
miRNAs as Therapeutic Tools in Alzheimer’s Disease


Abstract
:1. Introduction
2. Current Treatment for AD
2.1. Pharmacotherapy for Alzheimer’s Disease
2.2. Limitations of Drugs for AD
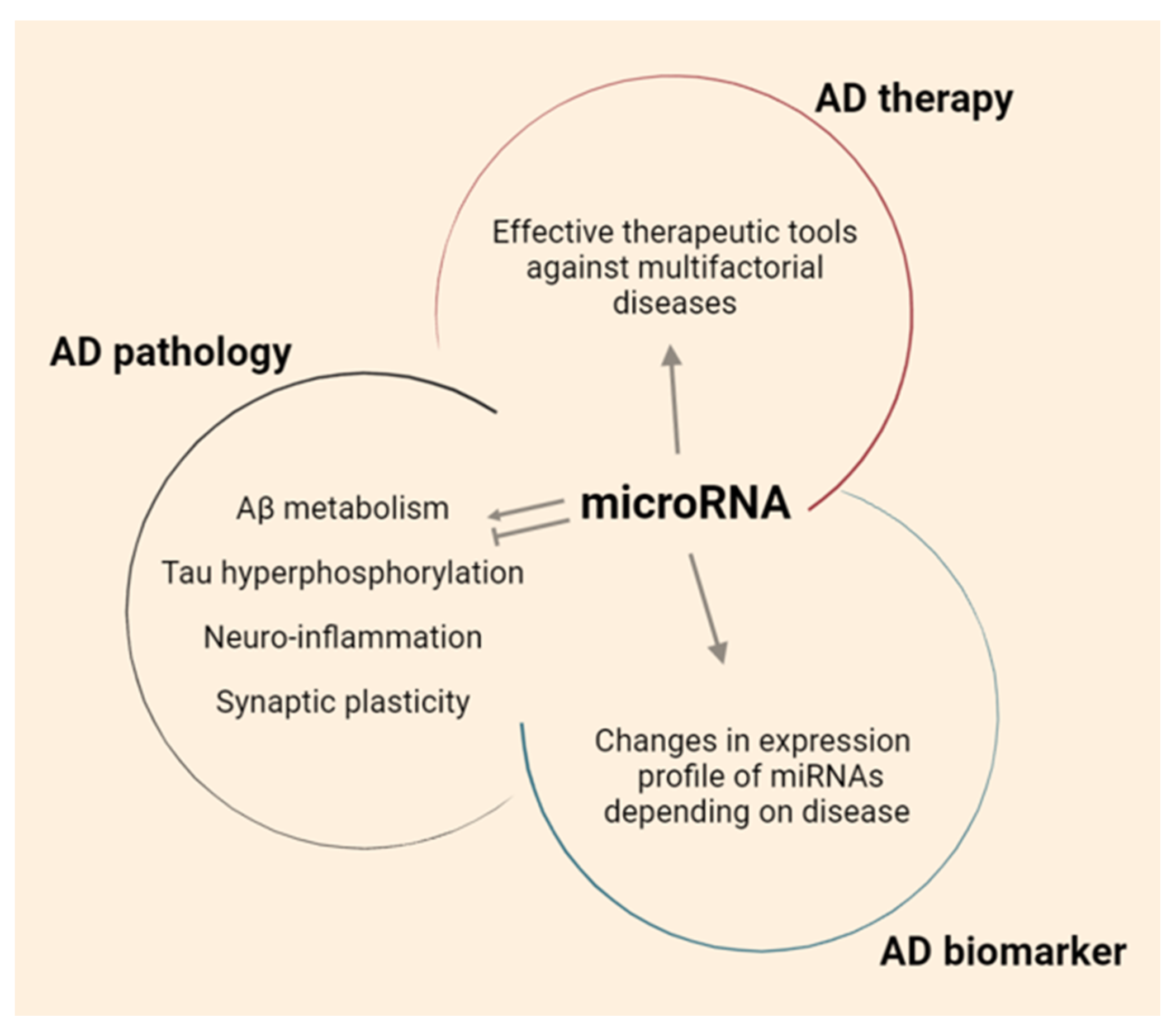
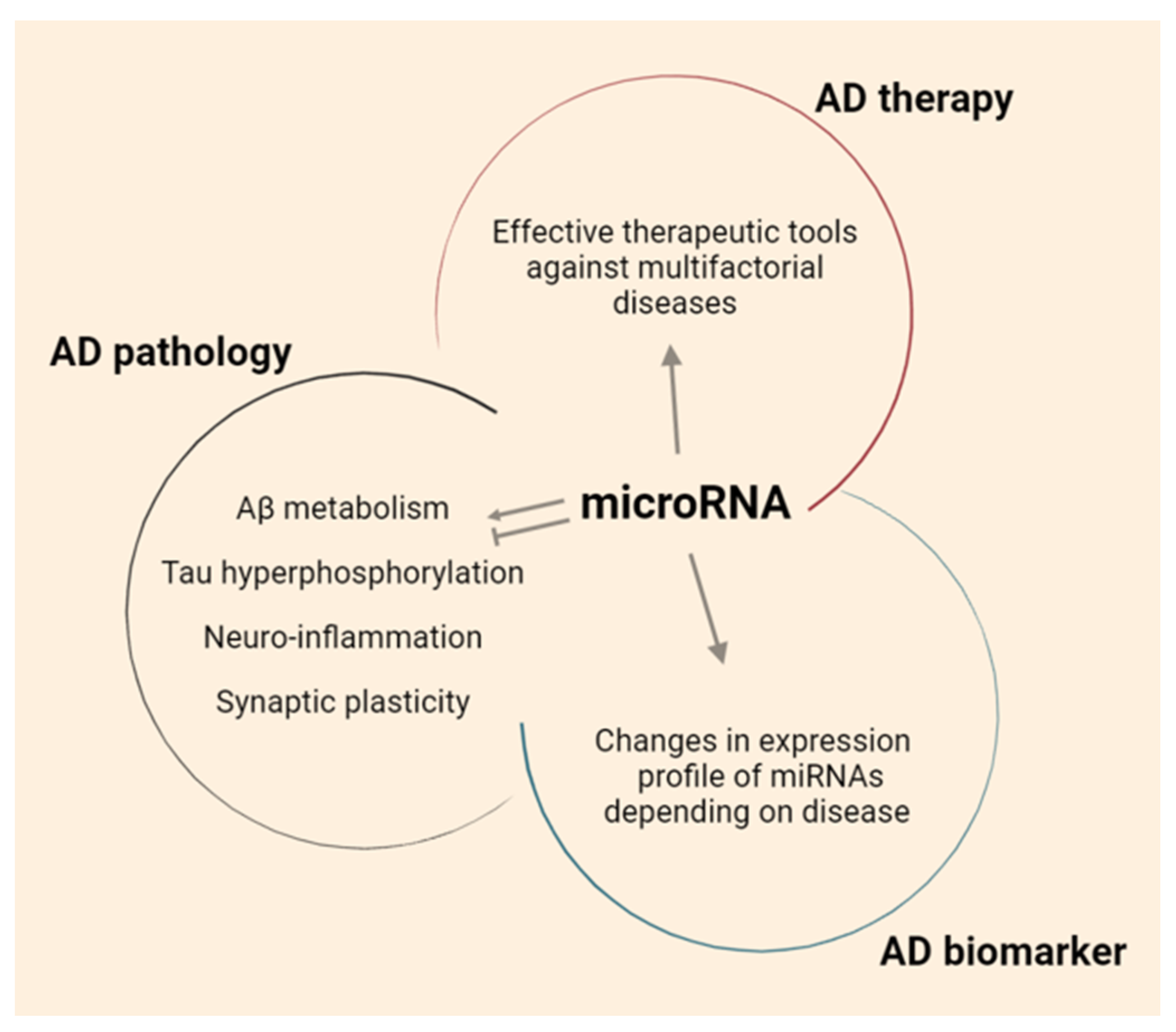
3. Pathological Characteristics of AD and the Role of miRNAs
3.1. miRNAs Regulate Aβ
3.2. miRNAs Regulate Tau
3.3. miRNAs Regulate Neuroinflammation
3.4. miRNAs Regulate Synaptic Function

{kind=link}
| AD Phenotype | miRNA | Target mRNA | Reference |
|---|---|---|---|
| Aβ | miR-16, miR-17, miR-20a, miR-101, miR-106a, miR-106b, miR-147, miR-153, miR-520c | APP | [57,58,59,60] |
| miR-15a, miR-15b, miR-29a, miR-29b, miR-29c, miR-103, miR-107, miR-298, miR-328, miR-195, miR-485 | BACE1 | [63,64,65,66,67] | |
| miR-128 | Cathepsin B, D, S | [74] | |
| Tau | miR-128 | BAG2 | [83] |
| miR-9 | SIRT1 | [85] | |
| miR-922, miR-181b | UCHL1 | [88,119] | |
| miR-124 | Caveolin-1 | [120] | |
| miR-132 | ITPKB | [121] | |
| miR-34a | Tau | [122] | |
| miR-26b | Rb1 | [86] | |
| Neuroinflammation | miR-7, miR-223 | NLRP3 | [98,123] |
| miR-9 | NLRP1 | [99] | |
| miR-22 | GSDMD | [102] | |
| let-7 family | TLR4 | [124] | |
| IL-6 | [125] | ||
| miR-485 | AKT3 | [126] | |
| Synaptic function | miR-26b, miR-206 | IGF1 | [103] |
| miR-132, miR-206 | BDNF | [104] | |
| miR-134 | LIMK1, PUM2 | [105] | |
| miR-9 | REST | [108] | |
| miR-210 | Snap25 | [109] | |
| miR-485 | SV2A | [110] | |
| miR-34c | VAMP2 | [111] | |
| miR-92, miR-137, miR-501 | GRIA1 | [113,114,115] | |
| miR-181a | GRIA2 | [116] | |
| miR-125b | GRIN2A | [117] | |
| miR-34a | GRIN2B | [118] |
4. miRNA as Diagnostic AD Biomarker
5. Limitation of mRNAs
6. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Apostolova, L.G. Alzheimer Disease. Continuum 2016, 22, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement. 2013, 9, 63–75.e2. [Google Scholar] [CrossRef]
- Fotenos, A.F.; Mintun, M.A.; Snyder, A.Z.; Morris, J.C.; Buckner, R.L. Brain volume decline in aging: Evidence for a relation between socioeconomic status, preclinical Alzheimer disease, and reserve. Arch. Neurol. 2008, 65, 113–120. [Google Scholar] [CrossRef]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Coon, K.D.; Myers, A.J.; Craig, D.W.; Webster, J.A.; Pearson, J.V.; Lince, D.H.; Zismann, V.L.; Beach, T.G.; Leung, D.; Bryden, L.; et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J. Clin. Psychiatry 2007, 68, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Wijesekara, N.; Ahrens, R.; Sabale, M.; Wu, L.; Ha, K.; Verdile, G.; Fraser, P.E. Amyloid-beta and islet amyloid pathologies link Alzheimer’s disease and type 2 diabetes in a transgenic model. FASEB J. 2017, 31, 5409–5418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.Y.; Yamauchi, Y.; Hasan, M.T.; Chang, C. Cellular cholesterol homeostasis and Alzheimer’s disease. J. Lipid Res. 2017, 58, 2239–2254. [Google Scholar] [CrossRef] [Green Version]
- Stampfer, M.J. Cardiovascular disease and Alzheimer’s disease: Common links. J. Intern. Med. 2006, 260, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Iqbal, K.; Alonso Adel, C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesulam, M.M. Neuroplasticity failure in Alzheimer’s disease: Bridging the gap between plaques and tangles. Neuron 1999, 24, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Idda, M.L.; Munk, R.; Abdelmohsen, K.; Gorospe, M. Noncoding RNAs in Alzheimer’s disease. Wiley Interdiscip. Rev. RNA 2018, 9, e1463. [Google Scholar] [CrossRef]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2001, 2, 919–929. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Kong, W.; He, L.; Zhao, J.J.; O’Donnell, J.D.; Wang, J.; Wenham, R.M.; Coppola, D.; Kruk, P.A.; Nicosia, S.V.; et al. