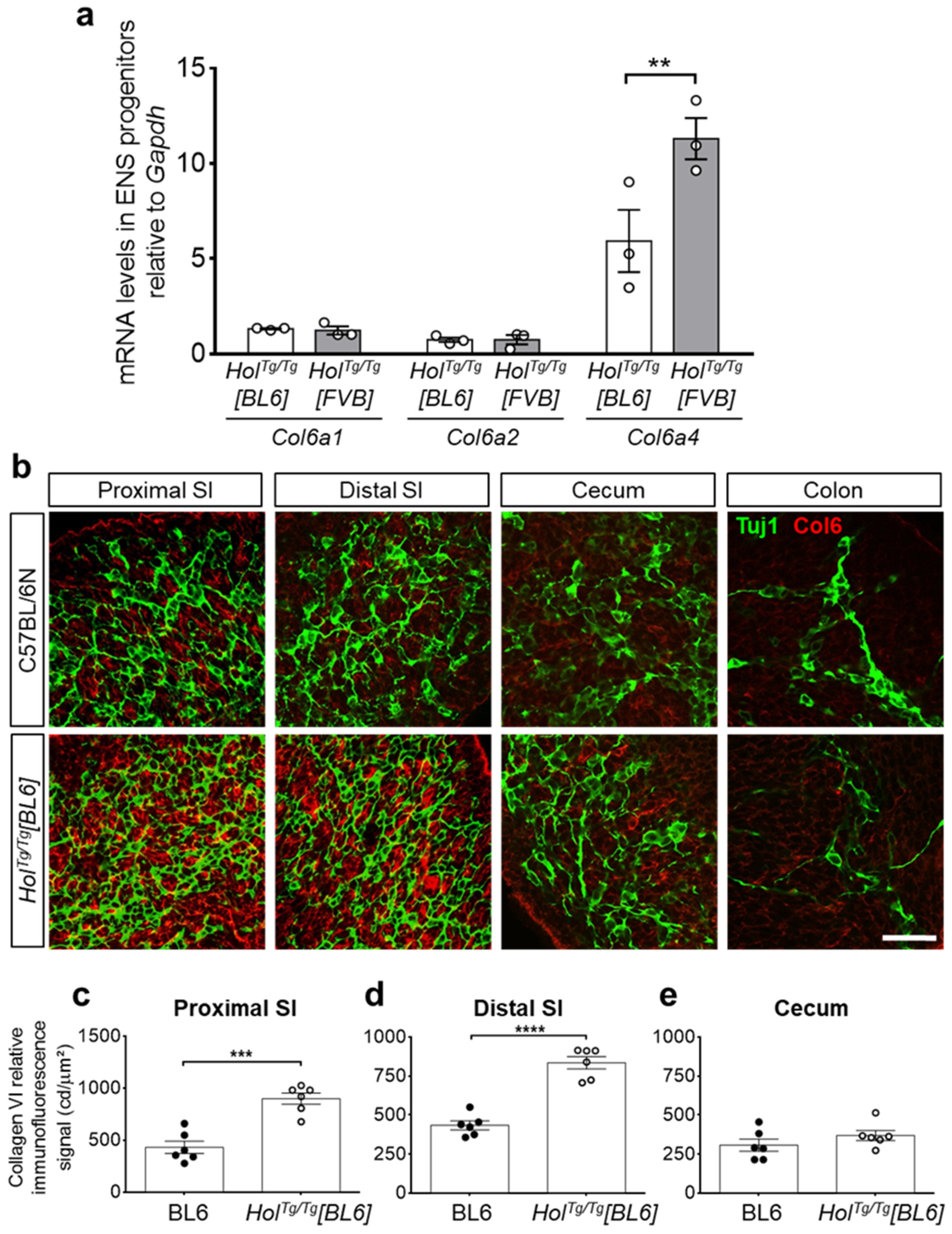
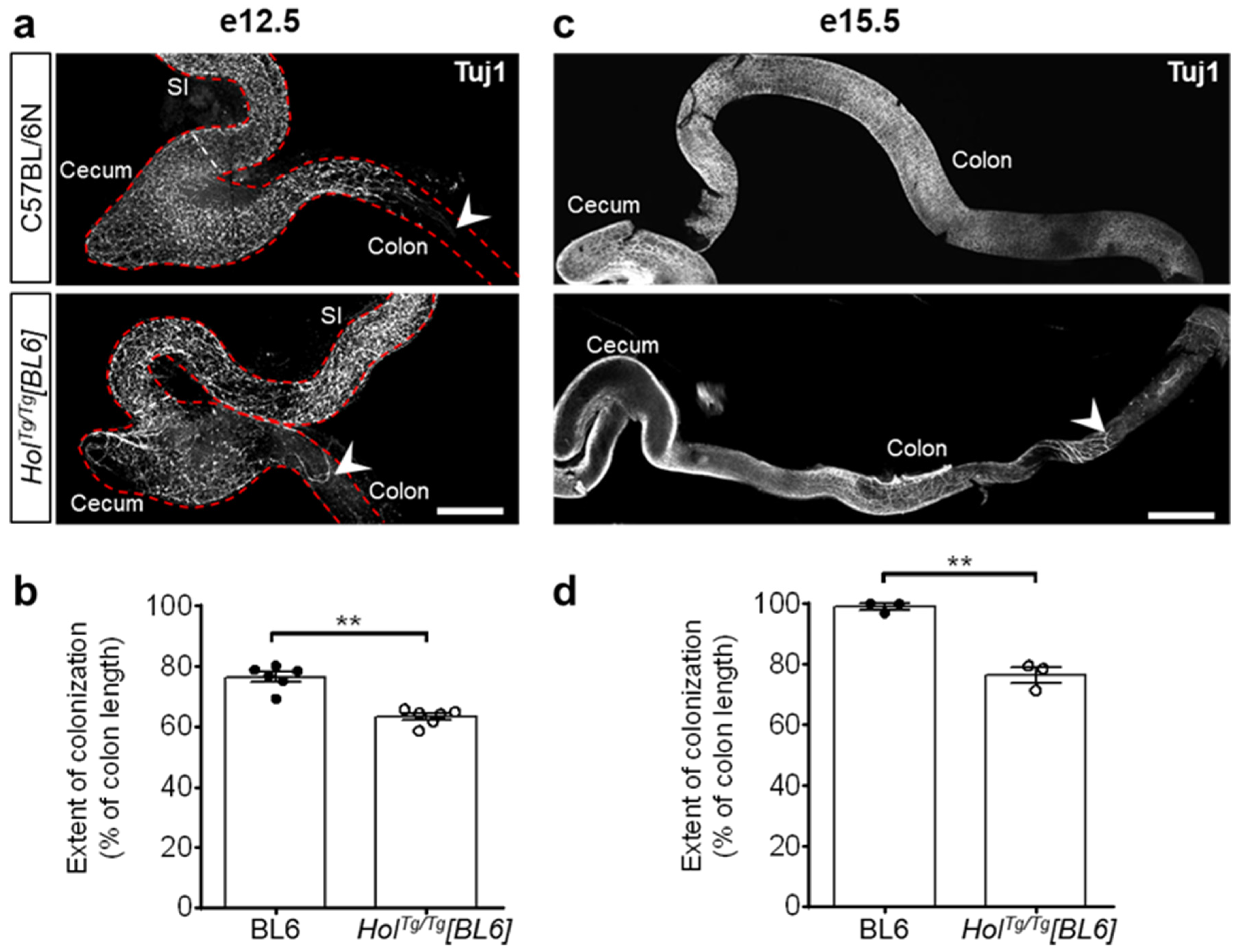
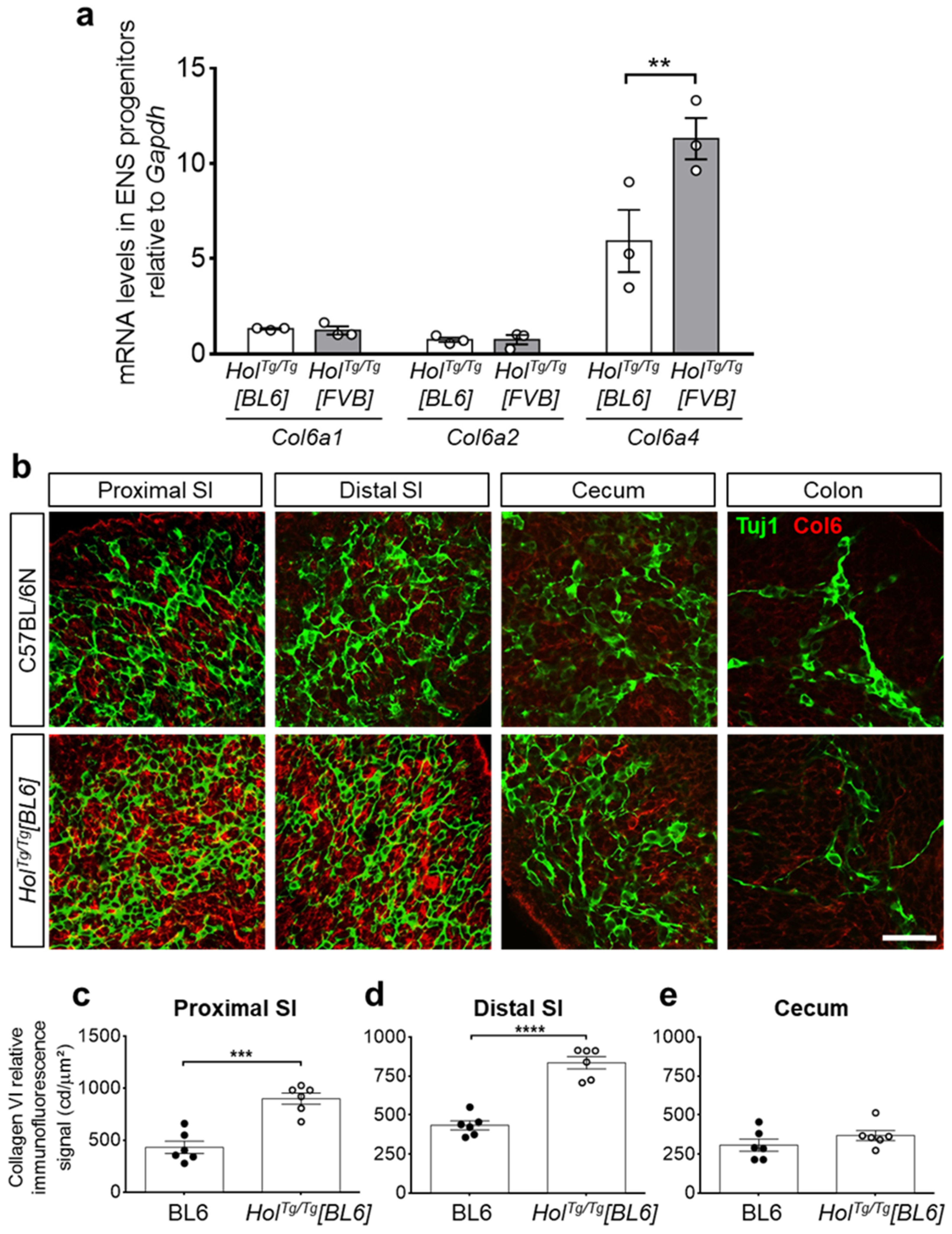
2.3. Col6a4 Overexpression Is Less Extensive in HolTg/Tg[BL6] than in HolTg/Tg[FVB] Mice
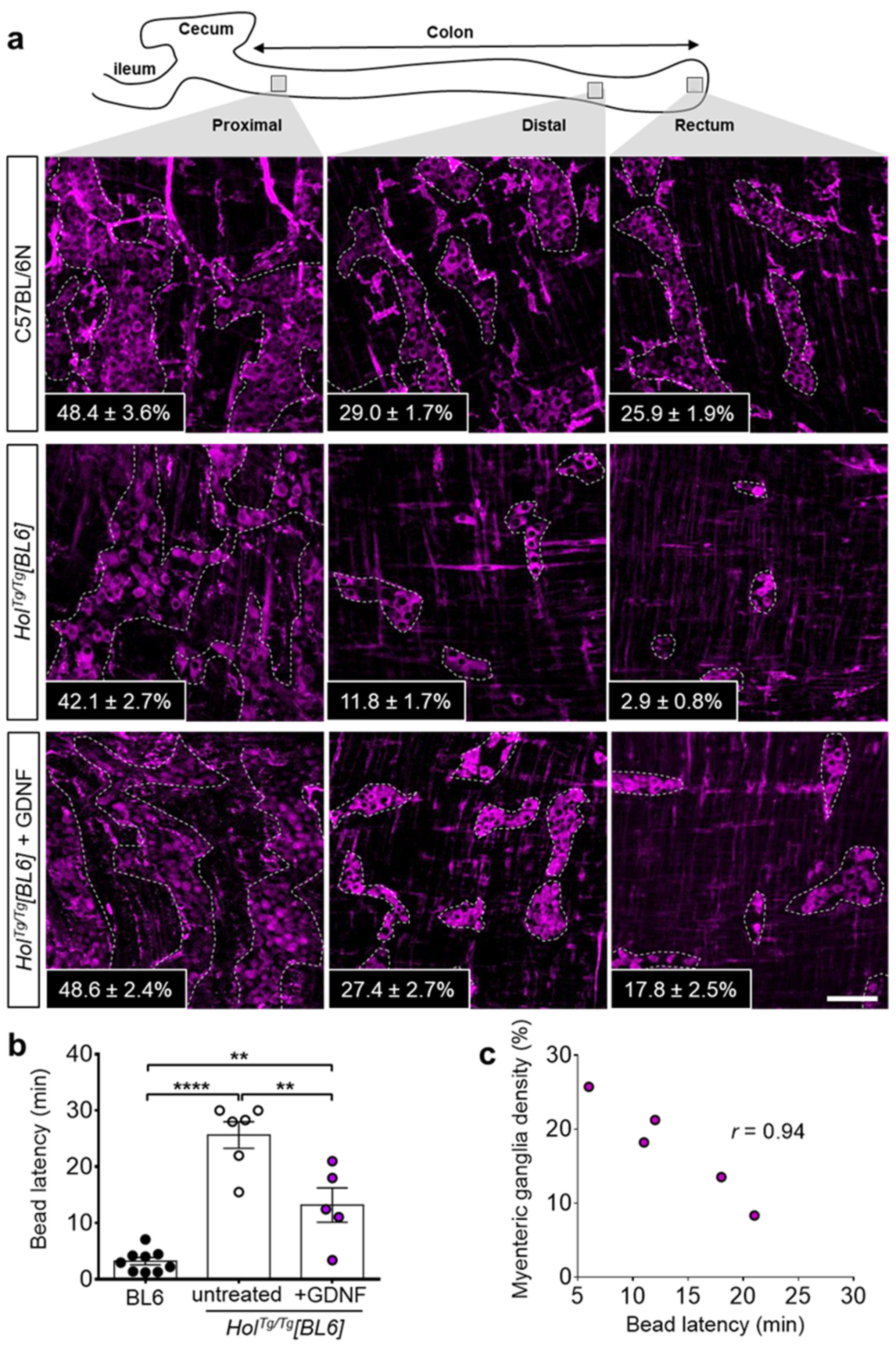
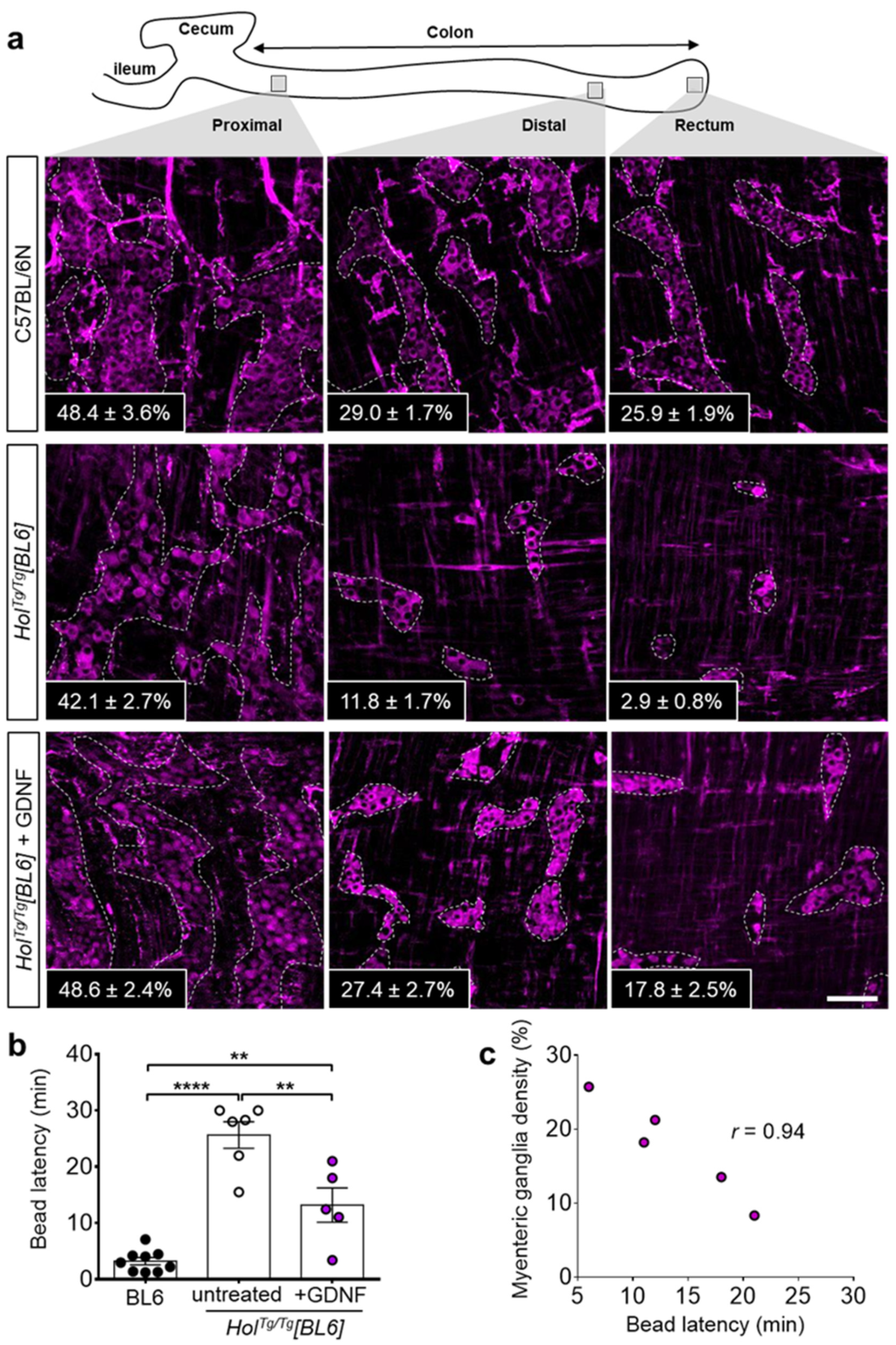
2.4. GDNF Enemas Restore Nearly Normal ENS Density and Function in HolTg/Tg[BL6] Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Tissue Staining and Imaging
4.3. Bead Latency Test
4.4. Fluorescence-Activated Cell Sorting (FACS) and RT-qPCR
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heuckeroth, R.O. Hirschsprung disease—Integrating basic science and clinical medicine to improve outcomes. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Pilon, N. Treatment and Prevention of Neurocristopathies. Trends Mol. Med. 2021, 27, 451–468. [Google Scholar] [CrossRef]
- Bergeron, K.F.; Silversides, D.W.; Pilon, N. The developmental genetics of Hirschsprung’s disease. Clin. Genet. 2013, 83, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Gosain, A.; Brinkman, A.S. Hirschsprung’s associated enterocolitis. Curr Opin Pediatr. 2015, 27, 364–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinhaus, S.; Boley, S.J.; Sheran, M.; Sieber, W.K. Hirschsprung’s disease—A survey of the members of the Surgical Section of the American Academy of Pediatrics. J. Pediatr. Surg. 1979, 14, 588–597. [Google Scholar] [CrossRef]
- Alves, M.M.; Sribudiani, Y.; Brouwer, R.W.; Amiel, J.; Antinolo, G.; Borrego, S.; Ceccherini, I.; Chakravarti, A.; Fernandez, R.M.; Garcia-Barcelo, M.M.; et al. Contribution of rare and common variants determine complex diseases-Hirschsprung disease as a model. Dev. Biol. 2013, 382, 320–329. [Google Scholar] [CrossRef]
