
Molecular Bases of Human Malformation Syndromes Involving the SHH Pathway: GLIA/R Balance and Cardinal Phenotypes



Abstract
1. Introduction
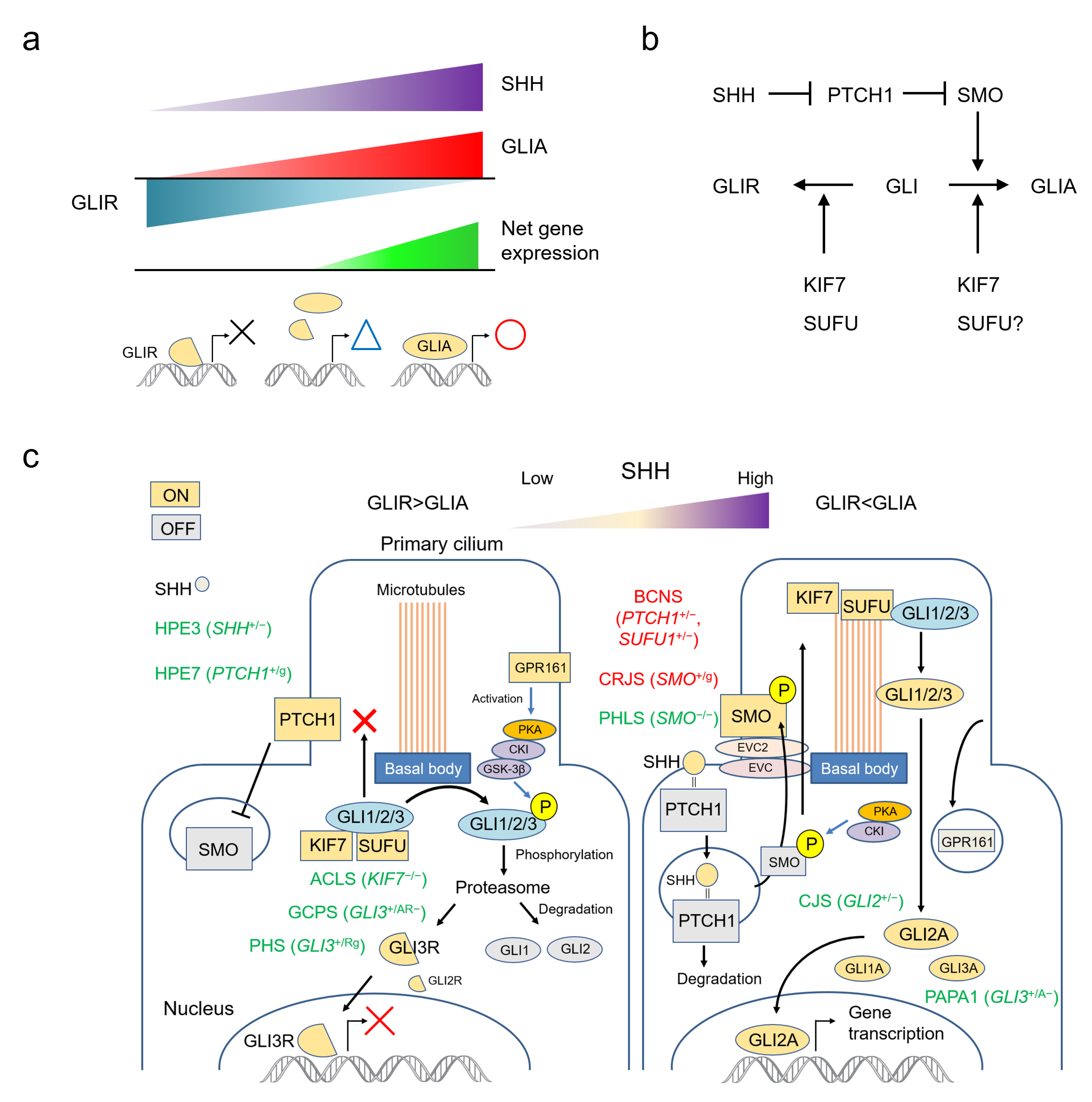
2. The SHH Pathway
2.1. SHH Acts by Establishing a Morphogen Gradient
2.2. The SHH Pathway and The Primary Cilium
3. Human Development and Malformations Related to the SHH Pathway
3.1. Brain Morphology and Size (Holoprosencephaly and Micro/Macro-Cephaly)
3.2. Craniofacial (Midface Anormalies)
3.3. Limbs and Digits (Polydactyly and Syndactyly)
3.4. Neoplasia (Basal Cell Carcinoma, Medulloblastoma)
4. Human Malformation Syndromes Caused by Major Genes in the SHH Pathway
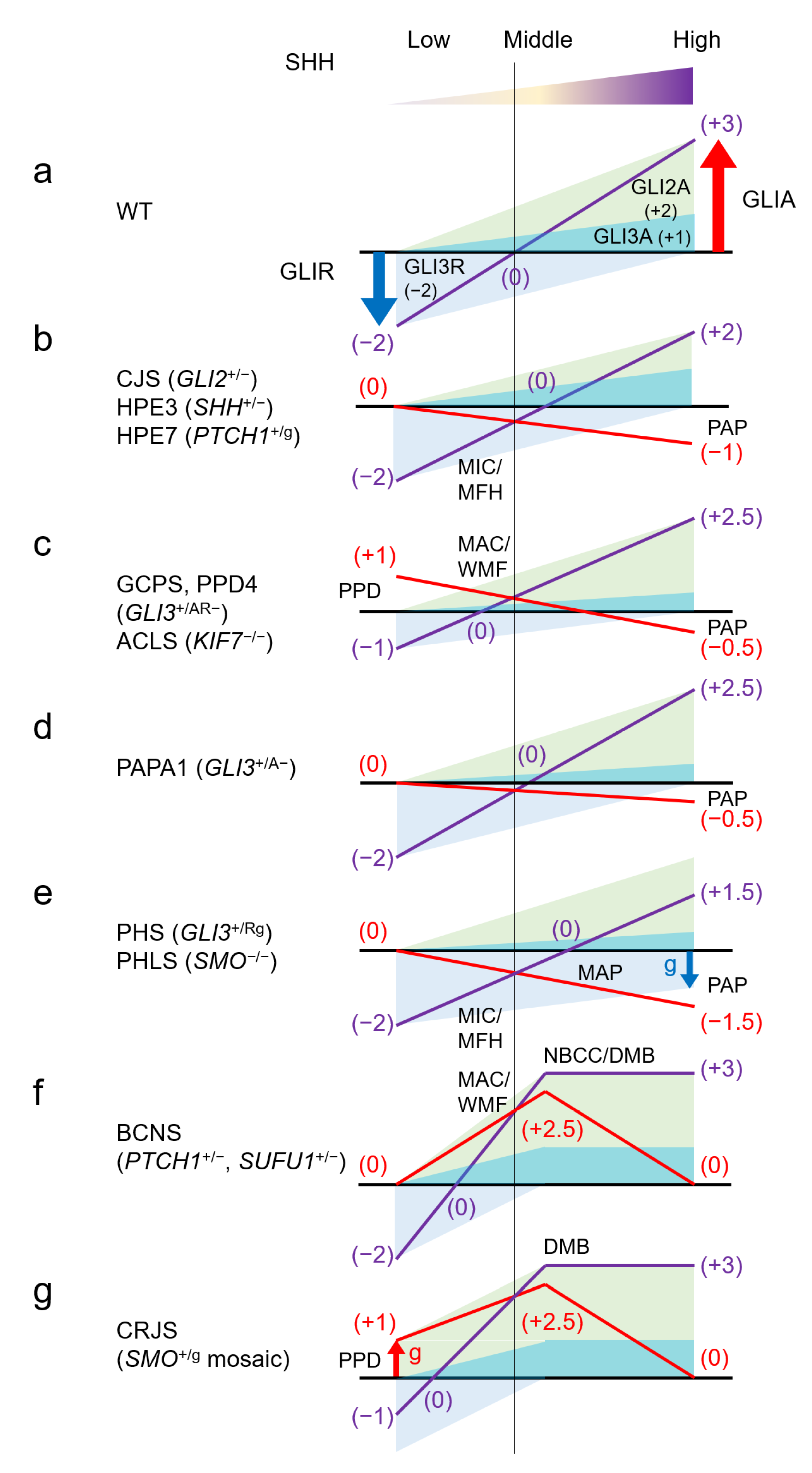
4.1. GLI2 (GLI Family Zinc Finger 2) *165230
4.1.1. Culler-Jones Syndrome; CJS #615849
4.1.2. Holoprosencephaly 9; HPE9 #610829
4.2. GLI3 (GLI Family Zinc Finger 3) *165240
4.2.1. Greig Cephalopolysyndactyly Syndrome; GCPS #175700
4.2.2. Polydactyly, Preaxial, Type IV; PPD4 #174700
4.2.3. Polydactyly, Postaxial, Types A1 and B; PAPA1 #174200
4.2.4. Pallister-Hall Syndrome; PHS #146510
4.3. SHH (Sonic Hedgehog Signalling Molecule) *600725
4.3.1. Holoprosencephaly 3; HPE3 #142945
4.3.2. Microphthalmia, Isolated, with Coloboma 5; MCOPCB5 #611638
4.3.3. Single Median Maxillary Central Incisor; SMMCI #147250 (SMMCI Syndrome Included)
4.4. PTCH1 (Patched 1) *601309
4.4.1. Basal Cell Nevus Syndrome; BCNS #109400
4.4.2. Holoprosencephaly 7; HPE7 #610828
4.5. SMO (Smoothened, Frizzled Class Receptor) *601500
4.5.1. Pallister-Hall-Like Syndrome; PHLS #241800
4.5.2. Curry-Jones Syndrome; CRJS #601707
4.6. SUFU (SUFU Negative Regulator of Hedgehog Signaling) *607035
4.6.1. Basal Cell Nevus Syndrome; BCNS #109400
4.6.2. Medulloblastoma, Desmoplastic; MDB #155255
4.6.3. Meningioma, Familial, Susceptibility to #607174
4.6.4. Joubert Syndrome 32; JBTS32 #617757 and Corelated Conditions
4.7. KIF7 (Kinesin Family Member 7) *611254
4.7.1. Acrocallosal Syndrome; ACLS #200990
4.7.2. Al-Gazali-Bakalinova Syndrome; AGBK #607131
4.7.3. Hydrolethalus Syndrome 2; HLS2 #614120
5. Miscellaneous Syndromes Related to SHH Pathways
5.1. Other Human GLI Genes
5.2. Other Human Hedgehog Genes
5.3. Ellis-Van Creveld Syndrome
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Riddle, R.D.; Johnson, R.L.; Laufer, E.; Tabin, C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 1993, 75, 1401–1416. [Google Scholar] [CrossRef]
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Krauss, S.; Concordet, J.P.; Ingham, P.W. A functionally conserved homolog of the Drosophila segment polarity gene hh is expressed in tissues with polarizing activity in zebrafish embryos. Cell 1993, 75, 1431–1444. [Google Scholar] [CrossRef]
- Roelink, H.; Augsburger, A.; Heemskerk, J.; Korzh, V.; Norlin, S.; Ruiz i Altaba, A.; Tanabe, Y.; Placzek, M.; Edlund, T.; Jessell, T.M. Floor plate and motor neuron induction by vhh-1, a vertebrate homolog of hedgehog expressed by the notochord. Cell 1994, 76, 761–775. [Google Scholar] [CrossRef]
- Chang, D.T.; López, A.; von Kessler, D.P.; Chiang, C.; Simandl, B.K.; Zhao, R.; Seldin, M.F.; Fallon, J.F.; Beachy, P.A. Products, genetic linkage and limb patterning activity of a murine hedgehog gene. Development 1994, 120, 3339–3353. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.L.; Laufer, E.; Riddle, R.D.; Tabin, C. Ectopic expression of Sonic hedgehog alters dorsal-ventral patterning of somites. Cell 1994, 79, 1165–1173. [Google Scholar] [CrossRef]
- Martí, E.; Takada, R.; Bumcrot, D.A.; Sasaki, H.; McMahon, A.P. Distribution of Sonic hedgehog peptides in the developing chick and mouse embryo. Development 1995, 121, 2537–2547. [Google Scholar] [CrossRef]
- Jessell, T.M. Neuronal specification in the spinal cord: Inductive signals and transcriptional codes. Nat. Rev. Genet. 2000, 1, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Xavier, G.M.; Seppala, M.; Barrell, W.; Birjandi, A.A.; Geoghegan, F.; Cobourne, M.T. Hedgehog receptor function during craniofacial development. Dev. Biol. 2016, 415, 198–215. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Bitgood, M.J.; Shen, L.; McMahon, A.P. Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr. Biol. 1996, 6, 298–304. [Google Scholar] [CrossRef]
