Mfn2 Overexpression Attenuates MPTP Neurotoxicity In Vivo


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
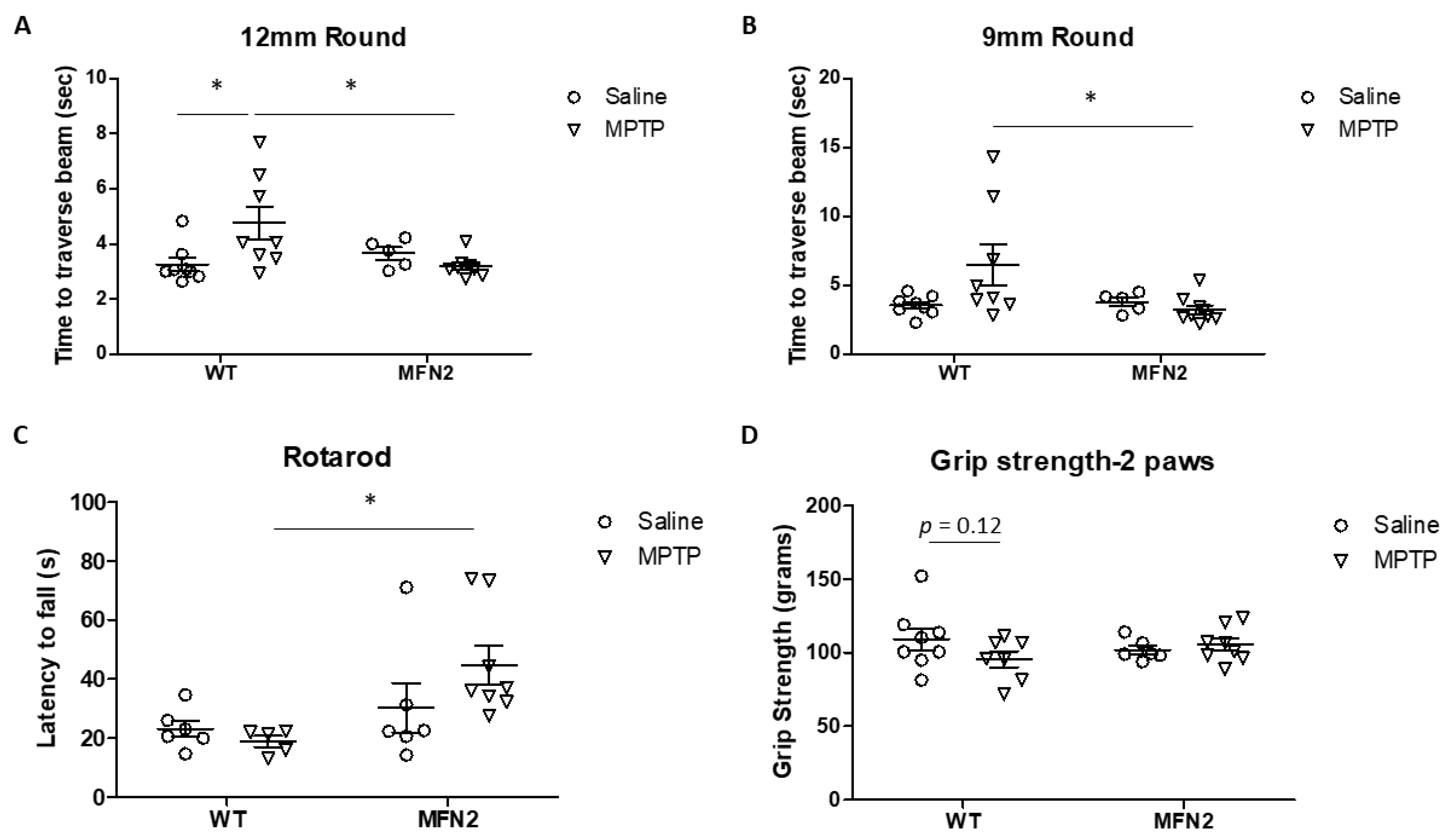
2.1. Mfn2 Overexpression (OE) Reduces MPTP-Induced Motor Deficits
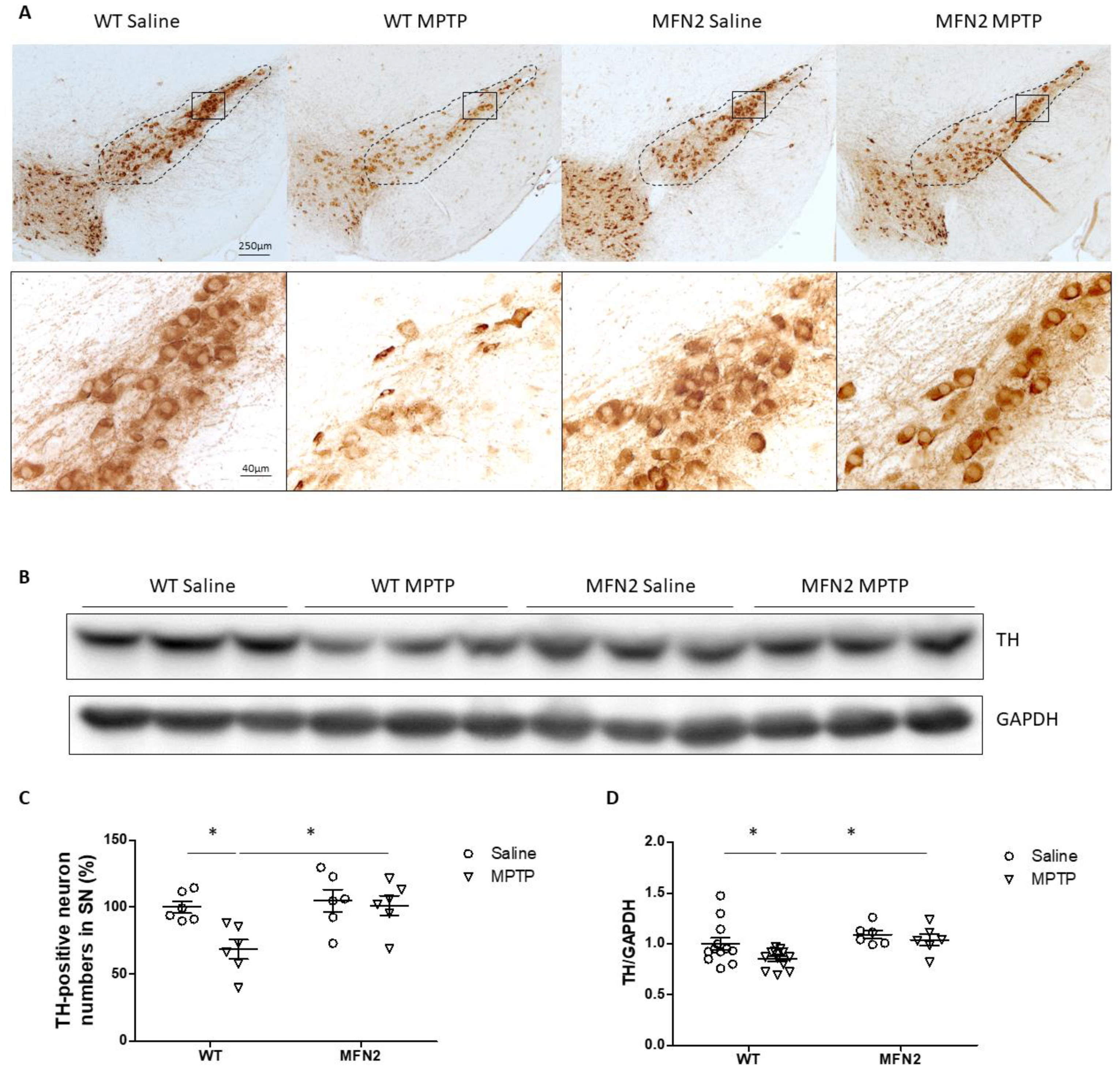
2.2. Mfn2 OE Protects DA Neurons from MPTP-Induced Degeneration in the SN
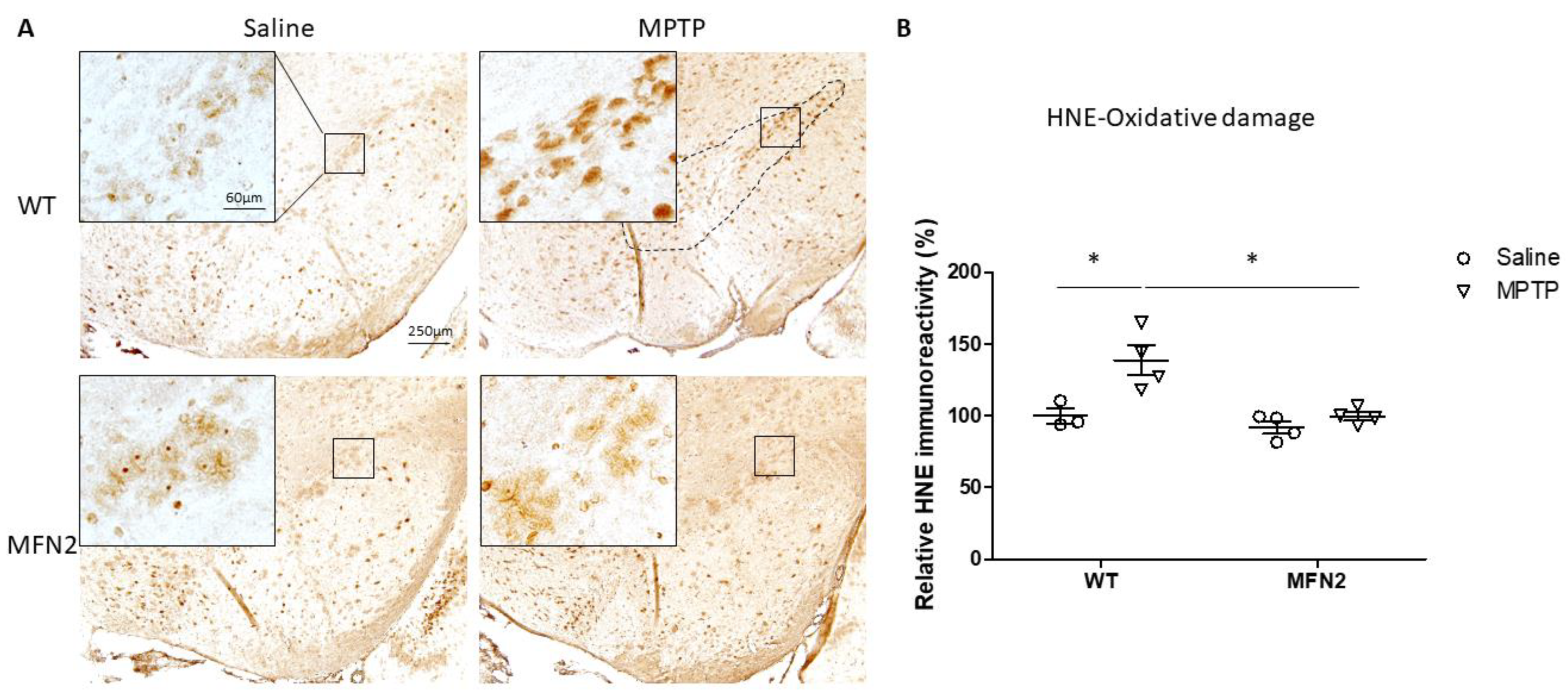
2.3. MFN2 OE Rescues MPTP-Induced Oxidative Stress in the SN
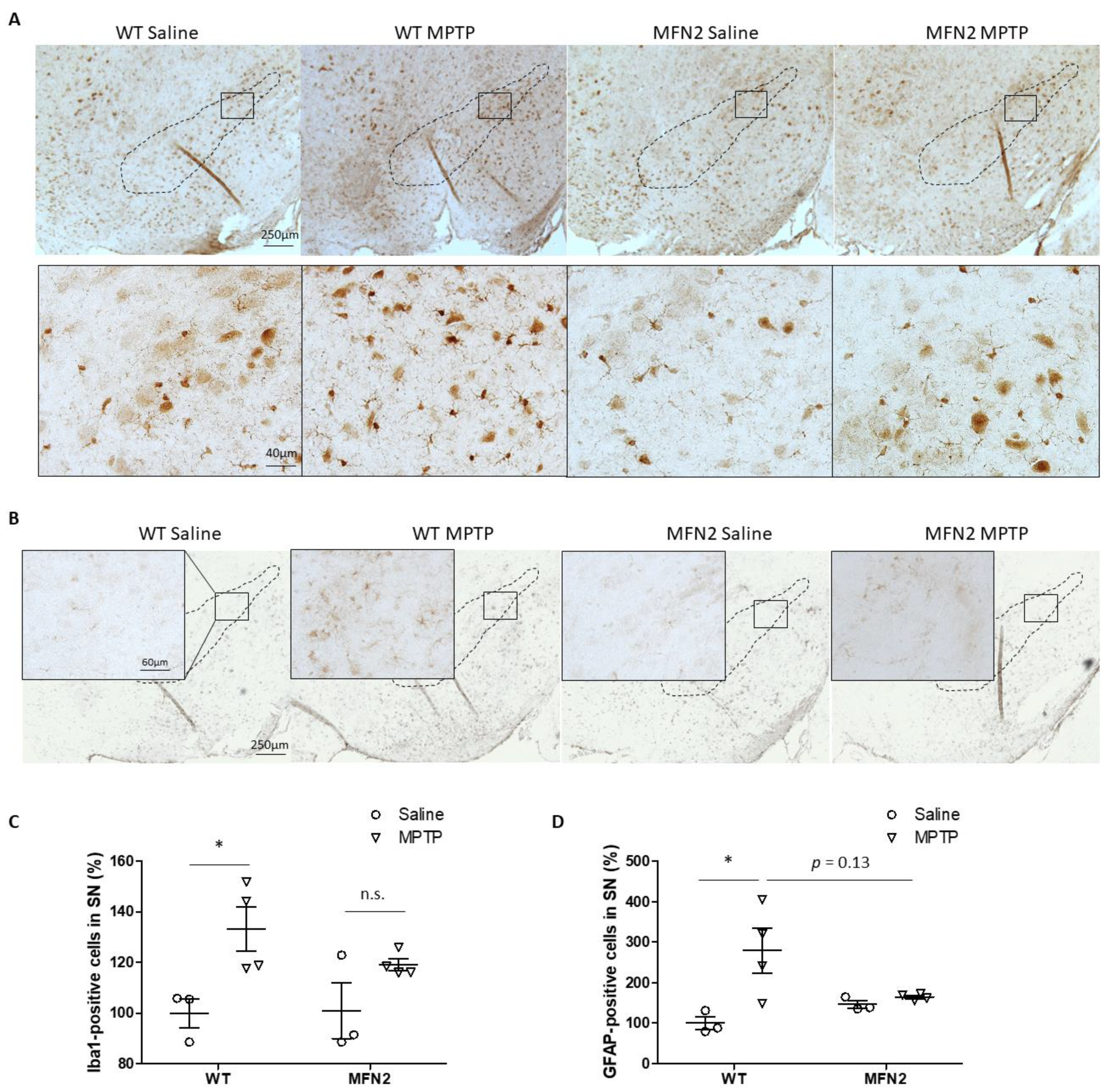
2.4. Mfn2 OE Inhibits MPTP-Induced Activation of Microglia and Astrocytes in SN
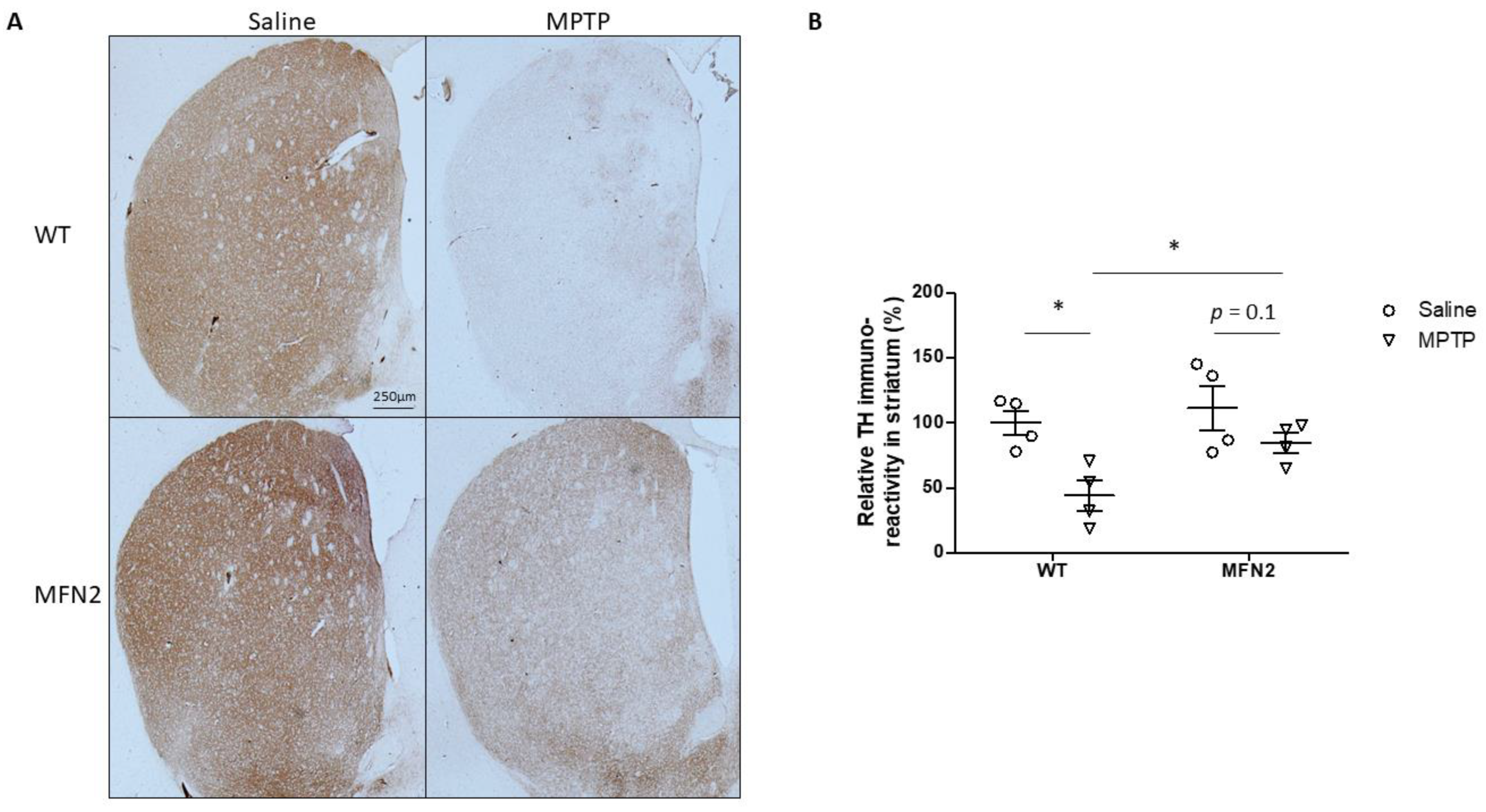
2.5. Mfn2 OE Alleviates MPTP-Caused Damage in the Striatum
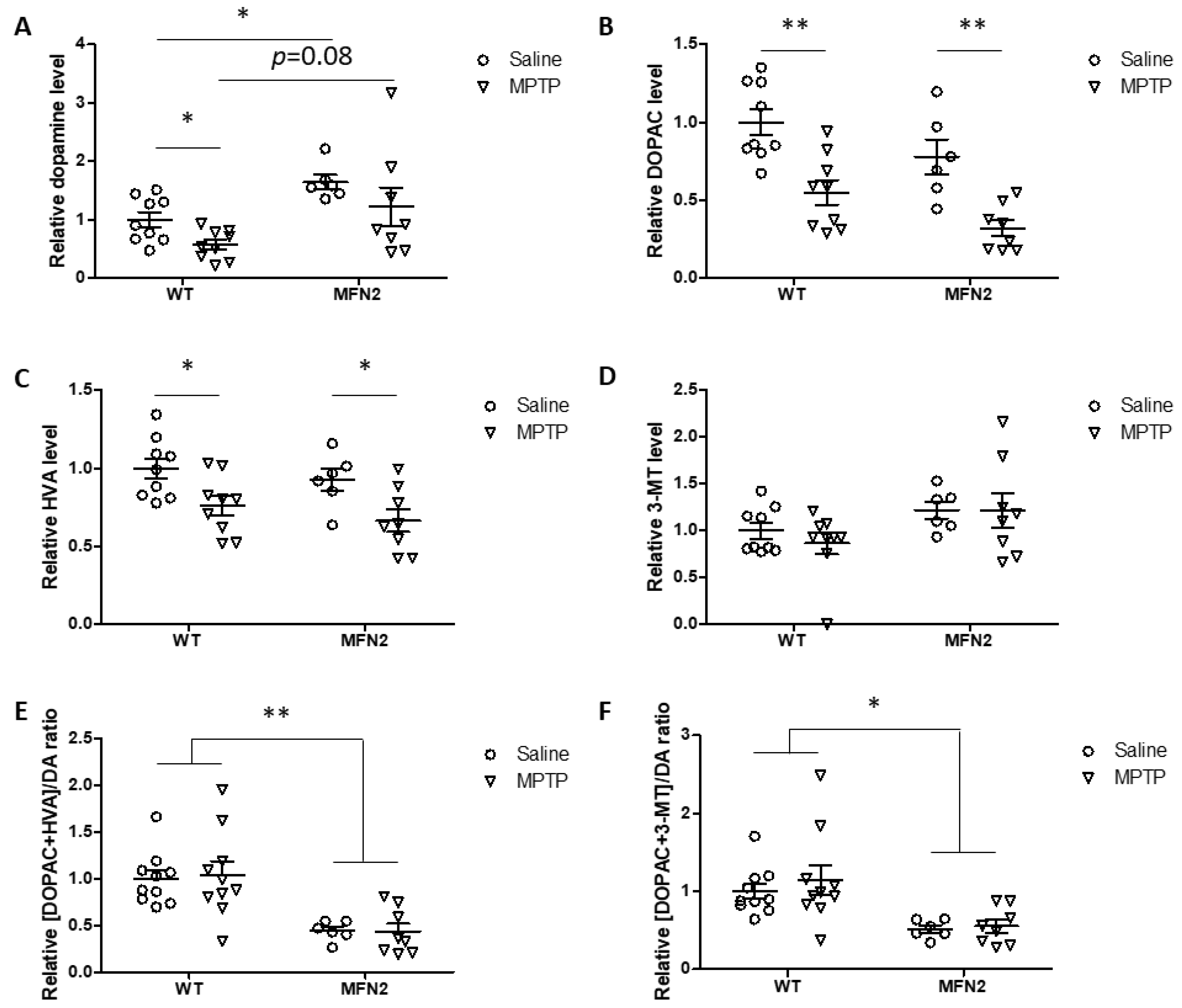
2.6. Mfn2 OE Rescues MPTP-Induced Striatal Dopamine Loss
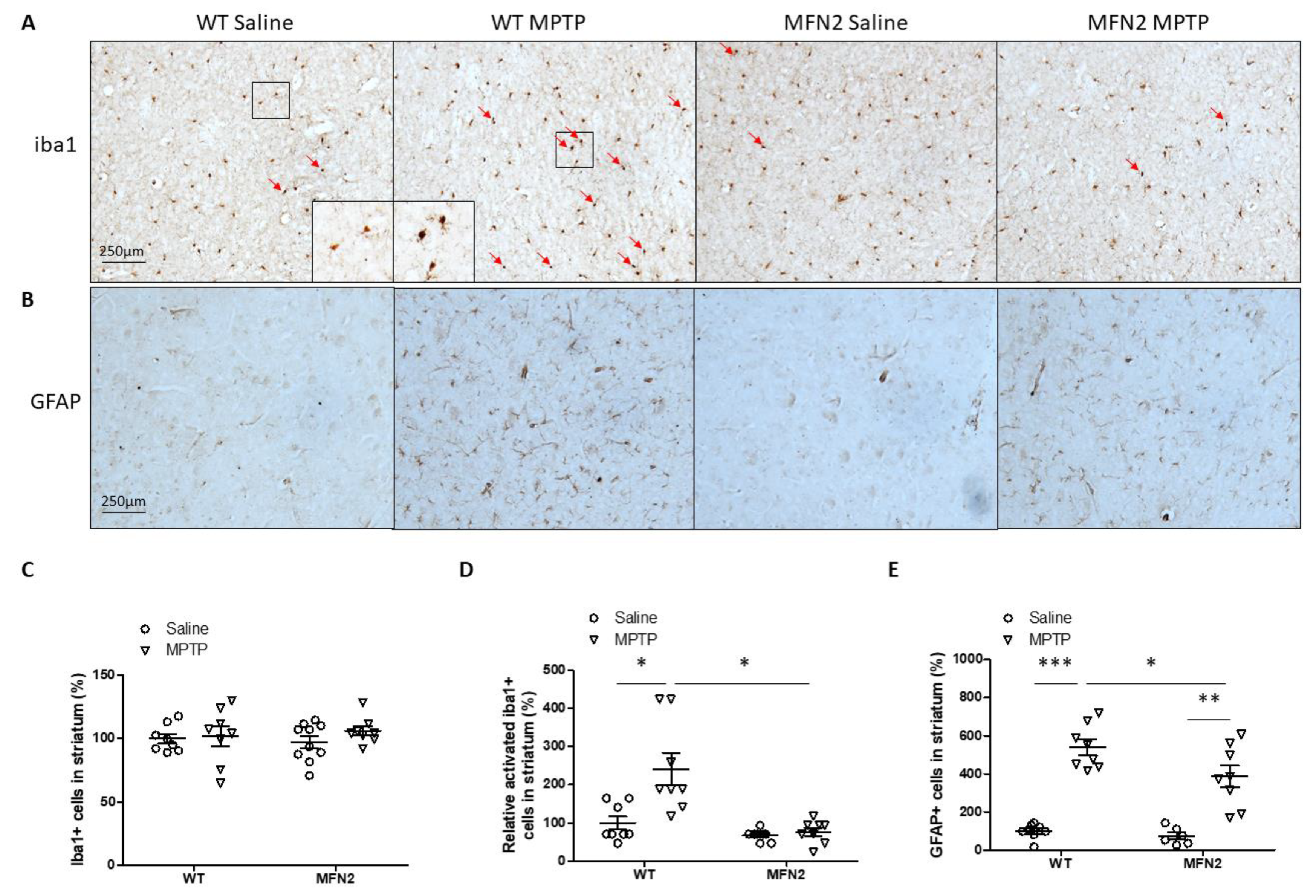
2.7. Mfn2 OE Inhibits MPTP-Induced Inflammation in the Striatum
3. Discussion