- Amiel, J.; Sproat-Emison, E.; Garcia-Barcelo, M.; Lantieri, F.; Burzynski, G.; Borrego, S.; Pelet, A.; Arnold, S.; Miao, X.; Griseri, P.; et al. Hirschsprung disease, associated syndromes and genetics: A review. J. Med. Genet. 2008, 45, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Attie, T.; Pelet, A.; Edery, P.; Eng, C.; Mulligan, L.M.; Amiel, J.; Boutrand, L.; Beldjord, C.; Nihoul-Fekete, C.; Munnich, A.; et al. Diversity of RET proto-oncogene mutations in familial and sporadic Hirschsprung disease. Hum. Mol. Genet. 1995, 4, 1381–1386. [Google Scholar] [CrossRef]
- Bolk, S.; Pelet, A.; Hofstra, R.M.; Angrist, M.; Salomon, R.; Croaker, D.; Buys, C.H.; Lyonnet, S.; Chakravarti, A. A human model for multigenic inheritance: Phenotypic expression in Hirschsprung disease requires both the RET gene and a new 9q31 locus. Proc. Natl. Acad. Sci. USA 2000, 97, 268–273. [Google Scholar] [CrossRef] [Green Version]
- Pingault, V.; Bondurand, N.; Kuhlbrodt, K.; Goerich, D.E.; Prehu, M.O.; Puliti, A.; Herbarth, B.; Hermans-Borgmeyer, I.; Legius, E.; Matthijs, G.; et al. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat. Genet. 1998, 18, 171–173. [Google Scholar] [CrossRef]
- Karim, A.; Tang, C.S.; Tam, P.K. The Emerging Genetic Landscape of Hirschsprung Disease and Its Potential Clinical Applications. Front. Pediatr. 2021, 9, 638093. [Google Scholar] [CrossRef]
- Luzon-Toro, B.; Villalba-Benito, L.; Torroglosa, A.; Fernandez, R.M.; Antinolo, G.; Borrego, S. What is new about the genetic background of Hirschsprung disease? Clin. Genet. 2020, 97, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Gui, H.; Schriemer, D.; Cheng, W.W.; Chauhan, R.K.; Antinolo, G.; Berrios, C.; Bleda, M.; Brooks, A.S.; Brouwer, R.W.; Burns, A.J.; et al. Whole exome sequencing coupled with unbiased functional analysis reveals new Hirschsprung disease genes. Genome Biol. 2017, 18, 48. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Nandakumar, P.; Auer, D.R.; Sosa, M.X.; Ross, H.; Bollinger, J.; Yan, J.; Berrios, C.; Hirschsprung Disease Research, C.; Chakravarti, A. Multiple, independent, common variants at RET, SEMA3 and NRG1 gut enhancers specify Hirschsprung disease risk in European ancestry subjects. J. Pediatr. Surg. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kuil, L.E.; MacKenzie, K.C.; Tang, C.S.; Windster, J.D.; Le, T.L.; Karim, A.; de Graaf, B.M.; van der Helm, R.; van Bever, Y.; Sloots, C.E.J.; et al. Size matters: Large copy number losses in Hirschsprung disease patients reveal genes involved in enteric nervous system development. PLoS Genet. 2021, 17, e1009698. [Google Scholar] [CrossRef]
- Mederer, T.; Schmitteckert, S.; Volz, J.; Martinez, C.; Roth, R.; Thumberger, T.; Eckstein, V.; Scheuerer, J.; Thoni, C.; Lasitschka, F.; et al. A complementary study approach unravels novel players in the pathoetiology of Hirschsprung disease. PLoS Genet. 2020, 16, e1009106. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.S.; Li, P.; Lai, F.P.; Fu, A.X.; Lau, S.T.; So, M.T.; Lui, K.N.; Li, Z.; Zhuang, X.; Yu, M.; et al. Identification of Genes Associated With Hirschsprung Disease, Based on Whole-Genome Sequence Analysis, and Potential Effects on Enteric Nervous System Development. Gastroenterology 2018, 155, 1908–1922. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.S.; Zhuang, X.; Lam, W.Y.; Ngan, E.S.; Hsu, J.S.; Michelle, Y.U.; Man-Ting, S.O.; Cherny, S.S.; Ngo, N.D.; Sham, P.C.; et al. Uncovering the genetic lesions underlying the most severe form of Hirschsprung disease by whole-genome sequencing. Eur. J. Hum. Genet. 2018, 26, 818–826. [Google Scholar] [CrossRef] [Green Version]
- Tilghman, J.M.; Ling, A.Y.; Turner, T.N.; Sosa, M.X.; Krumm, N.; Chatterjee, S.; Kapoor, A.; Coe, B.P.; Nguyen, K.H.; Gupta, N.; et al. Molecular Genetic Anatomy and Risk Profile of Hirschsprung’s Disease. N. Engl. J. Med. 2019, 380, 1421–1432. [Google Scholar] [CrossRef]
