
Mitochondrial Biogenesis in Neurons: How and Where


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mechanisms for Mitochondrial Biogenesis
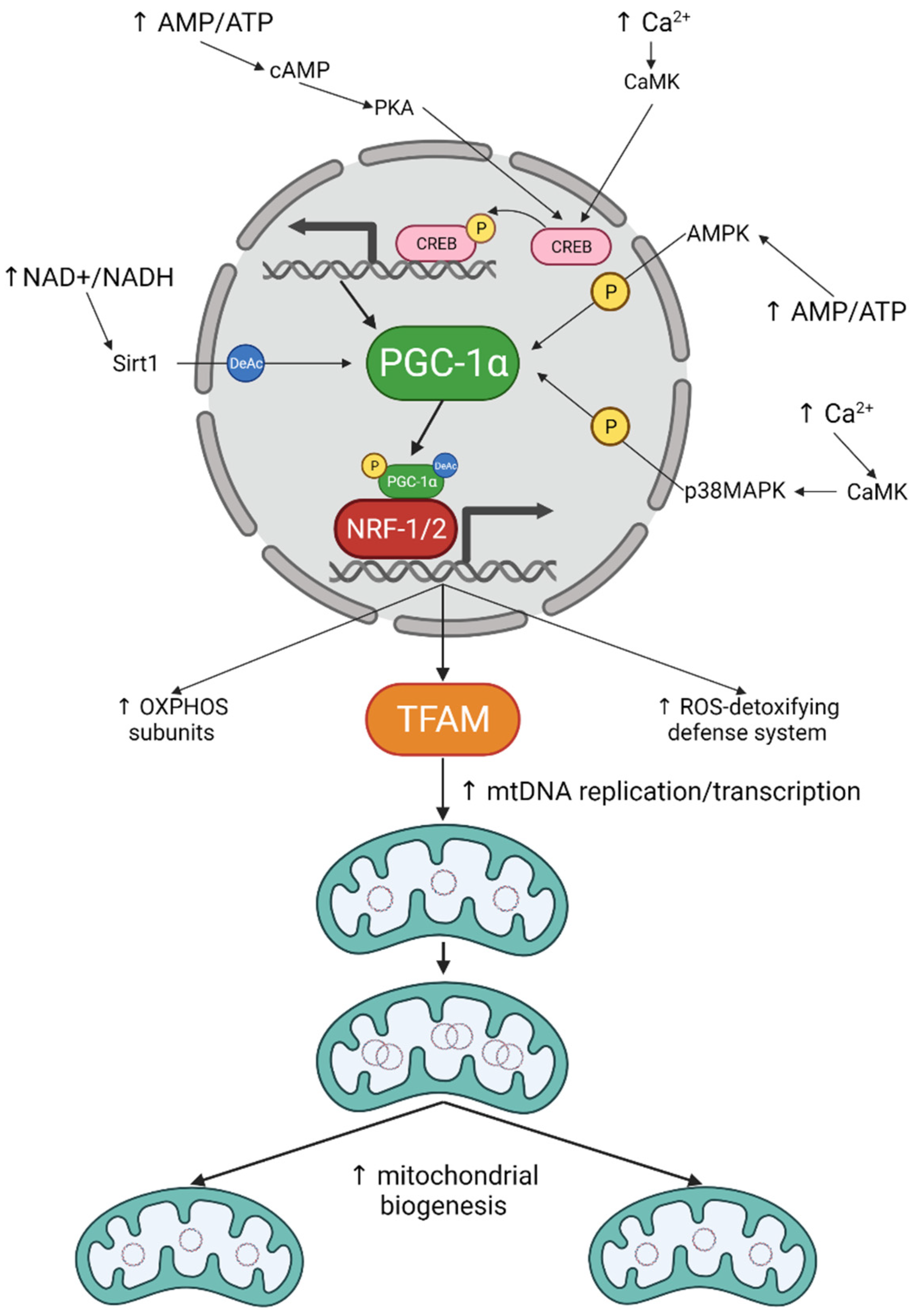
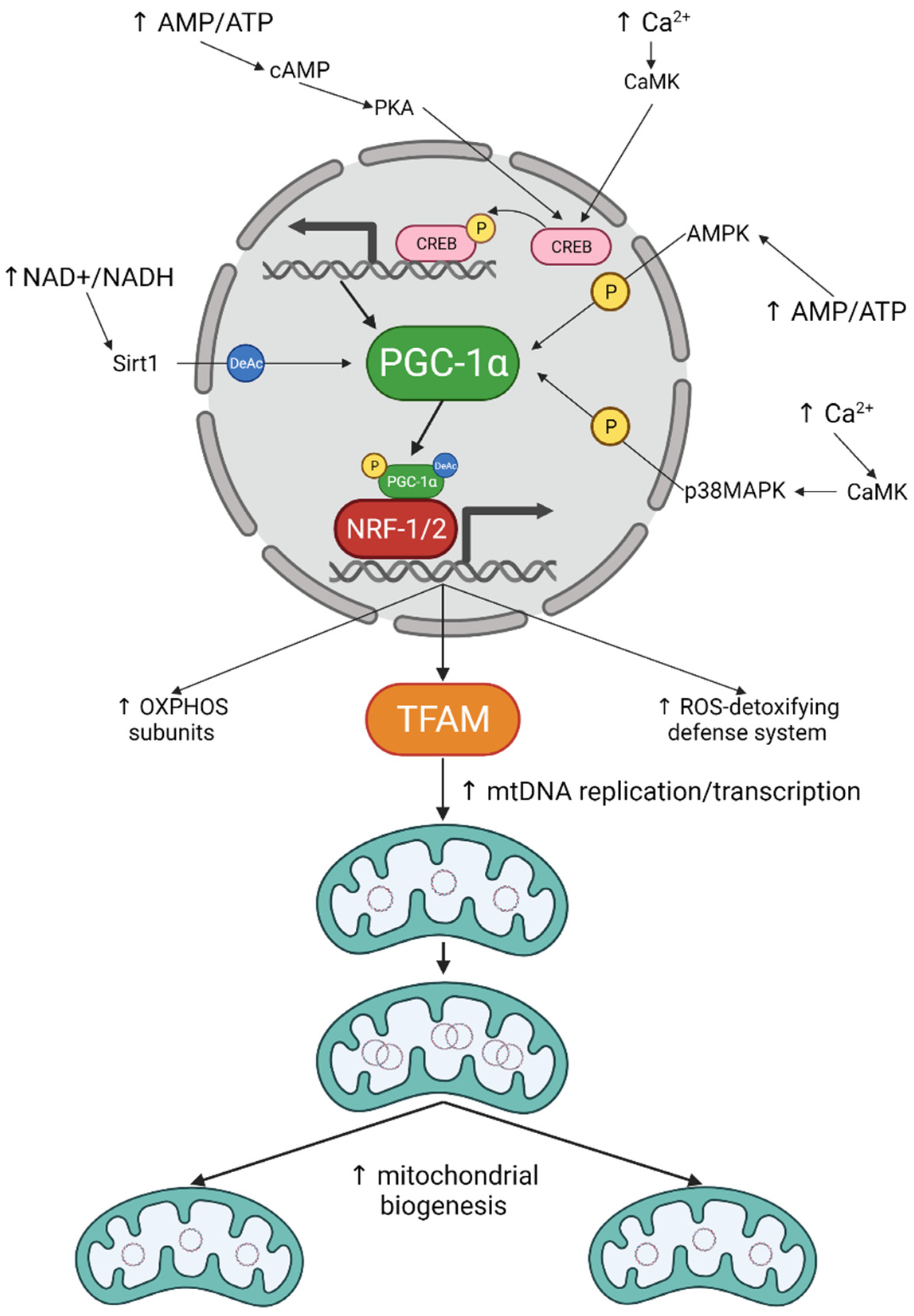
3. Mitochondrial Biogenesis in Neurons: The How
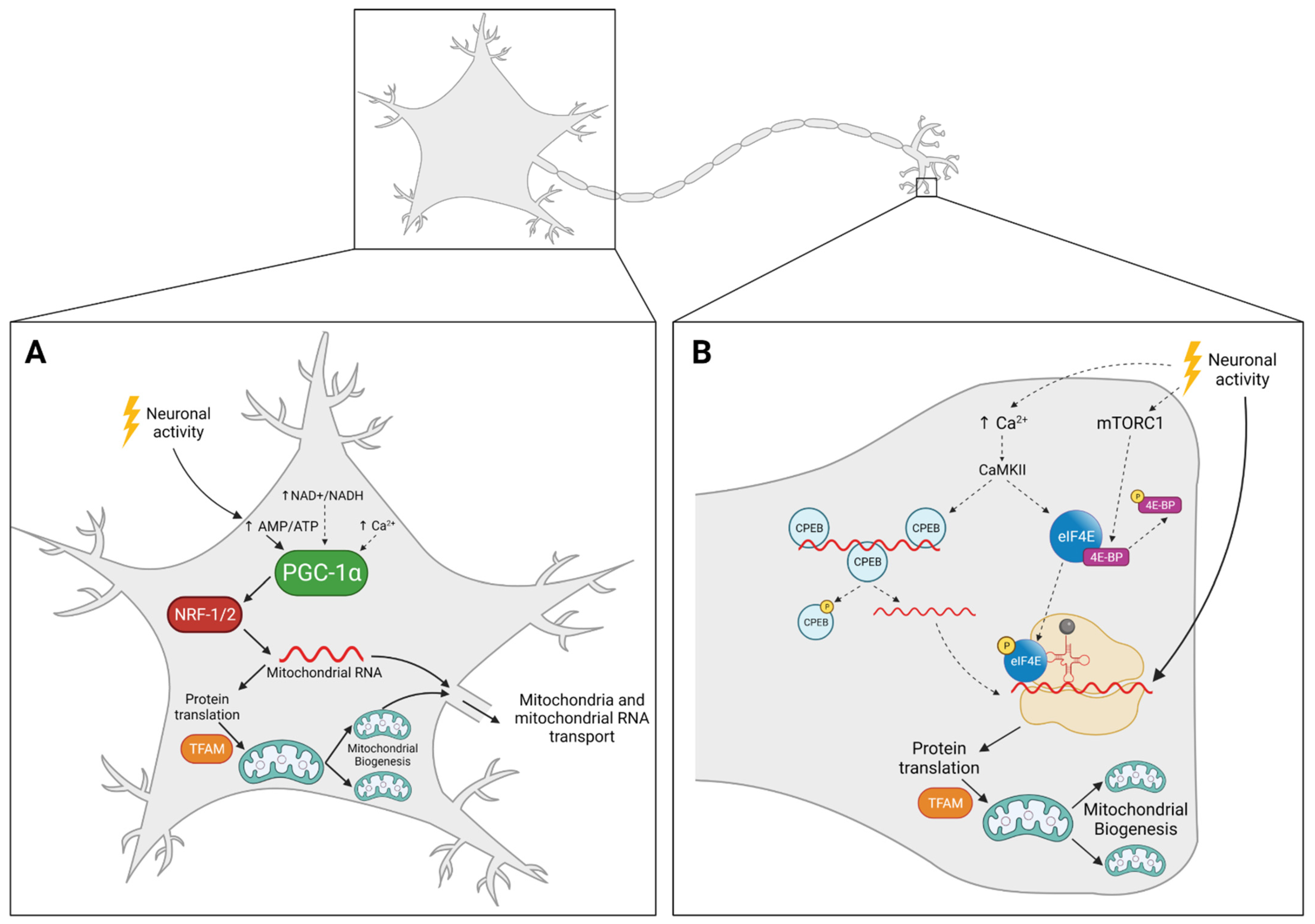
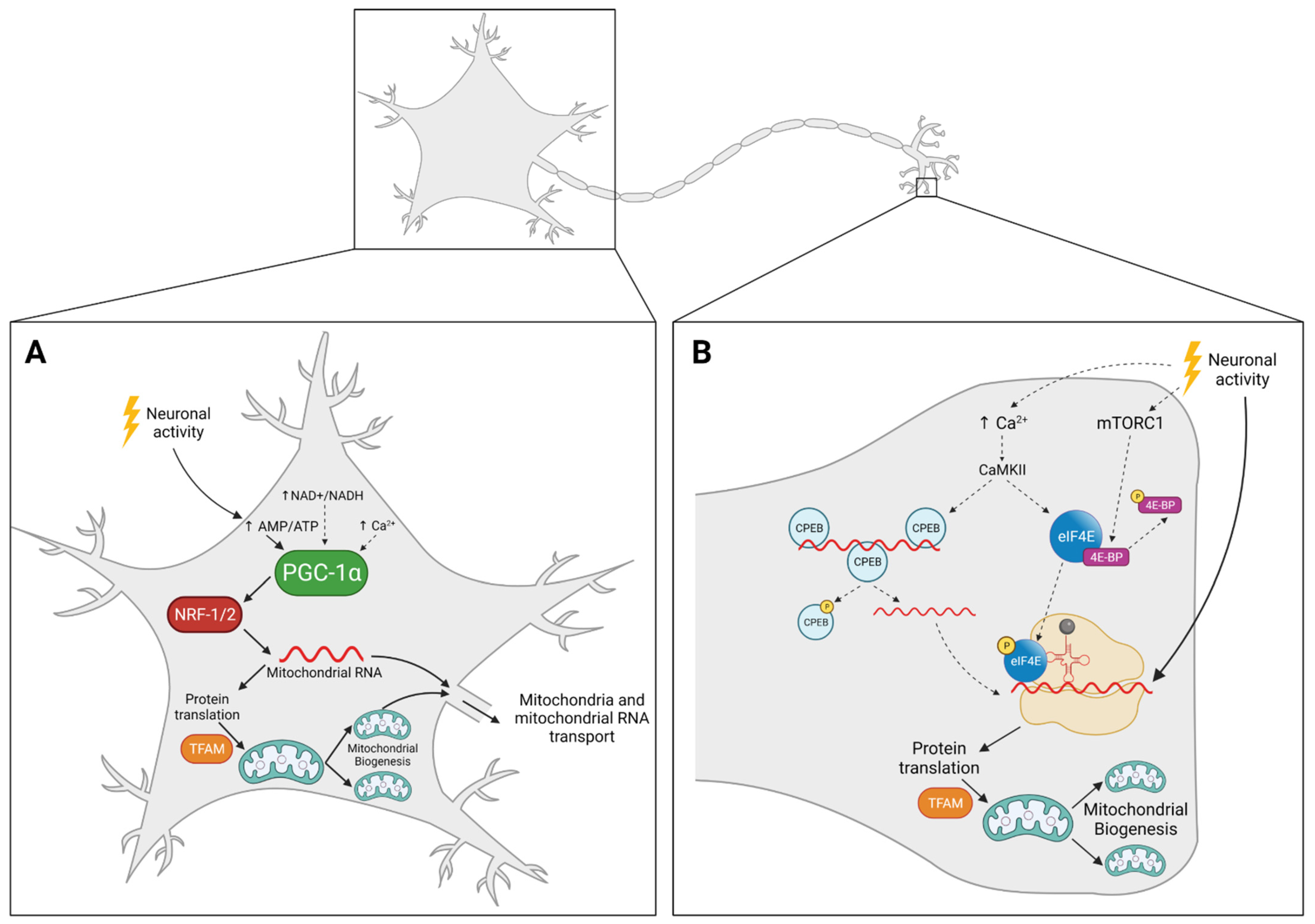
4. Mitochondrial Biogenesis in Neurons: The Where
4.1. Mitochondria Replicate in the Cell Body and Travel Back and Forth to Supply Distal Regions
4.2. Mitochondria Replicate Locally, Away from the Cell Body
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schon, E.A.; Przedborski, S. Mitochondria: The Next (Neurode) Generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauch, K.L.; Purnell, P.R.; Fox, H.S. Quantitative proteomics of synaptic and nonsynaptic mitochondria: Insights for synaptic mitochondrial vulnerability. J. Proteom. Res. 2014, 13, 2620–2636. [Google Scholar] [CrossRef]
- Völgyi, K.; Gulyássy, P.; Háden, K.; Kis, V.; Badics, K.; Kékesi, K.A.; Simor, A.; Györffy, B.; Tóth, E.A.; Lubec, G.; et al. Synaptic mitochondria: A brain mitochondria cluster with a specific proteome. J. Proteom. 2015, 120, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Kiebish, M.A.; Han, X.; Cheng, H.; Luncedord, A.; Clarke, C.F.; Moon, H.; Chuang, J.H.; Seyfried, T.N. Lipidomic analysis and electron transport chain activities in C57BL/6J mouse brain mitchondria. J. Neurochem. 2008, 106, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, 169–176. [Google Scholar] [CrossRef]
- Pilling, A.D.; Horiuchi, D.; Lively, C.M.; Saxton, W.M. Kinesin-1 and Dynein Are the Primary Motors for Fast Transport of Mitochondria in Drosophila Motor Axons. Mol. Biol. Cell 2006, 17, 2057–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.; Gerwin, C.; Sheng, Z.H. Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J. Cell Biol. 2005, 170, 959–969. [Google Scholar] [CrossRef] [Green Version]
- Brickley, K.; Stephenson, F.A. Trafficking Kinesin Protein (TRAK)-mediated Transport of Mitochondria in Axons of Hippocampal Neurons. J. Biol. Chem. 2011, 286, 18079–18092. [Google Scholar] [CrossRef] [Green Version]