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008, 68, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.; Lee, C.Y.; Park, J.H.; Park, M.S.; Maeng, L.S.; Yoon, C.S.; Lee, M.Y.; Hwang, K.C.; Chung, Y.A. Survival of hypoxic human mesenchymal stem cells is enhanced by a positive feedback loop involving miR-210 and hypoxia-inducible factor 1. J. Vet. Sci. 2013, 14, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Fu, G.; Brkic, J.; Hayder, H.; Peng, C. MicroRNAs in Human Placental Development and Pregnancy Complications. Int. J. Mol. Sci. 2013, 14, 5519–5544. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.Y.; Shin, S.; Lee, J.; Seo, H.H.; Lim, K.H.; Kim, H.; Choi, J.W.; Kim, S.W.; Lee, S.; Lim, S.; et al. MicroRNA-Mediated Down-Regulation of Apoptosis Signal-Regulating Kinase 1 (ASK1) Attenuates the Apoptosis of Human Mesenchymal Stem Cells (MSCs) Transplanted into Infarcted Heart. Int. J. Mol. Sci. 2016, 17, 1752. [Google Scholar] [CrossRef] [Green Version]
- Paul, P.; Chakraborty, A.; Sarkar, D.; Langthasa, M.; Rahman, M.; Bari, M.; Singha, R.S.; Malakar, A.K.; Chakraborty, S. Interplay between miRNAs and human diseases. J. Cell Physiol. 2018, 233, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Tufekci, K.U.; Oner, M.G.; Meuwissen, R.L.; Genc, S. The role of microRNAs in human diseases. Methods Mol. Biol. 2014, 1107, 33–50. [Google Scholar]
- Wang, M.; Qin, L.; Tang, B. MicroRNAs in Alzheimer’s Disease. Front. Genet. 2019, 10, 153. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal. Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Cheng, Y.; Yang, J.; Li, J.; Liu, X.; Wang, X.; Wang, D.; Krall, T.J.; Delphin, E.S.; Zhang, C. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J. Biol. Chem. 2009, 284, 29514–29525. [Google Scholar] [CrossRef] [Green Version]
- Kontaraki, J.E.; Marketou, M.E.; Zacharis, E.A.; Parthenakis, F.I.; Vardas, P.E. MicroRNA-9 and microRNA-126 expression levels in patients with essential hypertension: Potential markers of target-organ damage. J. Am. Soc. Hypertens. 2014, 8, 368–375. [Google Scholar] [CrossRef]
- Anaparti, V.; Smolik, I.; Meng, X.; Spicer, V.; Mookherjee, N.; El-Gabalawy, H. Whole blood microRNA expression pattern differentiates patients with rheumatoid arthritis, their seropositive first-degree relatives, and healthy unrelated control subjects. Arthritis Res. Ther. 2017, 19, 249. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.H.; Gao, Y.; Ren, A.J.; Zhao, S.H.; Zhong, M.; Peng, Y.J.; Shen, W.; Jing, M.; Liu, L. Altered microRNA expression profiles in retinas with diabetic retinopathy. Ophthalmic Res. 2012, 47, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.J.; Moreno-Navarrete, J.M.; Pardo, G.; Sabater, M.; Hummel, M.; Ferrer, A.; Rodriguez-Hermosa, J.I.; Ruiz, B.; Ricart, W.; Peral, B.; et al. MiRNA expression profile of human subcutaneous adipose and during adipocyte differentiation. PLoS ONE 2010, 5, e9022. [Google Scholar] [CrossRef] [Green Version]
- Fehlmann, T.; Lehallier, B.; Schaum, N.; Hahn, O.; Kahraman, M.; Li, Y.; Grammes, N.; Geffers, L.; Backes, C.; Balling, R.; et al. Common diseases alter the physiological age-related blood microRNA profile. Nat. Commun. 2020, 11, 5958. [Google Scholar] [CrossRef]
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 2015, 6, 37043–37053. [Google Scholar] [CrossRef] [Green Version]
- Sproviero, D.; Gagliardi, S.; Zucca, S.; Arigoni, M.; Giannini, M.; Garofalo, M.; Olivero, M.; Dell’Orco, M.; Pansarasa, O.; Bernuzzi, S.; et al. Different miRNA Profiles in Plasma Derived Small and Large Extracellular Vesicles from Patients with Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 2737. [Google Scholar] [CrossRef]
- Shamsuzzama; Kumar, L.; Haque, R.; Nazir, A. Role of MicroRNA Let-7 in Modulating Multifactorial Aspect of Neurodegenerative Diseases: An Overview. Mol. Neurobiol. 2016, 53, 2787–2793. [Google Scholar] [CrossRef]