- Yao, H.H.; Whoriskey, W.; Capel, B. Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev. 2002, 16, 1433–1440. [Google Scholar] [CrossRef]
- Wijgerde, M.; Ooms, M.; Hoogerbrugge, J.W.; Grootegoed, J.A. Hedgehog signaling in mouse ovary: Indian hedgehog and desert hedgehog from granulosa cells induce target gene expression in developing theca cells. Endocrinology 2005, 146, 3558–3566. [Google Scholar] [CrossRef] [PubMed]
- Allanson, J.E.; Biesecker, L.G.; Carey, J.C.; Hennekam, R.C. Elements of morphology: Introduction. Am. J. Med. Genet. A 2009, 149A, 2–5. [Google Scholar] [CrossRef]
- Allanson, J.E.; Cunniff, C.; Hoyme, H.E.; McGaughran, J.; Muenke, M.; Neri, G. Elements of morphology: Standard terminology for the head and face. Am. J. Med. Genet. A 2009, 149A, 6–28. [Google Scholar] [CrossRef]
- Hall, B.D.; Graham, J.M.; Cassidy, S.B.; Opitz, J.M. Elements of morphology: Standard terminology for the periorbital region. Am. J. Med. Genet. A 2009, 149A, 29–39. [Google Scholar] [CrossRef]
- Hunter, A.; Frias, J.L.; Gillessen-Kaesbach, G.; Hughes, H.; Jones, K.L.; Wilson, L. Elements of morphology: Standard terminology for the ear. Am. J. Med. Genet. A 2009, 149A, 40–60. [Google Scholar] [CrossRef]
- Hennekam, R.C.; Cormier-Daire, V.; Hall, J.G.; Méhes, K.; Patton, M.; Stevenson, R.E. Elements of morphology: Standard terminology for the nose and philtrum. Am. J. Med. Genet. A 2009, 149A, 61–76. [Google Scholar] [CrossRef]
- Carey, J.C.; Cohen, M.M.; Curry, C.J.; Devriendt, K.; Holmes, L.B.; Verloes, A. Elements of morphology: Standard terminology for the lips, mouth, and oral region. Am. J. Med. Genet. A 2009, 149A, 77–92. [Google Scholar] [CrossRef]
- Biesecker, L.G.; Aase, J.M.; Clericuzio, C.; Gurrieri, F.; Temple, I.K.; Toriello, H. Elements of morphology: Standard terminology for the hands and feet. Am. J. Med. Genet. A 2009, 149A, 93–127. [Google Scholar] [CrossRef] [PubMed]
- Klinger, G.; Merlob, P. Elements of morphology: Standard terminology for the ear—Additional features. Am. J. Med. Genet. A 2009, 149A, 1607. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef]
- Shimeld, S.M.; van den Heuvel, M.; Dawber, R.; Briscoe, J. An amphioxus Gli gene reveals conservation of midline patterning and the evolution of hedgehog signalling diversity in chordates. PLoS ONE 2007, 2, e864. [Google Scholar] [CrossRef]
- Ruppert, J.M.; Kinzler, K.W.; Wong, A.J.; Bigner, S.H.; Kao, F.T.; Law, M.L.; Seuanez, H.N.; O’Brien, S.J.; Vogelstein, B. The GLI-Kruppel family of human genes. Mol. Cell Biol. 1988, 8, 3104–3113. [Google Scholar]
- Villavicencio, E.H.; Walterhouse, D.O.; Iannaccone, P.M. The sonic hedgehog-patched-gli pathway in human development and disease. Am. J. Hum. Genet. 2000, 67, 1047–1054. [Google Scholar] [CrossRef]
- Gorojankina, T. Hedgehog signaling pathway: A novel model and molecular mechanisms of signal transduction. Cell Mol. Life Sci. 2016, 73, 1317–1332. [Google Scholar] [CrossRef]
- Kong, J.H.; Siebold, C.; Rohatgi, R. Biochemical mechanisms of vertebrate hedgehog signaling. Development 2019, 146, 6892. [Google Scholar] [CrossRef]
- Sasai, N.; Toriyama, M.; Kondo, T. Hedgehog Signal and Genetic Disorders. Front. Genet. 2019, 10, 1103. [Google Scholar] [CrossRef] [PubMed]
- Satir, P.; Pedersen, L.B.; Christensen, S.T. The primary cilium at a glance. J. Cell Sci. 2010, 123, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Ezquerro, J.J.; Münsterberg, A.E.; Stricker, S. Editorial: Signaling Pathways in Embryonic Development. Front. Cell Dev. Biol. 2017, 5, 76. [Google Scholar] [CrossRef]
- Oonuma, K.; Yamamoto, M.; Moritsugu, N.; Okawa, N.; Mukai, M.; Sotani, M.; Tsunemi, S.; Sugimoto, H.; Nakagome, E.; Hasegawa, Y.; et al. Evolution of Developmental Programs for the Midline Structures in Chordates: Insights From Gene Regulation in the Floor Plate and Hypochord Homologues of. Front. Cell Dev. Biol. 2021, 9, 704367. [Google Scholar] [CrossRef]
- Yabut, O.R.; Pleasure, S.J. Sonic Hedgehog Signaling Rises to the Surface: Emerging Roles in Neocortical Development. Brain Plast. 2018, 3, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, S.; Boglev, Y.; Owens, H.; Goldie, S.J. The Role of Sonic Hedgehog in Craniofacial Patterning, Morphogenesis and Cranial Neural Crest Survival. J. Dev. Biol. 2016, 4, 24. [Google Scholar] [CrossRef] [PubMed]
