Abstract
Alzheimer’s disease (AD) is one of the looming health crises of the near future. Increasing lifespans and better medical treatment for other conditions mean that the prevalence of this disease is expected to triple by 2050. The impact of AD includes both the large toll on individuals and their families as well as a large financial cost to society. So far, we have no way to prevent, slow, or cure the disease. Current medications can only alleviate some of the symptoms temporarily. Many animal models of AD have been created, with the first transgenic mouse model in 1995. Mouse models have been beset by challenges, and no mouse model fully captures the symptomatology of AD without multiple genetic mutations and/or transgenes, some of which have never been implicated in human AD. Over 25 years later, many mouse models have been given an AD-like disease and then ‘cured’ in the lab, only for the treatments to fail in clinical trials. This review argues that small animal models are insufficient for modelling complex disorders such as AD. In order to find effective treatments for AD, we need to create large animal models with brains and lifespan that are closer to humans, and underlying genetics that already predispose them to AD-like phenotypes.
1. Introduction
Alzheimer’s disease (AD) is a devastating neurodegenerative disease, behaviourally characterised by memory loss and cognitive decline, generally in later life, which is ultimately fatal [1]. The prevalence of AD is rapidly increasing due to an ageing population worldwide, and expected to triple between the years 2000 and 2050 [2,3]. Besides those affected, AD places a severe burden on families, carers, and the economy [4,5]. Alois Alzheimer discovered the neuropathological hallmarks of AD in 1906 [6]. Despite many decades of research since the 1900s, a cure has remained elusive with current therapies only offering temporary symptomatic relief.
Classically, AD is characterised by plaques and tangles, both of which contain insoluble protein deposits that progressively accumulate in the brain [7,8,9]. These pathological features develop over decades, and considerable effort has been devoted to their replication in short lived models. Implicit in these modelling efforts is that the relatively rare dominant genetic forms of AD represent the condition as a whole, and that the accelerated processes artificially engineered into these models accurately represents the mechanisms of a slow disease process in humans. Most models have been constructed to recapitulate the end stage pathological features, assuming that they represent the cause of the condition rather than the consequence. To a large extent, this pathology attainment strategy for AD animal model construction has driven the preclinical selection of compounds going through to human clinical trials. Well over 200 compounds have now failed to prevent, slow, or cure the disease, despite most being effective at ‘curing’ mouse models of AD [10,11,12]. This review summarises the modelling efforts up to November 2021. It briefly covers the extensive work undertaken to capture AD symptomatology predominantly in the mouse, and summarises the key factors that still need to be obtained. Finally, suggestions are given for an alternative modelling strategy through the use of large animals.
2. The Defining Pathological Features of AD; Plaques and Tangles
Plaques are extracellular deposits formed from the amyloid-beta (Aβ) peptide; cleavage products of the transmembrane amyloid precursor protein (APP) [7,13,14,15,16]. APP is cleaved in two regions, releasing peptide fragments (Figure 1). It is cleaved by either an alpha or beta secretase enzyme in the lumen closer to the C terminus. It is cleaved nearer the N-terminus by the gamma secretase complex in a membrane domain. The gamma secretase complex can cleave at multiple sites within its target region of the APP protein, creating different sized products [17]. If APP is cleaved by alpha secretase, the peptides released are shorter and appear to be harmless. When APP is cleaved by a beta secretase instead, usually the BACE1 enzyme, longer (Aβ) peptides are released. Amyloid beta has the potential to aggregate into plaques. Aβ1-–40 and Aβ1–42 are the longest peptides and have the strongest association with AD, with Aβ1–42 deemed to have a causal role [18].
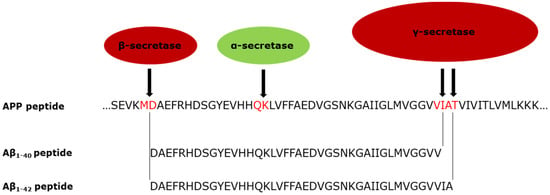
Figure 1.
Cleavage of APP by beta and gamma secretase (in red) to release the Aβ1–40 and Aβ1–42 peptides associated with AD, and showing the alternative cleavage position of alpha secretase (green) [17,19].