- Christopher, A.F.; Kaur, R.P.; Kaur, G.; Kaur, A.; Gupta, V.; Bansal, P. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect. Clin. Res. 2016, 7, 68–74. [Google Scholar]
- Birks, J.S.; Harvey, R.J. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst. Rev. 2018, 6, CD001190. [Google Scholar] [CrossRef]
- Shintani, E.Y.; Uchida, K.M. Donepezil: An anticholinesterase inhibitor for Alzheimer’s disease. Am. J. Health Syst. Pharm. 1997, 54, 2805–2810. [Google Scholar] [CrossRef]
- McShane, R.; Westby, M.J.; Roberts, E.; Minakaran, N.; Schneider, L.; Farrimond, L.E.; Maayan, N.; Ware, J.; Debarros, J. Memantine for dementia. Cochrane Database Syst. Rev. 2019, 3, CD003154. [Google Scholar] [CrossRef]
- Popik, P.; Wrobel, M.; Rygula, R.; Bisaga, A.; Bespalov, A.Y. Effects of memantine, an NMDA receptor antagonist, on place preference conditioned with drug and nondrug reinforcers in mice. Behav. Pharmacol. 2003, 14, 237–244. [Google Scholar] [CrossRef]
- Knowles, J. Donepezil in Alzheimer’s disease: An evidence-based review of its impact on clinical and economic outcomes. Core Evid. 2006, 1, 195–219. [Google Scholar]
- Tampi, R.R.; van Dyck, C.H. Memantine: Efficacy and safety in mild-to-severe Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2007, 3, 245–258. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Liu, K.Y.; Schneider, L.S.; Howard, R. The need to show minimum clinically important differences in Alzheimer’s disease trials. Lancet Psychiatry 2021, 8, 1013–1016. [Google Scholar] [CrossRef]
- Dunn, B.; Stein, P.; Cavazzoni, P. Approval of Aducanumab for Alzheimer Disease-The FDA’s Perspective. JAMA Intern. Med. 2021, 181, 1276–1278. [Google Scholar] [CrossRef]
- Mullard, A. Landmark Alzheimer’s drug approval confounds research community. Nature 2021, 594, 309–310. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Yu, J.T.; Hu, N.; Tan, L. Non-coding RNAs in Alzheimer’s disease. Mol. Neurobiol. 2013, 47, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport 2007, 18, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Hebert, S.S.; Horre, K.; Nicolai, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef] [Green Version]
- Miya Shaik, M.; Tamargo, I.A.; Abubakar, M.B.; Kamal, M.A.; Greig, N.H.; Gan, S.H. The Role of microRNAs in Alzheimer’s Disease and Their Therapeutic Potentials. Genes 2018, 9, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swarbrick, S.; Wragg, N.; Ghosh, S.; Stolzing, A. Systematic Review of miRNA as Biomarkers in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 6156–6167. [Google Scholar] [CrossRef] [Green Version]
- Schipper, H.M.; Maes, O.C.; Chertkow, H.M.; Wang, E. MicroRNA expression in Alzheimer blood mononuclear cells. Gene Regul. Syst. Biol. 2007, 1, 263–274. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Coronel, R.; Bernabeu-Zornoza, A.; Palmer, C.; Muniz-Moreno, M.; Zambrano, A.; Cano, E.; Liste, I. Role of Amyloid Precursor Protein (APP) and Its Derivatives in the Biology and Cell Fate Specification of Neural Stem Cells. Mol. Neurobiol. 2018, 55, 7107–7117. [Google Scholar] [CrossRef] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.Y.; Hyman, B.T.; Bacskai, B.J. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef] [Green Version]
- Hunt, D.L.; Castillo, P.E. Synaptic plasticity of NMDA receptors: Mechanisms and functional implications. Curr. Opin. Neurobiol. 2012, 22, 496–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, S.S.; Horre, K.; Nicolai, L.; Bergmans, B.; Papadopoulou, A.S.; Delacourte, A.; De Strooper, B. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol. Dis. 2009, 33, 422–428. [Google Scholar] [CrossRef]