- Edery, P.; Lyonnet, S.; Mulligan, L.M.; Pelet, A.; Dow, E.; Abel, L.; Holder, S.; Nihoul-Fekete, C.; Ponder, B.A.; Munnich, A. Mutations of the RET proto-oncogene in Hirschsprung’s disease. Nature 1994, 367, 378–380. [Google Scholar] [CrossRef]
- Romeo, G.; Ronchetto, P.; Luo, Y.; Barone, V.; Seri, M.; Ceccherini, I.; Pasini, B.; Bocciardi, R.; Lerone, M.; Kaariainen, H.; et al. Point mutations affecting the tyrosine kinase domain of the RET proto-oncogene in Hirschsprung’s disease. Nature 1994, 367, 377–378. [Google Scholar] [CrossRef] [PubMed]
- Friedmacher, F.; Puri, P. Hirschsprung’s disease associated with Down syndrome: A meta-analysis of incidence, functional outcomes and mortality. Pediatric Surg. Int. 2013, 29, 937–946. [Google Scholar] [CrossRef]
- Cantrell, V.A.; Owens, S.E.; Chandler, R.L.; Airey, D.C.; Bradley, K.M.; Smith, J.R.; Southard-Smith, E.M. Interactions between Sox10 and EdnrB modulate penetrance and severity of aganglionosis in the Sox10Dom mouse model of Hirschsprung disease. Hum. Mol. Genet. 2004, 13, 2289–2301. [Google Scholar] [CrossRef]
- McCallion, A.S.; Stames, E.; Conlon, R.A.; Chakravarti, A. Phenotype variation in two-locus mouse models of Hirschsprung disease: Tissue-specific interaction between Ret and Ednrb. Proc. Natl. Acad. Sci. USA 2003, 100, 1826–1831. [Google Scholar] [CrossRef] [Green Version]
- Southard-Smith, E.M.; Angrist, M.; Ellison, J.S.; Agarwala, R.; Baxevanis, A.D.; Chakravarti, A.; Pavan, W.J. The Sox10(Dom) mouse: Modeling the genetic variation of Waardenburg-Shah (WS4) syndrome. Genome Res. 1999, 9, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Ohara, Y.; Fujimura, L.; Sakamoto, A.; Teratake, Y.; Hiraoka, S.; Koseki, H.; Saito, T.; Terui, K.; Mitsunaga, T.; Nakata, M.; et al. Genetic background-dependent abnormalities of the enteric nervous system and intestinal function in Kif26a-deficient mice. Sci. Rep. 2021, 11, 3191. [Google Scholar] [CrossRef]