Tangles are dead or dying neurons containing intracellular aggregated hyper-phosphorylated TAU protein and other aggregated proteins [20]. In its native state, TAU is a structural component of axons within neurons, and it is unclear why it becomes hyper-phosphorylated [21]. The accumulation and density of tangles correlates better with neurological symptoms than plaques [22,23]. However, plaques begin forming before tangles, which led to the ‘amyloid cascade hypothesis’. The original amyloid cascade hypothesis proposed that the formation of Aβ plaques initiates the disease process and eventually leads to tangle formation, cognitive symptoms, and neurodegeneration [24]. It has subsequently been modified to suggest that an increase in the soluble toxic forms of amyloid beta, which precede plaque formation, initiates the disease process [25,26].
Cases of AD are broadly classified as early-onset Alzheimer’s disease (EOAD) or sporadic late-onset AD (LOAD). Some of the early onset cases can be further subdivided into a familial early onset group (FAD) caused by single genetic mutations, typically inherited in an autosomal dominant fashion [27]. LOAD is the most prevalent form of AD and is more complex, influenced by both genetic and environmental risk factors. The FAD patients (<5%) are of particular interest because the clear genetic aetiology offers an opportunity to understand the molecular mechanisms involved. It also allows the disease to be predicted before the onset of symptoms, and therefore mutation carriers are an ideal cohort for clinical testing [28]. Aside from age at onset, these two forms of AD (FAD and LOAD) are typically indistinguishable [29].
Over 200 mutations have been identified as responsible for FAD, all clustered into three genes; APP, Presenilin 1 (PSEN1), and Presenilin 2 (PSEN2). As already introduced, the APP protein is cleaved to form Aβ. The APP gene is found on chromosome 21 and Trisomy 21 carriers (Down’s syndrome) have a high frequency of early onset AD, presumably caused by an increased dosage of APP [13]. The PSEN1 and PSEN2 proteins form part of the gamma secretase complex that cleaves the APP protein [30]. Mutations in PSEN1 are the most common cause of FAD. Mutations in PSEN2 are relatively infrequent and generally result in a later onset and slower disease course that those in PSEN1 (see review [31]). In summary, mechanistically it is thought that mutations in APP, PSEN1, and PSEN2 favour the production of the more amyloidogenic, Aβ1–42 form of amyloid (reviewed elsewhere [32,33]).
Briefly, the risk of developing LOAD has been associated with variations in an increasing number of genes as the power of the genome wide association studies (GWAS) increases. Over 75 risk loci have now been detected [34], with twin studies revealing that up to 79% of the risk for LOAD is genetic [35]. The gene with the largest effect is Apolipoprotein E (APOE). The APOE gene has three variants (APOE2, 3, and 4). Inheritance of the APOE2 allele is protective, the APOE3 allele is a neutral effect on risk, and the APOE4 allele confers a very significant risk for LOAD in a dose-dependent manner. APOE4 heterozygotes have AD susceptibility an odds ratio of 3.5–4 and homozygotes 12–15 [36]. All of the genes that have so far been implicated in AD can be functionally linked to Aβ peptide homeostasis [37,38,39], implying a common mechanism that could shed light on the aetiology of the disease.
3. Modelling AD in Animals
One of the most effective ways of investigating the pathogenic process of a disease is via animal models. Animal models can also be used for biomarker discovery, which can allow for early detection of disease, and for screening and safety tolerance testing of therapeutic agents. There are three main aspects of animal modelling that need to be considered: the resulting face, construct, and predictive validities [40]. These relate to how well the model replicates symptoms, the biological causes, and responds to clinically effective therapeutics, respectively.
The earliest animal models of AD were created by disrupting the cholinergic system in various mammalian species using surgical methods, neurotoxins, immunotoxins, or pharmacological methods. The species targeted included mice and rats [41], rabbits [42], and monkeys such as the marmoset and crab eating macaque [43,44]. The cholinergic system in the basal forebrain degenerates early in the course of AD [45,46]. These models replicated some of the symptoms of AD such as memory impairments, and were helpful for testing the efficacy of cholinesterase inhibitors, which can offer some symptomatic relief early in the course of AD [41]. These models, of course, did not develop plaques or tangles, nor did they represent the progression of the complex biochemical and cellular-level changes in AD [47].
The rapid development of genetic technology and engineering from the 1980s to the present has enabled the construction of animal models that can theoretically recapitulate diseases from their underlying causes, thus increasing the construct validity of the model.
4. Small Animal Models of AD
4.1. Mouse Models
4.1.1. Plaque Pathology in Mouse Models
As a mammalian model system, mice have the advantages of a short lifespan and rapid reproduction, which facilitates timely completion of experimental protocols. They are also comparatively easy to maintain and breed in a laboratory environment. Numerous tools, data, and standardised behavioural tests have been established for assessing phenotypes in mice. The development of embryonal stem cells and targeted mutagenesis has enabled the production of models that more accurately recapitulate the aetiology of human disease state. These factors combined has resulted in mice being the most common animal models of AD.