- Patel, N.; Hoang, D.; Miller, N.; Ansaloni, S.; Huang, Q.; Rogers, J.T.; Lee, J.C.; Saunders, A.J. MicroRNAs can regulate human APP levels. Mol. Neurodegener. 2008, 3, 10. [Google Scholar] [CrossRef] [Green Version]
- Vilardo, E.; Barbato, C.; Ciotti, M.; Cogoni, C.; Ruberti, F. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J. Biol. Chem. 2010, 285, 18344–18351. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Deng, W.; Liu, Y.; Qin, C. MicroRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Res. 2012, 1455, 103–113. [Google Scholar] [CrossRef]
- Delay, C.; Calon, F.; Mathews, P.; Hebert, S.S. Alzheimer-specific variants in the 3’UTR of Amyloid precursor protein affect microRNA function. Mol. Neurodegener. 2011, 6, 70. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; He, P.; Yao, H.; Dong, Q.; Li, R.; Shen, Y. Occludin deficiency with BACE1 elevation in cerebral amyloid angiopathy. Neurology 2014, 82, 1707–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, F.; Gong, G.; Wang, Y.; Bian, M.; Yu, L.; Wei, C. MiR-124 acts as a target for Alzheimer’s disease by regulating BACE1. Oncotarget 2017, 8, 114065–114071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, N.; Wang, R.; Maloney, B.; Nho, K.; Beck, J.S.; Pourshafie, N.; Niculescu, A.; Saykin, A.J.; Rinaldi, C.; Counts, S.E.; et al. MicroRNA-298 reduces levels of human amyloid-beta precursor protein (APP), beta-site APP-converting enzyme 1 (BACE1) and specific tau protein moieties. Mol. Psychiatry 2020, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Ding, Y.; Hou, D. Research status of the regulation of miRNA on BACE1. Int. J. Neurosci. 2014, 124, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Zhao, Y.; Zhou, Y.; Liu, L.; Liu, Y.; Wang, D.; Zhang, S.; Yang, M. MiR-9 Regulates the Expression of BACE1 in Dementia Induced by Chronic Brain Hypoperfusion in Rats. Cell Physiol. Biochem. 2017, 42, 1213–1226. [Google Scholar] [CrossRef] [Green Version]
- Faghihi, M.A.; Zhang, M.; Huang, J.; Modarresi, F.; Van der Brug, M.P.; Nalls, M.A.; Cookson, M.R.; St-Laurent, G., 3rd; Wahlestedt, C. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010, 11, R56. [Google Scholar] [CrossRef] [Green Version]
- Boissonneault, V.; Plante, I.; Rivest, S.; Provost, P. MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J. Biol. Chem. 2009, 284, 1971–1981. [Google Scholar] [CrossRef] [Green Version]
- Gong, G.; An, F.; Wang, Y.; Bian, M.; Yu, L.J.; Wei, C. miR-15b represses BACE1 expression in sporadic Alzheimer’s disease. Oncotarget 2017, 8, 91551–91557. [Google Scholar] [CrossRef] [Green Version]
- Moncini, S.; Lunghi, M.; Valmadre, A.; Grasso, M.; Del Vescovo, V.; Riva, P.; Denti, M.A.; Venturin, M. The miR-15/107 Family of microRNA Genes Regulates CDK5R1/p35 with Implications for Alzheimer’s Disease Pathogenesis. Mol. Neurobiol. 2017, 54, 4329–4342. [Google Scholar] [CrossRef]
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.X. MicroRNA-195 downregulates Alzheimer’s disease amyloid-beta production by targeting BACE1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Lei, L.; Zhang, Z.; Zhang, Z.; Cheng, Y. Downregulated miR-29c correlates with increased BACE1 expression in sporadic Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2015, 8, 1565–1574. [Google Scholar] [PubMed]
- Sun, B.; Zhou, Y.; Halabisky, B.; Lo, I.; Cho, S.H.; Mueller-Steiner, S.; Devidze, N.; Wang, X.; Grubb, A.; Gan, L. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer’s disease. Neuron 2008, 60, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Tiribuzi, R.; Crispoltoni, L.; Porcellati, S.; Di Lullo, M.; Florenzano, F.; Pirro, M.; Bagaglia, F.; Kawarai, T.; Zampolini, M.; Orlacchio, A.; et al. miR128 up-regulation correlates with impaired amyloid beta(1-42) degradation in monocytes from patients with sporadic Alzheimer’s disease. Neurobiol. Aging 2014, 35, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Ishiguro, K.; Hisanaga, S. Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci. 2014, 7, 65. [Google Scholar] [CrossRef] [Green Version]