- Parisi, M.A.; Baldessari, A.E.; Iida, M.H.; Clarke, C.M.; Doggett, B.; Shirasawa, S.; Kapur, R.P. Genetic background modifies intestinal pseudo-obstruction and the expression of a reporter gene in Hox11L1-/- mice. Gastroenterology 2003, 125, 1428–1440. [Google Scholar] [CrossRef]
- Dang, R.; Torigoe, D.; Suzuki, S.; Kikkawa, Y.; Moritoh, K.; Sasaki, N.; Agui, T. Genetic background strongly modifies the severity of symptoms of Hirschsprung disease, but not hearing loss in rats carrying Ednrb(sl) mutations. PLoS ONE 2011, 6, e24086. [Google Scholar] [CrossRef]
- Pilon, N. Pigmentation-based insertional mutagenesis is a simple and potent screening approach for identifying neurocristopathy-associated genes in mice. Rare Dis. 2016, 4, e1156287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Methot, D.; Reudelhuber, T.L.; Silversides, D.W. Evaluation of tyrosinase minigene co-injection as a marker for genetic manipulations in transgenic mice. Nucleic Acids Res. 1995, 23, 4551–4556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overbeek, P.A.; Aguilar-Cordova, E.; Hanten, G.; Schaffner, D.L.; Patel, P.; Lebovitz, R.M.; Lieberman, M.W. Coinjection strategy for visual identification of transgenic mice. Transgenic Res. 1991, 1, 31–37. [Google Scholar] [CrossRef]
- Yokoyama, T.; Silversides, D.W.; Waymire, K.G.; Kwon, B.S.; Takeuchi, T.; Overbeek, P.A. Conserved cysteine to serine mutation in tyrosinase is responsible for the classical albino mutation in laboratory mice. Nucleic Acids Res. 1990, 18, 7293–7298. [Google Scholar] [CrossRef] [Green Version]
- Soret, R.; Mennetrey, M.; Bergeron, K.F.; Dariel, A.; Neunlist, M.; Grunder, F.; Faure, C.; Silversides, D.W.; Pilon, N. A collagen VI-dependent pathogenic mechanism for Hirschsprung’s disease. J. Clin. Investig. 2015, 125, 4483–4496. [Google Scholar] [CrossRef] [Green Version]