- Tickle, C.; Towers, M. Sonic Hedgehog Signaling in Limb Development. Front. Cell Dev. Biol. 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Monteagudo, A. Holoprosencephaly. Am. J. Obstet. Gynecol. 2020, 223, B13–B16. [Google Scholar] [CrossRef] [PubMed]
- Roessler, E.; Belloni, E.; Gaudenz, K.; Jay, P.; Berta, P.; Scherer, S.W.; Tsui, L.C.; Muenke, M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat. Genet. 1996, 14, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Kruszka, P.; Muenke, M. Syndromes associated with holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Loo, C.K.C.; Pearen, M.A.; Ramm, G.A. The Role of Sonic Hedgehog in Human Holoprosencephaly and Short-Rib Polydactyly Syndromes. Int. J. Mol. Sci. 2021, 22, 9854. [Google Scholar] [CrossRef] [PubMed]
- Palma, V.; Ruiz i Altaba, A. Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development 2004, 131, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Lien, W.H.; Klezovitch, O.; Fernandez, T.E.; Delrow, J.; Vasioukhin, V. alphaE-catenin controls cerebral cortical size by regulating the hedgehog signaling pathway. Science 2006, 311, 1609–1612. [Google Scholar] [CrossRef] [PubMed]
- Komada, M. Sonic hedgehog signaling coordinates the proliferation and differentiation of neural stem/progenitor cells by regulating cell cycle kinetics during development of the neocortex. Congenit. Anom. 2012, 52, 72–77. [Google Scholar] [CrossRef]
- Han, Y.G. Sonic hedgehog signaling: A conserved mechanism for the expansion of outer radial glia and intermediate progenitor cells and for the growth and folding of the neocortex. Neurogenesis 2016, 3, e1242957. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lumsden, A.; Sprawson, N.; Graham, A. Segmental origin and migration of neural crest cells in the hindbrain region of the chick embryo. Development 1991, 113, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Ahlgren, S.C.; Bronner-Fraser, M. Inhibition of sonic hedgehog signaling in vivo results in craniofacial neural crest cell death. Curr. Biol. 1999, 9, 1304–1314. [Google Scholar] [CrossRef]
- Brito, J.M.; Teillet, M.A.; Le Douarin, N.M. An early role for sonic hedgehog from foregut endoderm in jaw development: Ensuring neural crest cell survival. Proc. Natl. Acad. Sci. USA 2006, 103, 11607–11612. [Google Scholar] [CrossRef]
- Brito, J.M.; Teillet, M.A.; Le Douarin, N.M. Induction of mirror-image supernumerary jaws in chicken mandibular mesenchyme by Sonic Hedgehog-producing cells. Development 2008, 135, 2311–2319. [Google Scholar] [CrossRef] [PubMed]
- Moore-Scott, B.A.; Manley, N.R. Differential expression of Sonic hedgehog along the anterior-posterior axis regulates patterning of pharyngeal pouch endoderm and pharyngeal endoderm-derived organs. Dev. Biol. 2005, 278, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Bear, K.A.; Solomon, B.D.; Antonini, S.; Arnhold, I.J.; França, M.M.; Gerkes, E.H.; Grange, D.K.; Hadley, D.W.; Jääskeläinen, J.; Paulo, S.S.; et al. Pathogenic mutations in GLI2 cause a specific phenotype that is distinct from holoprosencephaly. J. Med. Genet. 2014, 51, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Dubourg, C.; Bendavid, C.; Pasquier, L.; Henry, C.; Odent, S.; David, V. Holoprosencephaly. Orphanet J. Rare Dis. 2007, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Richieri-Costa, A.; Ribeiro, L.A. Holoprosencephaly-like phenotype: Clinical and genetic perspectives. Am. J. Med. Genet. A 2006, 140, 2587–2593. [Google Scholar] [CrossRef]
- Abramyan, J. Hedgehog Signaling and Embryonic Craniofacial Disorders. J. Dev. Biol. 2019, 7, 9. [Google Scholar] [CrossRef]
- Malik, S. Polydactyly: Phenotypes, genetics and classification. Clin. Genet. 2014, 85, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Le, H.; Kleinerman, R.; Lerman, O.Z.; Brown, D.; Galiano, R.; Gurtner, G.C.; Warren, S.M.; Levine, J.P.; Saadeh, P.B. Hedgehog signaling is essential for normal wound healing. Wound Repair Regen. 2008, 16, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef]