- Jara, C.; Aranguiz, A.; Cerpa, W.; Tapia-Rojas, C.; Quintanilla, R.A. Genetic ablation of tau improves mitochondrial function and cognitive abilities in the hippocampus. Redox Biol. 2018, 18, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M.; Binder, L.I. Polymerization of microtubule-associated protein tau under near-physiological conditions. J. Biol. Chem. 1995, 270, 24306–24314. [Google Scholar] [CrossRef] [Green Version]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [Green Version]
- Callahan, L.M.; Vaules, W.A.; Coleman, P.D. Quantitative decrease in synaptophysin message expression and increase in cathepsin D message expression in Alzheimer disease neurons containing neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 1999, 58, 275–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.S.; Bloom, G.S. Tau: The Center of a Signaling Nexus in Alzheimer’s Disease. Front. Neurosci. 2016, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Carrettiero, D.C.; Hernandez, I.; Neveu, P.; Papagiannakopoulos, T.; Kosik, K.S. The cochaperone BAG2 sweeps paired helical filament- insoluble tau from the microtubule. J. Neurosci. 2009, 29, 2151–2161. [Google Scholar] [CrossRef]
- Hebert, S.S.; Sergeant, N.; Buee, L. MicroRNAs and the Regulation of Tau Metabolism. Int. J. Alzheimer’s Dis. 2012, 2012, 406561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, D.; Roy, U.; Garg, S.; Ghosh, S.; Pathak, S.; Kolthur-Seetharam, U. Sirt1 and mir-9 expression is regulated during glucose-stimulated insulin secretion in pancreatic beta-islets. FEBS J. 2011, 278, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Absalon, S.; Kochanek, D.M.; Raghavan, V.; Krichevsky, A.M. MiR-26b, upregulated in Alzheimer’s disease, activates cell cycle entry, tau-phosphorylation, and apoptosis in postmitotic neurons. J. Neurosci. 2013, 33, 14645–14659. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Tang, H.; Li, X.Y.; Deng, M.F.; Wei, N.; Wang, X.; Zhou, Y.F.; Wang, D.Q.; Fu, P.; Wang, J.Z.; et al. Targeting the HDAC2/HNF-4A/miR-101b/AMPK Pathway Rescues Tauopathy and Dendritic Abnormalities in Alzheimer’s Disease. Mol. Ther. 2017, 25, 752–764. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.B.; Wu, L.; Xiong, R.; Wang, L.L.; Zhang, B.; Wang, C.; Li, H.; Liang, L.; Chen, S.D. MicroRNA-922 promotes tau phosphorylation by downregulating ubiquitin carboxy-terminal hydrolase L1 (UCHL1) expression in the pathogenesis of Alzheimer’s disease. Neuroscience 2014, 275, 232–237. [Google Scholar] [CrossRef]
- Wang, X.; Tan, L.; Lu, Y.; Peng, J.; Zhu, Y.; Zhang, Y.; Sun, Z. MicroRNA-138 promotes tau phosphorylation by targeting retinoic acid receptor alpha. FEBS Lett. 2015, 589, 726–729. [Google Scholar] [CrossRef] [Green Version]
- Craft, J.M.; Watterson, D.M.; Van Eldik, L.J. Human amyloid beta-induced neuroinflammation is an early event in neurodegeneration. Glia 2006, 53, 484–490. [Google Scholar] [CrossRef]
- Pizza, V.; Agresta, A.; D’Acunto, C.W.; Festa, M.; Capasso, A. Neuroinflamm-aging and neurodegenerative diseases: An overview. CNS Neurol. Disord Drug Targets 2011, 10, 621–634. [Google Scholar] [CrossRef]
- Varnum, M.M.; Ikezu, T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch. Immunol. Ther. Exp. 2012, 60, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Fillit, H.; Ding, W.H.; Buee, L.; Kalman, J.; Altstiel, L.; Lawlor, B.; Wolf-Klein, G. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci. Lett. 1991, 129, 318–320. [Google Scholar] [CrossRef]