- Korbel, J.O.; Tirosh-Wagner, T.; Urban, A.E.; Chen, X.N.; Kasowski, M.; Dai, L.; Grubert, F.; Erdman, C.; Gao, M.C.; Lange, K.; et al. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc. Natl. Acad. Sci. USA 2009, 106, 12031–12036. [Google Scholar] [CrossRef] [Green Version]
- Bergeron, K.F.; Cardinal, T.; Toure, A.M.; Beland, M.; Raiwet, D.L.; Silversides, D.W.; Pilon, N. Male-Biased Aganglionic Megacolon in the TashT Mouse Line Due to Perturbation of Silencer Elements in a Large Gene Desert of Chromosome 10. PLoS Genet. 2015, 11, e1005093. [Google Scholar] [CrossRef] [Green Version]
- Cardinal, T.; Bergeron, K.F.; Soret, R.; Souchkova, O.; Faure, C.; Guillon, A.; Pilon, N. Male-biased aganglionic megacolon in the TashT mouse model of Hirschsprung disease involves upregulation of p53 protein activity and Ddx3y gene expression. PLoS Genet. 2020, 16, e1009008. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, K.; Hammer, R.E.; Richardson, J.A.; Baynash, A.G.; Cheung, J.C.; Giaid, A.; Yanagisawa, M. Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell 1994, 79, 1267–1276. [Google Scholar] [CrossRef]
- Soret, R.; Schneider, S.; Bernas, G.; Christophers, B.; Souchkova, O.; Charrier, B.; Righini-Grunder, F.; Aspirot, A.; Landry, M.; Kembel, S.W.; et al. Glial Cell Derived Neurotrophic Factor Induces Enteric Neurogenesis and Improves Colon Structure and Function in Mouse Models of Hirschsprung Disease. Gastroenterology 2020, 159, 1824–1838. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Ito, F.; Ando, H.; Seo, T.; Harada, T.; Kaneko, K.; Ishiguro, Y.; Kobayashi, S. Extrinsic nerve strands in the aganglionic segment of Hirschsprung’s disease. J. Pediatr. Surg. 1998, 33, 1233–1237. [Google Scholar] [CrossRef]
- Pilon, N.; Raiwet, D.; Viger, R.S.; Silversides, D.W. Novel pre- and post-gastrulation expression of Gata4 within cells of the inner cell mass and migratory neural crest cells. Dev. Dyn. 2008, 237, 1133–1143. [Google Scholar] [CrossRef]