- Kimonis, V.E.; Goldstein, A.M.; Pastakia, B.; Yang, M.L.; Kase, R.; DiGiovanna, J.J.; Bale, A.E.; Bale, S.J. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am. J. Med. Genet. 1997, 69, 299–308. [Google Scholar] [CrossRef]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef]
- Yang, L.; Xie, G.; Fan, Q.; Xie, J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 2010, 29, 469–481. [Google Scholar] [CrossRef]
- Low, J.A.; de Sauvage, F.J. Clinical experience with Hedgehog pathway inhibitors. J. Clin. Oncol. 2010, 28, 5321–5326. [Google Scholar] [CrossRef] [PubMed]
- França, M.M.; Jorge, A.A.; Carvalho, L.R.; Costalonga, E.F.; Vasques, G.A.; Leite, C.C.; Mendonca, B.B.; Arnhold, I.J. Novel heterozygous nonsense GLI2 mutations in patients with hypopituitarism and ectopic posterior pituitary lobe without holoprosencephaly. J. Clin. Endocrinol. Metab. 2010, 95, E384–E391. [Google Scholar] [CrossRef] [PubMed]
- Bertolacini, C.D.; Ribeiro-Bicudo, L.A.; Petrin, A.; Richieri-Costa, A.; Murray, J.C. Clinical findings in patients with GLI2 mutations—Phenotypic variability. Clin. Genet. 2012, 81, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Roessler, E.; Du, Y.Z.; Mullor, J.L.; Casas, E.; Allen, W.P.; Gillessen-Kaesbach, G.; Roeder, E.R.; Ming, J.E.; Ruiz i Altaba, A.; Muenke, M. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc. Natl. Acad. Sci. USA 2003, 100, 13424–13429. [Google Scholar] [CrossRef] [PubMed]
- Niida, Y.; Inoue, M.; Ozaki, M.; Takase, E. Human Malformation Syndromes of Defective GLI: Opposite Phenotypes of 2q14.2 (GLI2) and 7p14.2 (GLI3) Microdeletions and a GLIA/R Balance Model. Cytogenet. Genome Res. 2017, 153, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, P.; Schoumans, J.; Staaf, J.; Jönsson, G.; Carlsson, F.; Kristoffersson, U.; Borg, A.; Nordenskjöld, M.; Dahl, N. Hemizygosity for chromosome 2q14.2-q22.1 spanning the GLI2 and PROC genes associated with growth hormone deficiency, polydactyly, deep vein thrombosis and urogenital abnormalities. Clin. Genet. 2006, 69, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Kordaß, U.; Schröder, C.; Elbracht, M.; Soellner, L.; Eggermann, T. A familial GLI2 deletion (2q14.2) not associated with the holoprosencephaly syndrome phenotype. Am. J. Med. Genet. A 2015, 167A, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, I.J.; França, M.M.; Carvalho, L.R.; Mendonca, B.B.; Jorge, A.A. Role of GLI2 in hypopituitarism phenotype. J. Mol. Endocrinol. 2015, 54, R141–R150. [Google Scholar] [CrossRef] [PubMed]
- Greally, M.T.; Robinson, E.; Allen, N.M.; O’Donovan, D.; Crolla, J.A. De novo interstitial deletion 2q14.1q22.1: Is there a recognizable phenotype? Am. J. Med. Genet. A 2014, 164A, 3194–3202. [Google Scholar] [CrossRef]
- Kevelam, S.H.; van Harssel, J.J.; van der Zwaag, B.; Smeets, H.J.; Paulussen, A.D.; Lichtenbelt, K.D. A patient with a mild holoprosencephaly spectrum phenotype and heterotaxy and a 1.3 Mb deletion encompassing GLI2. Am. J. Med. Genet. A 2012, 158A, 166–173. [Google Scholar] [CrossRef]
- Al-Qattan, M.M.; Shamseldin, H.E.; Salih, M.A.; Alkuraya, F.S. GLI3-related polydactyly: A review. Clin. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G. The Greig cephalopolysyndactyly syndrome. Orphanet J. Rare Dis. 2008, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Kroisel, P.M.; Petek, E.; Wagner, K. Phenotype of five patients with Greig syndrome and microdeletion of 7p13. Am. J. Med. Genet. 2001, 102, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.J.; Olivos-Glander, I.; Killoran, C.; Elson, E.; Turner, J.T.; Peters, K.F.; Abbott, M.H.; Aughton, D.J.; Aylsworth, A.S.; Bamshad, M.J.; et al. Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister-Hall syndromes: Robust phenotype prediction from the type and position of GLI3 mutations. Am. J. Hum. Genet. 2005, 76, 609–622. [Google Scholar] [CrossRef]
- Fujioka, H.; Ariga, T.; Horiuchi, K.; Otsu, M.; Igawa, H.; Kawashima, K.; Yamamoto, Y.; Sugihara, T.; Sakiyama, Y. Molecular analysis of non-syndromic preaxial polydactyly: Preaxial polydactyly type-IV and preaxial polydactyly type-I. Clin. Genet. 2005, 67, 429–433. [Google Scholar] [CrossRef]