- Tan, M.S.; Yu, J.T.; Jiang, T.; Zhu, X.C.; Tan, L. The NLRP3 inflammasome in Alzheimer’s disease. Mol. Neurobiol. 2013, 48, 875–882. [Google Scholar] [CrossRef]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lu, M.; Du, R.H.; Qiao, C.; Jiang, C.Y.; Zhang, K.Z.; Ding, J.H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Zhang, H.; Lu, X.; Wang, J.; Zhang, X.; Sun, S.; Bao, Z.; Tian, W.; Ning, S.; Wang, L.; et al. Overexpression of MicroRNA-9a-5p Ameliorates NLRP1 Inflammasome-mediated Ischemic Injury in Rats Following Ischemic Stroke. Neuroscience 2020, 444, 106–117. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Guo, L.; Yang, Y.; Guan, Q.; Shen, H.; Sheng, Y.; Jiao, Q. Mechanism of microRNA-22 in regulating neuroinflammation in Alzheimer’s disease. Brain Behav. 2020, 10, e01627. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chu, W.; Gong, L.; Gao, X.; Wang, W. MicroRNA-26b is upregulated in a double transgenic mouse model of Alzheimer’s disease and promotes the expression of amyloid-beta by targeting insulin-like growth factor 1. Mol. Med. Rep. 2016, 13, 2809–2814. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.T.; Chu, K.; Jung, K.H.; Kim, J.H.; Huh, J.Y.; Yoon, H.; Park, D.K.; Lim, J.Y.; Kim, J.M.; Jeon, D.; et al. miR-206 regulates brain-derived neurotrophic factor in Alzheimer disease model. Ann. Neurol. 2012, 72, 269–277. [Google Scholar] [CrossRef]
- Park, I.; Kim, H.J.; Kim, Y.; Hwang, H.S.; Kasai, H.; Kim, J.H.; Park, J.W. Nanoscale imaging reveals miRNA-mediated control of functional states of dendritic spines. Proc. Natl. Acad. Sci. USA 2019, 116, 9616–9621. [Google Scholar] [CrossRef] [Green Version]
- Schratt, G.M.; Tuebing, F.; Nigh, E.A.; Kane, C.G.; Sabatini, M.E.; Kiebler, M.; Greenberg, M.E. A brain-specific microRNA regulates dendritic spine development. Nature 2006, 439, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Fiore, R.; Rajman, M.; Schwale, C.; Bicker, S.; Antoniou, A.; Bruehl, C.; Draguhn, A.; Schratt, G. MiR-134-dependent regulation of Pumilio-2 is necessary for homeostatic synaptic depression. EMBO J. 2014, 33, 2231–2246. [Google Scholar] [CrossRef] [Green Version]
- Giusti, S.A.; Vogl, A.M.; Brockmann, M.M.; Vercelli, C.A.; Rein, M.L.; Trumbach, D.; Wurst, W.; Cazalla, D.; Stein, V.; Deussing, J.M.; et al. MicroRNA-9 controls dendritic development by targeting REST. Elife 2014, 3, e02755. [Google Scholar] [CrossRef]
- Ren, Z.; Yu, J.; Wu, Z.; Si, W.; Li, X.; Liu, Y.; Zhou, J.; Deng, R.; Chen, D. MicroRNA-210-5p Contributes to Cognitive Impairment in Early Vascular Dementia Rat Model Through Targeting Snap25. Front. Mol. Neurosci. 2018, 11, 388. [Google Scholar] [CrossRef]
- Cohen, J.E.; Lee, P.R.; Chen, S.; Li, W.; Fields, R.D. MicroRNA regulation of homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 11650–11655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Wang, H.; Chen, K.; Cheng, P.; Gao, S.; Liu, J.; Li, X.; Sun, X. MicroRNA-34c Downregulation Ameliorates Amyloid-beta-Induced Synaptic Failure and Memory Deficits by Targeting VAMP2. J. Alzheimer’s Dis. 2015, 48, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Sim, S.E.; Lim, C.S.; Kim, J.I.; Seo, D.; Chun, H.; Yu, N.K.; Lee, J.; Kang, S.J.; Ko, H.G.; Choi, J.H.; et al. The Brain-Enriched MicroRNA miR-9-3p Regulates Synaptic Plasticity and Memory. J. Neurosci. 2016, 36, 8641–8652. [Google Scholar] [CrossRef] [PubMed]
- Letellier, M.; Elramah, S.; Mondin, M.; Soula, A.; Penn, A.; Choquet, D.; Landry, M.; Thoumine, O.; Favereaux, A. miR-92a regulates expression of synaptic GluA1-containing AMPA receptors during homeostatic scaling. Nat. Neurosci. 2014, 17, 1040–1042. [Google Scholar] [CrossRef]
- Hu, Z.; Zhao, J.; Hu, T.; Luo, Y.; Zhu, J.; Li, Z. miR-501-3p mediates the activity-dependent regulation of the expression of AMPA receptor subunit GluA1. J. Cell Biol. 2015, 208, 949–959. [Google Scholar] [CrossRef] [Green Version]