- Fransson, Å.; Ruusala, A.; Aspenström, P. Atypical Rho GTPases Have Roles in Mitochondrial Homeostasis and Apoptosis. J. Biol. Chem. 2003, 278, 6495–6502. [Google Scholar] [CrossRef] [Green Version]
- Schroer, T.A. DYNACTIN. Annu. Rev. Cell Dev. Biol. 2004, 20, 759–779. [Google Scholar] [CrossRef]
- Schwarzer, C.; Barnikol-Watanabe, S.; Thinnes, F.P.; Hilschmann, N. Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int. J. Biochem. Cell Biol. 2002, 34, 1059–1070. [Google Scholar] [CrossRef]
- Russo, G.J.; Louie, K.; Wellington, A.; Macleod, G.T.; Hu, F.; Panchumarthi, S.; Zinsmaier, K.E. Drosophila Miro Is Required for Both Anterograde and Retrograde Axonal Mitochondrial Transport. J. Neurosci. 2009, 29, 5443–5455. [Google Scholar] [CrossRef]
- Li, S.; Xiong, G.J.; Huang, N.; Sheng, Z.H. The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat. Metab. 2020, 2, 1077–1095. [Google Scholar] [CrossRef]
- Mironov, S.L. ADP Regulates Movements of Mitochondria in Neurons. Biophys. J. 2007, 92, 2944–2952. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Schwarz, T.L. The Mechanism of Ca2+-Dependent Regulation of Kinesin-Mediated Mitochondrial Motility. Cell 2009, 136, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 Mediate Sequential Steps in Mitochondrial Membrane Fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafi, G.; Schlehe, J.S.; LaVoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef]
- Cai, Q.; Zakaria, H.M.; Simone, A.; Sheng, Z.H. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr. Biol. 2012, 22, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.Y.; Cheng, X.T.; Tammineni, P.; Xie, Y.; Zhou, B.; Cai, Q.; Sheng, Z.H. Releasing Syntaphilin Removes Stressed Mitochondria from Axons Independent of Mitophagy under Pathophysiological Conditions. Neuron 2017, 94, 595–610.e6. [Google Scholar] [CrossRef] [Green Version]