- Biesecker, L.G.; Johnston, J. Syndromic and non-syndromic GLI3 phenotypes. Clin. Genet. 2005, 68, 285. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.J.; Sapp, J.C.; Turner, J.T.; Amor, D.; Aftimos, S.; Aleck, K.A.; Bocian, M.; Bodurtha, J.N.; Cox, G.F.; Curry, C.J.; et al. Molecular analysis expands the spectrum of phenotypes associated with GLI3 mutations. Hum. Mutat. 2010, 31, 1142–1154. [Google Scholar] [CrossRef]
- Nanni, L.; Ming, J.E.; Bocian, M.; Steinhaus, K.; Bianchi, D.W.; Die-Smulders, C.; Giannotti, A.; Imaizumi, K.; Jones, K.L.; Campo, M.D.; et al. The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum. Mol. Genet. 1999, 8, 2479–2488. [Google Scholar] [CrossRef]
- Schimmenti, L.A.; de la Cruz, J.; Lewis, R.A.; Karkera, J.D.; Manligas, G.S.; Roessler, E.; Muenke, M. Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. Am. J. Med. Genet. A 2003, 116A, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Ming, J.E.; Du, Y.; Hall, R.K.; Aldred, M.; Bankier, A.; Muenke, M. SHH mutation is associated with solitary median maxillary central incisor: A study of 13 patients and review of the literature. Am. J. Med. Genet. 2001, 102, 1–10. [Google Scholar] [CrossRef]
- Marini, M.; Cusano, R.; De Biasio, P.; Caroli, F.; Lerone, M.; Silengo, M.; Ravazzolo, R.; Seri, M.; Camera, G. Previously undescribed nonsense mutation in SHH caused autosomal dominant holoprosencephaly with wide intrafamilial variability. Am. J. Med. Genet. A 2003, 117A, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.A.; Chrousos, G.P.; Chandra, R.; Evangelista, R.S.; Gilbert, J.C.; Nobuhara, K.; Zhuang, Z.; Vortmeyer, A.O. Two-hit model for tumorigenesis of nevoid basal cell carcinoma (Gorlin) syndrome-associated hepatic mesenchymal tumor. Am. J. Med. Genet. 2002, 109, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Tate, G.; Kishimoto, K.; Mitsuya, T. Biallelic disruption of the PTCH1 gene in multiple basal cell carcinomas in Japanese patients with nevoid basal cell carcinoma syndrome. Acta. Med. Okayama 2014, 68, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Akizawa, Y.; Miyashita, T.; Sasaki, R.; Nagata, R.; Aoki, R.; Ishitani, K.; Nagashima, Y.; Matsui, H.; Saito, K. Gorlin syndrome with an ovarian leiomyoma associated with a PTCH1 second hit. Am. J. Med. Genet. A 2016, 170A, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Smyth, I.; Narang, M.A.; Evans, T.; Heimann, C.; Nakamura, Y.; Chenevix-Trench, G.; Pietsch, T.; Wicking, C.; Wainwright, B.J. Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene inbasal cell carcinoma and medulloblastoma on chromosome 1p32. Hum. Mol. Genet. 1999, 8, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Metzis, V.; Courtney, A.D.; Kerr, M.C.; Ferguson, C.; Rondón Galeano, M.C.; Parton, R.G.; Wainwright, B.J.; Wicking, C. Patched1 is required in neural crest cells for the prevention of orofacial clefts. Hum. Mol. Genet. 2013, 22, 5026–5035. [Google Scholar] [CrossRef]
- Ming, J.E.; Kaupas, M.E.; Roessler, E.; Brunner, H.G.; Golabi, M.; Tekin, M.; Stratton, R.F.; Sujansky, E.; Bale, S.J.; Muenke, M. Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum. Genet. 2002, 110, 297–301. [Google Scholar] [CrossRef]
- Ribeiro, L.A.; Murray, J.C.; Richieri-Costa, A. PTCH mutations in four Brazilian patients with holoprosencephaly and in one with holoprosencephaly-like features and normal MRI. Am. J. Med. Genet. A 2006, 140, 2584–2586. [Google Scholar] [CrossRef]
- Derwińska, K.; Smyk, M.; Cooper, M.L.; Bader, P.; Cheung, S.W.; Stankiewicz, P. PTCH1 duplication in a family with microcephaly and mild developmental delay. Eur. J. Hum. Genet. 2009, 17, 267–271. [Google Scholar] [CrossRef]
- Rubino, S.; Qian, J.; Pinheiro-Neto, C.D.; Kenning, T.J.; Adamo, M.A. A familial syndrome of hypothalamic hamartomas, polydactyly, and SMO mutations: A clinical report of 2 cases. J. Neurosurg. Pediatr. 2018, 23, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Le, T.L.; Sribudiani, Y.; Dong, X.; Huber, C.; Kois, C.; Baujat, G.; Gordon, C.T.; Mayne, V.; Galmiche, L.; Serre, V.; et al. Bi-allelic Variations of SMO in Humans Cause a Broad Spectrum of Developmental Anomalies Due to Abnormal Hedgehog Signaling. Am. J. Hum. Genet. 2020, 106, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Twigg, S.R.F.; Hufnagel, R.B.; Miller, K.A.; Zhou, Y.; McGowan, S.J.; Taylor, J.; Craft, J.; Taylor, J.C.; Santoro, S.L.; Huang, T.; et al. A Recurrent Mosaic Mutation in SMO, Encoding the Hedgehog Signal Transducer Smoothened, Is the Major Cause of Curry-Jones Syndrome. Am. J. Hum. Genet. 2016, 98, 1256–1265. [Google Scholar] [CrossRef]