- Bergeron, K.F.; Nguyen, C.M.; Cardinal, T.; Charrier, B.; Silversides, D.W.; Pilon, N. Upregulation of the Nr2f1-A830082K12Rik gene pair in murine neural crest cells results in a complex phenotype reminiscent of waardenburg syndrome type 4. Dis. Models Mech. 2016, 9, 1283–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gara, S.K.; Grumati, P.; Urciuolo, A.; Bonaldo, P.; Kobbe, B.; Koch, M.; Paulsson, M.; Wagener, R. Three novel collagen VI chains with high homology to the alpha3 chain. J. Biol. Chem. 2008, 283, 10658–10670. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.A.; Chang, M.M.; Huang, W.; Ooi, W.F.; Xing, M.; Tan, P.; Skanderup, A.J. Mutation hotspots at CTCF binding sites coupled to chromosomal instability in gastrointestinal cancers. Nat. Commun. 2018, 9, 1520. [Google Scholar] [CrossRef] [Green Version]
- Essien, K.; Vigneau, S.; Apreleva, S.; Singh, L.N.; Bartolomei, M.S.; Hannenhalli, S. CTCF binding site classes exhibit distinct evolutionary, genomic, epigenomic and transcriptomic features. Genome Biol. 2009, 10, R131. [Google Scholar] [CrossRef]
- Bespalov, M.M.; Sidorova, Y.A.; Tumova, S.; Ahonen-Bishopp, A.; Magalhaes, A.C.; Kulesskiy, E.; Paveliev, M.; Rivera, C.; Rauvala, H.; Saarma, M. Heparan sulfate proteoglycan syndecan-3 is a novel receptor for GDNF, neurturin, and artemin. J. Cell Biol. 2011, 192, 153–169. [Google Scholar] [CrossRef] [Green Version]
- Bondarenko, O.; Saarma, M. Neurotrophic Factors in Parkinson’s Disease: Clinical Trials, Open Challenges and Nanoparticle-Mediated Delivery to the Brain. Front. Cell. Neurosci. 2021, 15, 682597. [Google Scholar] [CrossRef]