- Seibler, P.; Graziotto, J.; Jeong, H.; Simunovic, F.; Klein, C.; Krainc, D. Mitochondrial Parkin Recruitment Is Impaired in Neurons Derived from Mutant PINK1 Induced Pluripotent Stem Cells. J. Neurosci. 2011, 31, 5970–5976. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Diaz, F.; Moraes, C.T. Mitochondrial biogenesis and turnover. Cell Calcium 2008, 44, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Wu, P.H.; Tarr, P.T.; Lindenberg, K.S.; St-Pierre, J.; Zhang, C.Y.; Mootha, V.K.; Jäger, S.; Vianna, C.R.; Reznick, R.M.; et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell 2004, 119, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Amiri, M.; Hollenbeck, P.J. Mitochondrial biogenesis in the axons of vertebrate peripheral neurons. Dev. Neurobiol. 2008, 68, 1348–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Laar, V.S.; Arnold, B.; Howlett, E.H.; Calderon, M.J.; Croix, C.M.S.; Greenamyre, J.T.; Sanders, L.H.; Berman, S.B. Evidence for Compartmentalized Axonal Mitochondrial Biogenesis: Mitochondrial DNA Replication Increases in Distal Axons As an Early Response to Parkinson’s Disease-Relevant Stress. J. Neurosci. 2018, 38, 7505–7515. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [Green Version]
- Wright, D.C.; Geiger, P.C.; Han, D.H.; Jones, T.E.; Holloszy, J.O. Calcium induces increases in peroxisome proliferator-activated receptor γ coactivator-1α and mitochondrial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation. J. Biol. Chem. 2007, 282, 18793–18799. [Google Scholar] [CrossRef] [Green Version]
- Falkenberg, M. Mitochondrial DNA replication in mammalian cells: Overview of the pathway. Essays Biochem. 2018, 62, 287–296. [Google Scholar] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator-activated receptor γ coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Leone, T.C.; Lehman, J.J.; Finck, B.N.; Schaeffer, P.J.; Wende, A.R.; Boudina, S.; Courtois, M.; Wozniak, D.F.; Sambandam, N.; Bernal-Mizrachi, C.; et al. PGC-1α deficiency causes multi-system energy metabolic derangements: Muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005, 3, 672–687. [Google Scholar] [CrossRef] [Green Version]