There have been a large number of mouse models constructed in various ways, far too many to include here. We have selected a representative group of models that were either notable because they were novel at the time or have been widely used in the field. Table 1 lists these selected mouse models.

Table 1.
Selected key mouse models of AD and their major phenotypes.
Plaques and tangles are the two main pathological hallmarks of AD, followed by neurodegeneration. In order to create models with high face validity, these phenotypes have been highly sought after. The first reported mouse models that developed plaque pathology were created via transgenesis (TG). Researchers introduced the human APP gene (hAPP) containing mutations known to cause FAD. The first mouse model, the PDAPP line created in 1995, overexpressed the V717F Indiana mutation hAPP with the Platelet-Derived Growth Factor (PDGF) promoter via a minigene construct. Around 40 copies of the transgene were randomly inserted in this line at a single site, and all three major splice variants of hAPP (695, 751, and 770) were expressed. These mice developed both dense and diffuse plaque pathology by eight months of age in the entorhinal cortex, cingulate cortex, and hippocampus. By 18 months, the amyloid burden in these brain regions was thought to be greater than that seen in end stage human disease. This model also showed signs of synaptic loss, microgliosis, and astrocytosis, but no tau/tangle pathology or neurodegeneration [48,58].
The next, and still commonly used mouse model, was the Tg2576 line, which overexpressed the K670M/N671L Swedish mutation in a transgene containing the 695 isoform of human hAPP transgene driven by the Prion Protein (PrP) promoter. Tg2576 mice develop plaques and memory deficits in a progressive manner. Similar to the PDAPP mice, they do not show the tangles or neurodegeneration [58,59]. These mouse models developed memory deficits and synaptic loss preceding the accumulation of insoluble plaques, providing evidence for the hypothesis that it is the smaller soluble forms of Aβ that cause these symptoms [60,61]. Several further mouse lines were also created by introducing the hAPP gene with various FAD causing mutations; most exhibited plaques and memory deficits in an age-dependant manner as well as some level of synaptotoxicity (reviewed in [62]).
Some of these mouse lines were subsequently crossed to produce mouse lines with multiple APP transgenes; the result was usually a similar phenotype that appeared at an earlier age, which shows that these mutations have cumulative phenotypic effects. One example is the TgCRND8 line, engineered with a single transgene to contain the hAPP isoform 695 with both the Swedish and Indiana mutations under the control of the Prp promoter. These mice develop plaque pathology by three months of age, with earlier signs of cognitive impairment relative to the models with a transgene carrying a single AD mutation. The brain concentration of Aβ1–42 in this compound model at six months was equivalent to the original PDAPP mouse line at 16 months. This compound model also showed an increase in Aβ1–42 to Aβ1–40 ratio (now considered to be an important indication of amyloidogenesis) [50]. However, these mice still did not exhibit the other major neuropathological hallmarks of AD such as the tangles and neurodegeneration.
Some of the APP overexpression mouse lines were later crossed with mice carrying a human PSEN1 transgene (hPSEN1) with various mutations responsible for FAD. Interestingly, mice overexpressing hPSEN1 mutations do not develop plaques or other symptoms, but do exhibit an increased ratio of the more amyloidogenic Aβ1–42 relative to Aβ1–40 in the brain [63,64,65]. Crossing transgenic mice that overexpressed APP with PSEN1 transgenic mice greatly increased amyloid pathology. An example is the crossing of Tg2576 mice (APP Swedish mutation) with both the PS-1 line (PSEN1 M146L mutation) [51,65] and the PSEN1 A246E line [63,66]. Taken together, this animal model work helped confirm that APP metabolism, and in particular, the production of the Aβ1–42 peptide, was affected by mutations in APP and PSEN1, and that these mutations are likely acting on a single pathway. This work also provided supporting evidence for the hypothesis that the Aβ1–42 fragment is more toxic than Aβ1–40.
Attempts to confirm the role of individual Aβ peptides led to the creation of transgenic mouse lines that selectively expressed either the Aβ1–40 or Aβ1–42 amyloid fragment in the absence of the hAPP transgene (BRI-Aβ40 and BRI-Aβ42) [52]. These models showed that high expression of Aβ1–40 caused no overt plaque pathology, but even low expression levels of Aβ1–42 was sufficient to cause plaque formation in both parenchymal brain tissue and blood vessels (cerebral amyloid angiopathy).