- Pastorino, L.; Ghiorzo, P.; Nasti, S.; Battistuzzi, L.; Cusano, R.; Marzocchi, C.; Garrè, M.L.; Clementi, M.; Scarrà, G.B. Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am. J. Med. Genet. A 2009, 149A, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Beetz, C.; Williams, S.G.; Bhaskar, S.S.; O’Sullivan, J.; Anderson, B.; Daly, S.B.; Urquhart, J.E.; Bholah, Z.; Oudit, D.; et al. Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J. Clin. Oncol. 2014, 32, 4155–4161. [Google Scholar] [CrossRef] [PubMed]
- Schulman, J.M.; Oh, D.H.; Sanborn, J.Z.; Pincus, L.; McCalmont, T.H.; Cho, R.J. Multiple Hereditary Infundibulocystic Basal Cell Carcinoma Syndrome Associated With a Germline SUFU Mutation. JAMA Dermatol. 2016, 152, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.; Stavrou, T.; Liu, L.; Taylor, M.D.; Gold, B.; Dean, M.; Kelley, M.J.; Dubovsky, E.C.; Vezina, G.; Nicholson, H.S.; et al. Retrospective family study of childhood medulloblastoma. Am. J. Med. Genet. A 2005, 134, 399–403. [Google Scholar] [CrossRef]
- Slade, I.; Murray, A.; Hanks, S.; Kumar, A.; Walker, L.; Hargrave, D.; Douglas, J.; Stiller, C.; Izatt, L.; Rahman, N. Heterogeneity of familial medulloblastoma and contribution of germline PTCH1 and SUFU mutations to sporadic medulloblastoma. Fam. Cancer 2011, 10, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.C.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef]
- Brugières, L.; Remenieras, A.; Pierron, G.; Varlet, P.; Forget, S.; Byrde, V.; Bombled, J.; Puget, S.; Caron, O.; Dufour, C.; et al. High frequency of germline SUFU mutations in children with desmoplastic/nodular medulloblastoma younger than 3 years of age. J. Clin. Oncol. 2012, 30, 2087–2093. [Google Scholar] [CrossRef] [PubMed]
- Brugières, L.; Pierron, G.; Chompret, A.; Paillerets, B.B.; Di Rocco, F.; Varlet, P.; Pierre-Kahn, A.; Caron, O.; Grill, J.; Delattre, O. Incomplete penetrance of the predisposition to medulloblastoma associated with germ-line SUFU mutations. J. Med. Genet. 2010, 47, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Aavikko, M.; Li, S.P.; Saarinen, S.; Alhopuro, P.; Kaasinen, E.; Morgunova, E.; Li, Y.; Vesanen, K.; Smith, M.J.; Evans, D.G.; et al. Loss of SUFU function in familial multiple meningioma. Am. J. Hum. Genet. 2012, 91, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.F.; Yu, K.P.; Brueckner, M.; Brailey, L.L.; Johnson, L.; McGrath, J.M.; Bale, A.E. Cardiac and CNS defects in a mouse with targeted disruption of suppressor of fused. Development 2005, 132, 4407–4417. [Google Scholar] [CrossRef] [PubMed]
- Svärd, J.; Heby-Henricson, K.; Henricson, K.H.; Persson-Lek, M.; Rozell, B.; Lauth, M.; Bergström, A.; Ericson, J.; Toftgård, R.; Teglund, S. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev. Cell 2006, 10, 187–197. [Google Scholar] [CrossRef] [PubMed]
- De Mori, R.; Romani, M.; D’Arrigo, S.; Zaki, M.S.; Lorefice, E.; Tardivo, S.; Biagini, T.; Stanley, V.; Musaev, D.; Fluss, J.; et al. Hypomorphic Recessive Variants in SUFU Impair the Sonic Hedgehog Pathway and Cause Joubert Syndrome with Cranio-facial and Skeletal Defects. Am. J. Hum. Genet. 2017, 101, 552–563. [Google Scholar] [CrossRef]
- Serpieri, V.; D’Abrusco, F.; Dempsey, J.C.; Cheng, Y.H.; Arrigoni, F.; Baker, J.; Battini, R.; Bertini, E.S.; Borgatti, R.; Christman, A.K.; et al. SUFU haploinsufficiency causes a recognisable neurodevelopmental phenotype at the mild end of the Joubert syndrome spectrum. J. Med. Genet. 2021. [Google Scholar] [CrossRef]
- Kim, J.J.; Gill, P.S.; Rotin, L.; van Eede, M.; Henkelman, R.M.; Hui, C.C.; Rosenblum, N.D. Suppressor of fused controls mid-hindbrain patterning and cerebellar morphogenesis via GLI3 repressor. J. Neurosci. 2011, 31, 1825–1836. [Google Scholar] [CrossRef]