- Ongwijitwat, S.; Wong-Riley, M.T.T. Is nuclear respiratory factor 2 a master transcriptional coordinator for all ten nuclear-encoded cytochrome c oxidase subunits in neurons? Gene 2005, 360, 65–77. [Google Scholar] [CrossRef]
- Bergeron, R.; Ren, J.M.; Cadman, K.S.; Moore, I.K.; Perret, P.; Pypaert, M.; Young, L.H.; Semenkovich, C.F.; Shulman, G.I. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol.-Endocrinol. Metab. 2001, 281, E1340–E1346. [Google Scholar] [CrossRef] [PubMed]
- Marin, T.L.; Gongol, B.; Zhang, F.; Martin, M.; Johnson, D.A.; Xiao, H.; Wang, Y.; Subramaniam, S.; Chien, S.; Shyy, J.Y.J. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci. Signal. 2017, 10, eaaf7478. [Google Scholar] [CrossRef] [Green Version]
- Delghandi, M.P.; Johannessen, M.; Moens, U. The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell. Signal. 2005, 17, 1343–1351. [Google Scholar] [CrossRef]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001, 413, 179–183. [Google Scholar] [CrossRef]
- Handschin, C.; Rhee, J.; Lin, J.; Tarr, P.T.; Spiegelman, B.M. An autoregulatory loop controls peroxisome proliferator-activated receptor γ coactivator 1α expression in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 7111–7116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP Produced inside Mitochondria Regulates Oxidative Phosphorylation. Cell Metab. 2009, 9, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kim, C.H.; Simon, D.K.; Aminova, L.R.; Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Lonze, B.E.; Kim, K.S.; Ginty, D.D.; et al. Mitochondrial cyclic AMP response element-binding protein (CREB) mediates mitochondrial gene expression and neuronal survival. J. Biol. Chem. 2005, 280, 40398–40401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojuka, E.O.; Jones, T.E.; Han, D.-H.; Chen, M.; Holloszy, J.O. Raising Ca2+ in L6 myotubes mimics effects of exercise on mitochondrial biogenesis in muscle. FASEB J. 2003, 17, 675–681. [Google Scholar] [CrossRef]