- Ji, Y.; Tam, P.K.; Tang, C.S. Roles of Enteric Neural Stem Cell Niche and Enteric Nervous System Development in Hirschsprung Disease. Int. J. Mol. Sci. 2021, 22, 9659. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, H.; Araki, T.; Jackman, A.; Heuckeroth, R.O.; Snider, W.D.; Johnson, E.M., Jr.; Milbrandt, J. GFR alpha1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron 1998, 21, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Toure, A.M.; Charrier, B.; Pilon, N. Male-specific colon motility dysfunction in the TashT mouse line. Neurogastroenterol. Motil. 2016, 28, 1494–1507. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soret, R.; Lassoued, N.; Bonnamour, G.; Bernas, G.; Barbe, A.; Pelletier, M.; Aichi, M.; Pilon, N. Genetic Background Influences Severity of Colonic Aganglionosis and Response to GDNF Enemas in the Holstein Mouse Model of Hirschsprung Disease. Int. J. Mol. Sci. 2021, 22, 13140. https://doi.org/10.3390/ijms222313140
Soret R, Lassoued N, Bonnamour G, Bernas G, Barbe A, Pelletier M, Aichi M, Pilon N. Genetic Background Influences Severity of Colonic Aganglionosis and Response to GDNF Enemas in the Holstein Mouse Model of Hirschsprung Disease. International Journal of Molecular Sciences. 2021; 22(23):13140. https://doi.org/10.3390/ijms222313140
Chicago/Turabian StyleSoret, Rodolphe, Nejia Lassoued, Grégoire Bonnamour, Guillaume Bernas, Aurélie Barbe, Mélanie Pelletier, Manon Aichi, and Nicolas Pilon. 2021. "Genetic Background Influences Severity of Colonic Aganglionosis and Response to GDNF Enemas in the Holstein Mouse Model of Hirschsprung Disease" International Journal of Molecular Sciences 22, no. 23: 13140. https://doi.org/10.3390/ijms222313140
APA StyleSoret, R., Lassoued, N., Bonnamour, G., Bernas, G., Barbe, A., Pelletier, M., Aichi, M., & Pilon, N. (2021). Genetic Background Influences Severity of Colonic Aganglionosis and Response to GDNF Enemas in the Holstein Mouse Model of Hirschsprung Disease. International Journal of Molecular Sciences, 22(23), 13140. https://doi.org/10.3390/ijms222313140