4. Materials and Methods
4.1. Animals and Treatment
4.2. Measurement of Motor Function
4.3. Antibodies and Chemicals
4.4. Immunohistochemistry
4.5. Western Blot Analysis
4.6. HPLC Measurements of Striatal Dopamine and Metabolites
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alexander, G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004, 6, 259–280. [Google Scholar] [PubMed]
- DeMaagd, G.; Philip, A. Parkinson’s disease and its management: Part 1: Disease entity, risk factors, pathophysiology, clinical presentation, and diagnosis. P T 2015, 40, 504–532. [Google Scholar] [PubMed]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed]
- Lill, C.M. Genetics of Parkinson’s disease. Mol. Cell Probes. 2016, 30, 386–396. [Google Scholar] [CrossRef]
- Pan-Montojo, F.; Reichmann, H. Considerations on the role of environmental toxins in idiopathic Parkinson’s disease pathophysiology. Transl. Neurodegener. 2014, 3, 10. [Google Scholar] [CrossRef]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Parkinsons Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316. [Google Scholar] [CrossRef]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef]
- Helley, M.P.; Pinnell, J.; Sportelli, C.; Tieu, K. Mitochondria: A common target for genetic mutations and environmental toxicants in Parkinson’s disease. Front. Genet. 2017, 8, 177. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics--fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta 2010, 1802, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, X.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochim. Biophys. Acta 2010, 1802, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Perier, C.; Vila, M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009332. [Google