- Akimoto, T.; Pohnert, S.C.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.B.; Williams, R.S.; Yan, Z. Exercise stimulates Pgc-1α transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef] [Green Version]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M.; et al. Adiponectin and AdipoR1 regulate PGC-1α and mitochondria by Ca2+ and AMPK/SIRT1. Nature 2010, 464, 1313–1319. [Google Scholar] [CrossRef]
- Wareski, P.; Vaarmann, A.; Choubey, V.; Safiulina, D.; Liiv, J.; Kuum, M.; Kaasik, A. PGC-1α and PGC-1β regulate mitochondrial density in neurons. J. Biol. Chem. 2009, 284, 21379–21385. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Yang, S.J. AMP-activated protein kinase mediates activity-dependent regulation of peroxisome proliferator-activated receptor γ coactivator-1α and nuclear respiratory factor 1 expression in rat visual cortical neurons. Neuroscience 2010, 169, 23–38. [Google Scholar] [CrossRef]
- Cheng, A.; Wan, R.; Yang, J.L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3, 1250. [Google Scholar] [CrossRef] [Green Version]
- Spillane, M.; Ketschek, A.; Merianda, T.T.; Twiss, J.L.; Gallo, G. Mitochondria Coordinate Sites of Axon Branching through Localized Intra-axonal Protein Synthesis. Cell Rep. 2013, 5, 1564–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial Biogenesis: A Therapeutic Target for Neurodevelopmental Disorders and Neurodegenerative Diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional Repression of PGC-1α by Mutant Huntingtin Leads to Mitochondrial Dysfunction and Neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Li, S.; Lucas, E.K.; Cowell, R.M.; Lin, J.D. Neuronal inactivation of peroxisome proliferator-activated receptor γ coactivator 1α(PGC-1α) protects mice from diet-induced obesity and leads to degenerative lesions. J. Biol. Chem. 2010, 285, 39087–39095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [Green Version]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.-I.; Bower, A.; Jiang, H.; Kang, S.U.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin loss leads to Paris-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.-I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Stevens, D.A.; Kang, S.U.; Jiang, H.; Lee, Y.