- Olde Loohuis, N.F.; Ba, W.; Stoerchel, P.H.; Kos, A.; Jager, A.; Schratt, G.; Martens, G.J.; van Bokhoven, H.; Nadif Kasri, N.; Aschrafi, A. MicroRNA-137 Controls AMPA-Receptor-Mediated Transmission and mGluR-Dependent LTD. Cell Rep. 2015, 11, 1876–1884. [Google Scholar] [CrossRef] [Green Version]
- Baby, N.; Alagappan, N.; Dheen, S.T.; Sajikumar, S. MicroRNA-134-5p inhibition rescues long-term plasticity and synaptic tagging/capture in an Abeta(1-42)-induced model of Alzheimer’s disease. Aging Cell 2020, 19, e13046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edbauer, D.; Neilson, J.R.; Foster, K.A.; Wang, C.F.; Seeburg, D.P.; Batterton, M.N.; Tada, T.; Dolan, B.M.; Sharp, P.A.; Sheng, M. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 2010, 65, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Engler-Chiurazzi, E.B.; Cavendish, J.Z.; Povroznik, J.M.; Russell, A.E.; Quintana, D.D.; Mathers, P.H.; Simpkins, J.W. Over-expression of miR-34a induces rapid cognitive impairment and Alzheimer’s disease-like pathology. Brain Res. 2019, 1721, 146327. [Google Scholar] [CrossRef]
- Peng, Z.; Li, J.; Li, Y.; Yang, X.; Feng, S.; Han, S.; Li, J. Downregulation of miR-181b in mouse brain following ischemic stroke induces neuroprotection against ischemic injury through targeting heat shock protein A5 and ubiquitin carboxyl-terminal hydrolase isozyme L1. J. Neurosci. Res. 2013, 91, 1349–1362. [Google Scholar] [CrossRef]
- Kang, Q.; Xiang, Y.; Li, D.; Liang, J.; Zhang, X.; Zhou, F.; Qiao, M.; Nie, Y.; He, Y.; Cheng, J.; et al. MiR-124-3p attenuates hyperphosphorylation of Tau protein-induced apoptosis via caveolin-1-PI3K/Akt/GSK3beta pathway in N2a/APP695swe cells. Oncotarget 2017, 8, 24314–24326. [Google Scholar] [CrossRef]
- Salta, E.; Sierksma, A.; Vanden Eynden, E.; De Strooper, B. miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in Alzheimer’s brain. EMBO Mol. Med. 2016, 8, 1005–1018. [Google Scholar] [CrossRef]
- Dickson, J.R.; Kruse, C.; Montagna, D.R.; Finsen, B.; Wolfe, M.S. Alternative polyadenylation and miR-34 family members regulate tau expression. J. Neurochem. 2013, 127, 739–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauernfeind, F.; Rieger, A.; Schildberg, F.A.; Knolle, P.A.; Schmid-Burgk, J.L.; Hornung, V. NLRP3 inflammasome activity is negatively controlled by miR-223. J. Immunol. 2012, 189, 4175–4181. [Google Scholar] [CrossRef] [Green Version]
- Teng, G.G.; Wang, W.H.; Dai, Y.; Wang, S.J.; Chu, Y.X.; Li, J. Let-7b is involved in the inflammation and immune responses associated with Helicobacter pylori infection by targeting Toll-like receptor 4. PLoS ONE 2013, 8, e56709. [Google Scholar]
- Schulte, L.N.; Eulalio, A.; Mollenkopf, H.J.; Reinhardt, R.; Vogel, J. Analysis of the host microRNA response to Salmonella uncovers the control of major cytokines by the let-7 family. EMBO J. 2011, 30, 1977–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Li, H.; Liu, W.; Zhang, L.; Tian, Q.; Li, H.; Li, M. MiR-485-3p serves as a biomarker and therapeutic target of Alzheimer’s disease via regulating neuronal cell viability and neuroinflammation by targeting AKT3. Mol. Genet. Genom. Med. 2021, 9, e1548. [Google Scholar] [CrossRef]
- Norton, S.; Matthews, F.E.; Barnes, D.E.; Yaffe, K.; Brayne, C. Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol. 2014, 13, 788–794. [Google Scholar] [CrossRef] [Green Version]
- Dolgin, E. Alzheimer’s disease is getting easier to spot. Nature 2018, 559, S10–S12. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Bhatnagar, S.; Chertkow, H.; Schipper, H.M.; Yuan, Z.; Shetty, V.; Jenkins, S.; Jones, T.; Wang, E. Increased microRNA-34c abundance in Alzheimer’s disease circulating blood plasma. Front. Mol. Neurosci. 2014, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chen, C.; Zhang, Y. An investigation of microRNA-103 and microRNA-107 as potential blood-based biomarkers for disease risk and progression of Alzheimer’s disease. J. Clin. Lab. Anal. 2020, 34, e23006. [Google Scholar] [CrossRef] [Green Version]