Scholar] [CrossRef]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Fujioka, H.; Hoppel, C.; Whone, A.L.; Caldwell, M.A.; Cullen, P.J.; Liu, J.; Zhu, X. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 2016, 22, 54–63. [Google Scholar] [CrossRef]
- Wang, X.; Petrie, T.G.; Liu, Y.; Liu, J.; Fujioka, H.; Zhu, X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J. Neurochem. 2012, 121, 830–839. [Google Scholar] [CrossRef]
- Pozo Devoto, V.M.; Falzone, T.L. Mitochondrial dynamics in Parkinson’s disease: A role for alpha-synuclein? Dis. Model. Mech. 2017, 10, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Wang, W.; Wang, C.; Siedlak, S.L.; Fujioka, H.; Tang, B.; Zhu, X. Mfn2 protects dopaminergic neurons exposed to paraquat both in vitro and in vivo: Implications for idiopathic Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Liu, W.; He, X.; Gao, Y.; Castellani, R.J.; Perry, G.; Smith, M.A.; Zhu, X. DLP1-dependent mitochondrial fragmentation mediates 1-methyl-4-phenylpyridinium toxicity in neurons: Implications for Parkinson’s disease. Aging Cell 2011, 10, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef]
- Burke, R.E.; O’Malley, K. Axon degeneration in Parkinson’s disease. Exp. Neurol. 2013, 246, 72–83. [Google Scholar] [CrossRef]
- Joniec, I.; Ciesielska, A.; Kurkowska-Jastrzebska, I.; Przybylkowski, A.; Czlonkowska, A.; Czlonkowski, A. Age- and sex-differences in the nitric oxide synthase expression and dopamine concentration in the murine model of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Brain Res. 2009, 1261, 7–19. [Google Scholar] [CrossRef]
- Nanasi, N.; Veres, G.; Cseh, E.K.; Martos, D.; Hadady, L.; Klivenyi, P.; Vecsei, L.; Zadori, D. The assessment of possible gender-related effect of endogenous striatal alpha-tocopherol level on MPTP neurotoxicity in mice. Heliyon 2020, 6, e04425. [Google Scholar] [CrossRef]