-I.; Ko, H.S.; Scarffe, L.A.; Umanah, G.E.; Kang, H.; Ham, S.; et al. PINK1 Primes Parkin-Mediated Ubiquitination of PARIS in Dopaminergic Neuronal Survival. Cell Rep. 2017, 18, 918–932. [Google Scholar] [CrossRef] [Green Version]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Perry, G.; Moreira, P.I.; Aliev, G.; Cash, A.D.; Hirai, K.; Smith, M.A. Mitochondrial abnormalities and oxidative imbalance in Alzheimer disease. J. Alzheimer’s Dis. 2006, 9, 147–153. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [Green Version]
- Fedorovich, S.V.; Waseem, T.V.; Puchkova, L.V. Biogenetic and morphofunctional heterogeneity of mitochondria: The case of synaptic mitochondria. Rev. Neurosci. 2017, 28, 363–373. [Google Scholar] [CrossRef]
- Bros, H.; Hauser, A.; Paul, F.; Niesner, R.; Infante-Duarte, C. Assessing Mitochondrial Movement within Neurons: Manual versus Automated Tracking Methods. Traffic 2015, 16, 906–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligon, L.A.; Steward, O. Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J. Comp. Neurol. 2000, 427, 351–361. [Google Scholar] [CrossRef]
- Miller, N.; Shi, H.; Zelikovich, A.S.; Ma, Y.C. Motor neuron mitochondrial dysfunction in spinal muscular atrophy. Hum. Mol. Genet. 2016, 25, 3395–3406. [Google Scholar] [CrossRef] [Green Version]
- Lewis, T.L.; Turi, G.F.; Kwon, S.K.; Losonczy, A.; Polleux, F. Progressive Decrease of Mitochondrial Motility during Maturation of Cortical Axons In Vitro and In Vivo. Curr. Biol. 2016, 26, 2602–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit-Rigter, L.; Rajendran, R.; Silva, C.A.P.; Spierenburg, L.; Groeneweg, F.; Ruimschotel, E.M.; van Versendaal, D.; van der Togt, C.; Eysel, U.T.; Heimel, J.A.; et al. Mitochondrial Dynamics in Visual Cortex Are Limited In Vivo and Not Affected by Axonal Structural Plasticity. Curr. Biol. 2016, 26, 2609–2616. [Google Scholar] [CrossRef] [Green Version]
- Misgeld, T.; Kerschensteiner, M.; Bareyre, F.M.; Burgess, R.W.; Lichtman, J.W. Imaging axonal transport of mitochondria in vivo. Nat. Methods 2007, 4, 559–561. [Google Scholar] [CrossRef]