- Souza, V.C.; Morais, G.S., Jr.; Henriques, A.D.; Machado-Silva, W.; Perez, D.I.V.; Brito, C.J.; Camargos, E.F.; Moraes, C.F.; Nobrega, O.T. Whole-Blood Levels of MicroRNA-9 Are Decreased in Patients with Late-Onset Alzheimer Disease. Am. J. Alzheimer’s Dis. Other Dement. 2020, 35, 1533317520911573. [Google Scholar] [CrossRef] [PubMed]
- Loganantharaj, R.; Randall, T.A. The Limitations of Existing Approaches in Improving MicroRNA Target Prediction Accuracy. Methods Mol. Biol. 2017, 1617, 133–158. [Google Scholar]
- Guo, L.; Zhang, Q.; Ma, X.; Wang, J.; Liang, T. miRNA and mRNA expression analysis reveals potential sex-biased miRNA expression. Sci. Rep. 2017, 7, 39812. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Yang, W.; Shi, J.; Zhou, Y.; Yang, J.; Cui, Q.; Zhou, Y. Identification and Analysis of Human Sex-biased MicroRNAs. Genom. Proteom. Bioinform. 2018, 16, 200–211. [Google Scholar] [CrossRef]
- Huan, T.; Chen, G.; Liu, C.; Bhattacharya, A.; Rong, J.; Chen, B.H.; Seshadri, S.; Tanriverdi, K.; Freedman, J.E.; Larson, M.G.; et al. Age-associated microRNA expression in human peripheral blood is associated with all-cause mortality and age-related traits. Aging Cell 2018, 17, e12687. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Abdelmohsen, K.; Gorospe, M.; Ejiogu, N.; Zonderman, A.B.; Evans, M.K. microRNA expression patterns reveal differential expression of target genes with age. PLoS ONE 2010, 5, e10724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Qin, Y.W.; Brewer, G.; Jing, Q. MicroRNA degradation and turnover: Regulating the regulators. Wiley Interdiscip. Rev. RNA 2012, 3, 593–600. [Google Scholar] [CrossRef] [Green Version]
- Godoy, P.M.; Bhakta, N.R.; Barczak, A.J.; Cakmak, H.; Fisher, S.; MacKenzie, T.C.; Patel, T.; Price, R.W.; Smith, J.F.; Woodruff, P.G.; et al. Large Differences in Small RNA Composition Between Human Biofluids. Cell Rep. 2018, 25, 1346–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshiol, J.; Wang, E.; Zhao, Y.; Marincola, F.; Landi, M.T. Strengths and limitations of laboratory procedures for microRNA detection. Cancer Epidemiol. Biomark. Prev. 2010, 19, 907–911. [Google Scholar] [CrossRef] [Green Version]
| Drug Name | Active Ingredient | Action | Indication | FDA Approval |
|---|---|---|---|---|
| Aricept | Donepezil | AChE inhibition | Mild to severe AD | 1996 |
| Exelon | Rivastigmine | AChE and BuChe inhibition | Mild to moderate AD | 1997 |
| Razadyne | Galantamine | AChE inhibition Nicotinic receptor-like structure | Mild to moderate AD | 2001 |
| Ebixa | Memantine | NMDA receptor antagonists Inhibition of hyperactivity of glutamate | Moderate to severe AD | 2003 |
| Aduhelm | Aducanumab | Aβ-directed monoclonal antibody | Mild cognitive impairment or mild dementia | 2021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.Y.; Ryu, I.S.; Ryu, J.-H.; Cho, H.-J. miRNAs as Therapeutic Tools in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 13012. https://doi.org/10.3390/ijms222313012
Lee CY, Ryu IS, Ryu J-H, Cho H-J. miRNAs as Therapeutic Tools in Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(23):13012. https://doi.org/10.3390/ijms222313012
Chicago/Turabian StyleLee, Chang Youn, In Soo Ryu, Jin-Hyeob Ryu, and Hyun-Jeong Cho. 2021. "miRNAs as Therapeutic Tools in Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 23: 13012. https://doi.org/10.3390/ijms222313012
APA StyleLee, C. Y., Ryu, I. S., Ryu, J.-H., & Cho, H.-J. (2021). miRNAs as Therapeutic Tools in Alzheimer’s Disease. International Journal of Molecular Sciences, 22(23), 13012. https://doi.org/10.3390/ijms222313012