- Chen, M.H.; Wilson, C.W.; Li, Y.J.; Law, K.K.; Lu, C.S.; Gacayan, R.; Zhang, X.; Hui, C.C.; Chuang, P.T. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009, 23, 1910–1928. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.; Li, Y.; Yigit, G.; Altmüller, J.; Bader, I.; Bevot, A.; Biskup, S.; Dreha-Kulaczewski, S.; Christoph Korenke, G.; Kottke, R.; et al. Heterozygous truncating variants in SUFU cause congenital ocular motor apraxia. Genet. Med. 2021, 23, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.J.; Olivos-Glander, I.; Turner, J.; Aleck, K.; Bird, L.M.; Mehta, L.; Schimke, R.N.; Heilstedt, H.; Spence, J.E.; Blancato, J.; et al. Clinical and molecular delineation of the Greig cephalopolysyndactyly contiguous gene deletion syndrome and its distinction from acrocallosal syndrome. Am. J. Med. Genet. A 2003, 123A, 236–242. [Google Scholar] [CrossRef]
- Ali, B.R.; Silhavy, J.L.; Akawi, N.A.; Gleeson, J.G.; Al-Gazali, L. A mutation in KIF7 is responsible for the autosomal recessive syndrome of macrocephaly, multiple epiphyseal dysplasia and distinctive facial appearance. Orphanet J. Rare Dis. 2012, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.H.; Zhang, X.; Yu, C.; Li, Z.J.; Wunder, J.S.; Hui, C.C.; Alman, B.A. Kif7 promotes hedgehog signaling in growth plate chondrocytes by restricting the inhibitory function of Sufu. Development 2011, 138, 3791–3801. [Google Scholar] [CrossRef] [PubMed]
- Putoux, A.; Thomas, S.; Coene, K.L.; Davis, E.E.; Alanay, Y.; Ogur, G.; Uz, E.; Buzas, D.; Gomes, C.; Patrier, S.; et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet. 2011, 43, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Umair, M.; Ahmad, F.; Ahmad, S.; Alam, Q.; Rehan, M.; Alqosaibi, A.I.; Alnamshan, M.M.; Rafeeq, M.M.; Haque, S.; Sain, Z.M.; et al. A Novel Homozygous Missense Mutation in the Zinc Finger DNA Binding Domain of GLI1 Causes Recessive Post-Axial Polydactyly. Front. Genet. 2021, 12, 746949. [Google Scholar] [CrossRef] [PubMed]
- Palencia-Campos, A.; Martínez-Fernández, M.L.; Altunoglu, U.; Soto-Bielicka, P.; Torres, A.; Marín, P.; Aller, E.; Şentürk, L.; Berköz, Ö.; Yıldıran, M.; et al. Heterozygous pathogenic variants in GLI1 are a common finding in isolated postaxial polydactyly A/B. Hum. Mutat. 2020, 41, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Palencia-Campos, A.; Ullah, A.; Nevado, J.; Yildirim, R.; Unal, E.; Ciorraga, M.; Barruz, P.; Chico, L.; Piceci-Sparascio, F.; Guida, V.; et al. GLI1 inactivation is associated with developmental phenotypes overlapping with Ellis-van Creveld syndrome. Hum. Mol. Genet. 2017, 26, 4556–4571. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Umair, M.; Majeed, A.I.; Abdullah; Jan, A.; Ahmad, W. A novel homozygous sequence variant in GLI1 underlies first case of autosomal recessive pre-axial polydactyly. Clin. Genet. 2019, 95, 540–541. [Google Scholar] [CrossRef]
- Zhu, G.; Ke, X.; Liu, Q.; Li, J.; Chen, B.; Shao, C.; Gong, Y. Recurrence of the D100N mutation in a Chinese family with brachydactyly type A1: Evidence for a mutational hot spot in the Indian hedgehog gene. Am. J. Med. Genet. A 2007, 143A, 1246–1248. [Google Scholar] [CrossRef] [PubMed]
- Lodder, E.M.; Hoogeboom, A.J.; Coert, J.H.; de Graaff, E. Deletion of 1 amino acid in Indian hedgehog leads to brachydactylyA1. Am. J. Med. Genet. A 2008, 146A, 2152–2154. [Google Scholar] [CrossRef] [PubMed]
- Hellemans, J.; Coucke, P.J.; Giedion, A.; De Paepe, A.; Kramer, P.; Beemer, F.; Mortier, G.R. Homozygous mutations in IHH cause acrocapitofemoral dysplasia, an autosomal recessive disorder with cone-shaped epiphyses in hands and hips. Am. J. Hum. Genet. 2003, 72, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Zhang, X.M.; Karp, S.; Yang, Y.; McMahon, A.P. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 2001, 128, 5099–5108. [Google Scholar] [CrossRef] [PubMed]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef] [PubMed]
- Canto, P.; Söderlund, D.; Reyes, E.; Méndez, J.P. Mutations in the desert hedgehog (DHH) gene in patients with 46,XY complete pure gonadal dysgenesis. J. Clin. Endocrinol. Metab. 2004, 89, 4480–4483. [Google Scholar] [CrossRef] [PubMed]