- Shigeoka, T.; Jung, H.; Jung, J.; Turner-Bridger, B.; Ohk, J.; Lin, J.Q.; Amieux, P.S.; Holt, C.E. Dynamic Axonal Translation in Developing and Mature Visual Circuits. Cell 2016, 166, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Aschrafi, A.; Kar, A.N.; Gale, J.R.; Elkahloun, A.G.; Vargas, J.N.S.; Sales, N.; Wilson, G.; Tompkins, M.; Gioio, A.E.; Kaplan, B.B. A heterogeneous population of nuclear-encoded mitochondrial mRNAs is present in the axons of primary sympathetic neurons. Mitochondrion 2016, 30, 18–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzniewska, B.; Cysewski, D.; Wasilewski, M.; Sakowska, P.; Milek, J.; Kulinski, T.M.; Winiarski, M.; Kozielewicz, P.; Knapska, E.; Dadlez, M.; et al. Mitochondrial protein biogenesis in the synapse is supported by local translation. EMBO Rep. 2020, 21, e48882. [Google Scholar] [CrossRef] [PubMed]
- Cioni, J.M.; Lin, J.Q.; Holtermann, A.V.; Koppers, M.; Jakobs, M.A.H.; Azizi, A.; Turner-Bridger, B.; Shigeoka, T.; Franze, K.; Harris, W.A.; et al. Late Endosomes Act as mRNA Translation Platforms and Sustain Mitochondria in Axons. Cell 2019, 176, 56–72.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, B.C.; Jung, H.; Dwivedy, A.; O’Hare, C.M.; Zivraj, K.H.; Holt, C.E. Local translation of extranuclear lamin B promotes axon maintenance. Cell 2012, 148, 752–764. [Google Scholar] [CrossRef] [Green Version]
- Cosker, K.E.; Fenstermacher, S.J.; Pazyra-Murphy, M.F.; Elliott, H.L.; Segal, R.A. The RNA-binding protein SFPQ orchestrates an RNA regulon to promote axon viability. Nat. Neurosci. 2016, 19, 690–696. [Google Scholar] [CrossRef]
- Hillefors, M.; Gioio, A.E.; Mameza, M.G.; Kaplan, B.B. Axon viability and mitochondrial function are dependent on local protein synthesis in sympathetic neurons. Cell. Mol. Neurobiol. 2007, 27, 701–716. [Google Scholar] [CrossRef]
- Cajigas, I.J.; Will, T.; Schuman, E.M. Protein homeostasis and synaptic plasticity. EMBO J. 2010, 29, 2746–2752. [Google Scholar] [CrossRef]