- Unzeta, M.; Baron, S.; Perez, V.; Ambrosio, S.; Mahy, N. Sex-related effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydro-pyridine treatment may be related to differences in monoamine oxidase B. Neurosci. Lett. 1994, 176, 235–238. [Google Scholar] [CrossRef]
- Ramonet, D.; Perier, C.; Recasens, A.; Dehay, B.; Bove, J.; Costa, V.; Scorrano, L.; Vila, M. Optic atrophy 1 mediates mitochondria remodeling and dopaminergic neurodegeneration linked to complex I deficiency. Cell Death Differ. 2013, 20, 77–85. [Google Scholar] [CrossRef]
- Filichia, E.; Hoffer, B.; Qi, X.; Luo, Y. Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson’s disease model induced by MPTP. Sci. Rep. 2016, 6, 32656. [Google Scholar] [CrossRef] [PubMed]
- Rappold, P.M.; Cui, M.; Grima, J.C.; Fan, R.Z.; de Mesy-Bentley, K.L.; Chen, L.; Zhuang, X.; Bowers, W.J.; Tieu, K. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat. Commun. 2014, 5, 5244. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Sterky, F.H.; Mourier, A.; Terzioglu, M.; Cullheim, S.; Olson, L.; Larsson, N.G. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet. 2012, 21, 4827–4835. [Google Scholar] [CrossRef]
- Fujita, K.; Seike, T.; Yutsudo, N.; Ohno, M.; Yamada, H.; Yamaguchi, H.; Sakumi, K.; Yamakawa, Y.; Kido, M.A.; Takaki, A.; et al. Hydrogen in drinking water reduces dopaminergic neuronal loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. PLoS ONE 2009, 4, e7247. [Google Scholar] [CrossRef]
- Wood, J.; LaPalombara, Z.; Ahmari, S.E. Monoamine abnormalities in the SAPAP3 knockout model of obsessive-compulsive disorder-related behaviour. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 23. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, F.; Austria, Q.; Wang, W.; Zhu, X. Mfn2 Overexpression Attenuates MPTP Neurotoxicity In Vivo. Int. J. Mol. Sci. 2021, 22, 601. https://doi.org/10.3390/ijms22020601
Zhao F, Austria Q, Wang W, Zhu X. Mfn2 Overexpression Attenuates MPTP Neurotoxicity In Vivo. International Journal of Molecular Sciences. 2021; 22(2):601. https://doi.org/10.3390/ijms22020601
Chicago/Turabian StyleZhao, Fanpeng, Quillan Austria, Wenzhang Wang, and Xiongwei Zhu. 2021. "Mfn2 Overexpression Attenuates MPTP Neurotoxicity In Vivo" International Journal of Molecular Sciences 22, no. 2: 601. https://doi.org/10.3390/ijms22020601
APA StyleZhao, F., Austria, Q., Wang, W., & Zhu, X. (2021). Mfn2 Overexpression Attenuates MPTP Neurotoxicity In Vivo. International Journal of Molecular Sciences, 22(2), 601. https://doi.org/10.3390/ijms22020601