- Umehara, F.; Tate, G.; Itoh, K.; Yamaguchi, N.; Douchi, T.; Mitsuya, T.; Osame, M. A novel mutation of desert hedgehog in a patient with 46,XY partial gonadal dysgenesis accompanied by minifascicular neuropathy. Am. J. Hum. Genet. 2000, 67, 1302–1305. [Google Scholar] [CrossRef] [PubMed]
- Parmantier, E.; Lynn, B.; Lawson, D.; Turmaine, M.; Namini, S.S.; Chakrabarti, L.; McMahon, A.P.; Jessen, K.R.; Mirsky, R. Schwann cell-derived Desert hedgehog controls the development of peripheral nerve sheaths. Neuron 1999, 23, 713–724. [Google Scholar] [CrossRef]
- Ruiz-Perez, V.L.; Tompson, S.W.; Blair, H.J.; Espinoza-Valdez, C.; Lapunzina, P.; Silva, E.O.; Hamel, B.; Gibbs, J.L.; Young, I.D.; Wright, M.J.; et al. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am. J. Hum. Genet. 2003, 72, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Taylor, S.P.; Ennis, H.A.; Forlenza, K.N.; Duran, I.; Li, B.; Sanchez, J.A.O.; Nevarez, L.; Nickerson, D.A.; Bamshad, M.; et al. Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies. Hum. Mutat. 2018, 39, 152–166. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene OMIM# | *165230 | *165240 | *600725 | *601309 | *601500 | *607035 | *611254 | ||||||||
| Gene | GLI2 | GLI3 | SHH | PTCH1 | SMO | SUFU | KIF7 | ||||||||
| Location | 2q14.2 | 7p14.1 | 7q36.3 | 9q22.32 | 7q32.1 | 10q24.32 | 15q26.1 | ||||||||
| Phenotype OMIM# | #615849 | #610829 | #175700 | #174200 | #146510 | #142945 | #109400 | #610828 | #241800 | #601707 | #109400 | #617757 | #200990 | #607131 | #614120 |
| Disease | CJS | HPE9 | GCPS | PAPA1 | PHS | HPE3 | BCNS | HPE7 | PHLS | CRJS | BCNS | JBTS32 | ACLS | AGBK | HLS2 |
| Inheritance | AD | AD | AD | AD | AD | AD | AD | AD | AR | Mos | AD | AR | AR | AR | AR |
| Mutation type | LOF | LOF | LOF 1 | LOF | GOF | LOF 2 | LOF | GOF | LOF | GOF | LOF 3 | partial LOF | LOF | LOF | LOF |
| Height | Short | Short | Short | Short | Tall | Short | Normal | ||||||||
| hypopituitalism | Y | Y | Y | Y | Y | ||||||||||
| Head size | Micro | Micro | Macro | Micro | Macro | Macro | Micro | Macro | Macro | Macro | Macro | ||||
| Holoprosencephaly | less common | variable degree | less common | variable degree | variable degree | Anencephaly | |||||||||
| Intellectual disability | some patients | Y | Normal, mild (rare) | Y | less common | Y | Speech delay | mild to moderate | less common | mild | Severe | Y | |||
| Eyes (telorism) | Hypo | Hypo | Hyper | Normal | Hypo | Hypo | Hyper | Hypo | Hyper | Hyper | Hyper | Hyper | |||
| Microphthalmia | Y | Y | Synophthalmia (in some) | Y | |||||||||||
| Mid-Face | hypo | hypo | wide | hypo | wide | hypo | wide | wide | |||||||
| Cleft lip/palate | both | both | both | both | both (5%) | both | Cleft palate | both (5%) | both | Cleft palate | |||||
| SMMCI | Y | Y | Y | ||||||||||||
| Hands Polydactyly | Post-Ax (some) | Post-Ax | Post/Pre-Ax | Post/Pre-Ax | Post-Ax | Post-Ax | Pre-Ax | Post-Ax | Post/Pre-Ax | Post-Ax | |||||
| Hand Syndactyly | Y | Y | Y | ||||||||||||
| Feet polydactyly | Post-Ax (in some) | Post-Ax | Post/Pre-Ax | Post/Pre-Ax | Post-Ax | Post-Ax | Pre-Ax (in some) | Post-Ax | Post/Pre-Ax | Post/Pre-Ax | |||||
| Feet syndactyly | Y | Y | Y | ||||||||||||
| Tumor | HTH | NBCC/DMB/OC | HTH | DMB | NBCC/DMB | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niida, Y.; Togi, S.; Ura, H. Molecular Bases of Human Malformation Syndromes Involving the SHH Pathway: GLIA/R Balance and Cardinal Phenotypes. Int. J. Mol. Sci. 2021, 22, 13060. https://doi.org/10.3390/ijms222313060
Niida Y, Togi S, Ura H. Molecular Bases of Human Malformation Syndromes Involving the SHH Pathway: GLIA/R Balance and Cardinal Phenotypes. International Journal of Molecular Sciences. 2021; 22(23):13060. https://doi.org/10.3390/ijms222313060
Chicago/Turabian StyleNiida, Yo, Sumihito Togi, and Hiroki Ura. 2021. "Molecular Bases of Human Malformation Syndromes Involving the SHH Pathway: GLIA/R Balance and Cardinal Phenotypes" International Journal of Molecular Sciences 22, no. 23: 13060. https://doi.org/10.3390/ijms222313060
APA StyleNiida, Y., Togi, S., & Ura, H. (2021). Molecular Bases of Human Malformation Syndromes Involving the SHH Pathway: GLIA/R Balance and Cardinal Phenotypes. International Journal of Molecular Sciences, 22(23), 13060. https://doi.org/10.3390/ijms222313060