- Sutton, M.A.; Schuman, E.M. Dendritic Protein Synthesis, Synaptic Plasticity, and Memory. Cell 2006, 127, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangaraju, V.; Lauterbach, M.; Schuman, E.M. Spatially Stable Mitochondrial Compartments Fuel Local Translation during Plasticity. Cell 2019, 176, 73–84.e15. [Google Scholar] [CrossRef] [Green Version]
- Atkins, C.M.; Nozaki, N.; Shigeri, Y.; Soderling, T.R. Cytoplasmic polyadenylation element binding protein-dependent protein synthesis is regulated by calcium/calmodulin-dependent protein kinase II. J. Neurosci. 2004, 24, 5193–5201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mockett, B.G.; Guévremont, D.; Wutte, M.; Hulme, S.R.; Williams, J.M.; Abraham, W.C. Calcium/Calmodulin-Dependent Protein Kinase II Mediates Group I Metabotropic Glutamate Receptor-Dependent Protein Synthesis and Long-Term Depression in Rat Hippocampus. J. Neurosci. 2011, 31, 7380–7391. [Google Scholar] [CrossRef] [PubMed]
- Takei, N.; Inamura, N.; Kawamura, M.; Namba, H.; Hara, K.; Yonezawa, K.; Nawa, H. Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J. Neurosci. 2004, 24, 9760–9769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizuka, Y.; Kakiya, N.; Witters, L.A.; Oshiro, N.; Shirao, T.; Nawa, H.; Takei, N. AMP-activated protein kinase counteracts brain-derived neurotrophic factor-induced mammalian target of rapamycin complex 1 signaling in neurons. J. Neurochem. 2013, 127, 66–77. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardanho-Ramos, C.; Morais, V.A. Mitochondrial Biogenesis in Neurons: How and Where. Int. J. Mol. Sci. 2021, 22, 13059. https://doi.org/10.3390/ijms222313059
Cardanho-Ramos C, Morais VA. Mitochondrial Biogenesis in Neurons: How and Where. International Journal of Molecular Sciences. 2021; 22(23):13059. https://doi.org/10.3390/ijms222313059
Chicago/Turabian StyleCardanho-Ramos, Carlos, and Vanessa Alexandra Morais. 2021. "Mitochondrial Biogenesis in Neurons: How and Where" International Journal of Molecular Sciences 22, no. 23: 13059. https://doi.org/10.3390/ijms222313059
APA StyleCardanho-Ramos, C., & Morais, V. A. (2021). Mitochondrial Biogenesis in Neurons: How and Where. International Journal of Molecular Sciences, 22(23), 13059. https://doi.org/10.3390/ijms222313059