Attempts to capture a more complete AD phenotype led to crossing transgenic mice or creating constructs to overexpress multiple transgenes and mutations within these genes. Cell loss and neurodegeneration was ultimately achieved in the 5XFAD mouse model that expressed three APP (Swedish K670M/N671L, Florida I716V, and London V717I) and two PSEN1 (M146L and L286V) mutations under the murine Thy-1 promoter [53,67]. The severe phenotype again supported the hypothesis that FAD mutations have an additive effect. However tangles, which are the other main hallmark of AD, were absent in these mice.
4.1.2. Replicating AD Tau Pathology
Interestingly, unlike other mammalian species (see below), wild type mice do not develop tangles as they age [68]. Mutations in the human MAPT gene (microtubule associated protein tau), which codes for the human TAU (hTAU) protein, cause frontotemporal dementia (FTD), but not AD [69]. However tangle pathology, neurodegeneration, and memory loss were seen in transgenic mice models expressing human MAPT (hMAPT) with FTD causing mutations. The first mouse model with this phenotype was the JNPL3 line, which expressed the 4R0N isoform of hMAPT with the P301L mutation [54]. Subsequently, a hTAU expression tetracycline repressible mouse line (rTg4510) demonstrated that the smaller soluble forms of oligomeric TAU caused memory loss and neurodegeneration [70,71]. Many overexpression hMAPT transgenic lines have been produced and some have been crossed with transgenic mouse lines overexpressing FAD mutations in APP and/or PSEN1. The resulting lines demonstrated that the mechanisms leading to amyloid and TAU pathology interact. The 3xTg mice (Swedish mutation in APP, M146V in PSEN1, and P301L in MAPT) develop plaques before tangles [71,72], as observed in AD patients. A line developed by crossing the aforementioned APP mutant mice Tg2576 with the MAPT JNPL3 mice (called the TAPP line) altered the spatial distribution of tangles in the brain relative to original TAU expressing strain, with TAPP mice exhibiting tangles in the subiculum, hippocampus, and isocortex that were not present in JNPL3 mice. TAPP mice also had greatly increased numbers of tangles in the olfactory cortex, entorhinal cortex, and amygdala. This suggests that Aβ fibril deposition can alter the amount and distribution of insoluble TAU as tangles [57].
4.1.3. Construct Validity of Transgenic Mouse Models of AD
Although mice expressing a transgene with a single FAD mutation display some symptoms of the disease, it is evident from the literature that three or more AD and FTD associated mutations are required to replicate the majority of the human pathology. In contrast, multiple mutations have not been reported in humans with AD, and in nearly all cases of FAD, only a single mutation is required to develop the entire phenotype.
There are good reasons for the requirement of a compound approach to create equivalent AD pathology. Unlike human hAPP, the proteolytic cleavage products of murine App (mApp) do not naturally form plaques. This is due to three amino acid substitutions in the amyloid beta sequence compared to human (Figure 2), which reduces the ability of murine Aβ peptides to aggregate [72]. In addition, murine β-secretase enzymes typically cleave mApp to form Aβ11-x, even though it cleaves hAPP to form Aβ1-x [73,74]. Deposition of cleavage products from mApp is only apparent in models with high expression levels of mApp and only after an extended period. This is one of the reasons why hAPP is typically used instead [75].

Figure 2.
A comparison of the human and mouse Aβ peptide sequence, showing the three amino acid substitutions responsible for the functional difference between the two.
The ratio of hAPP isoforms differs within brain regions and also in other organs. The ratio also changes during the course of development and ageing [76,77]. The two longer isoforms of hAPP, 751 and 770, are more prevalent in the AD brain relative to healthy controls [78]. Overexpressing the hAPP 751 isoform also causes more obvious amyloid pathology in mice than overexpressing the short (APP695) isoform [79]. The pathology generated in a mouse model therefore depends on which of the three isoforms of hAPP is overexpressed, or whether the full hAPP gene sequence is used.
Several different promoters have been used to drive overexpression of hAPP in mouse models of AD including the promoters for PDGF-B (platelet derived growth factor B-chain) and the PrP (prion protein gene motifs). Different promoters drive different levels and spatial patterns of expression including outside the brain. For example, the PDGF-B and Thy-1 (thymocyte differentiation antigen 1) promoters are neuron-specific [80,81], while the PrP promoter has less specificity, also driving expression in glial cells and other non-brain tissue [82]. The Thy-1 promoter included in the construct to make the APP23 model (Swedish mutation in APP) is active only after birth, preventing potential developmental effects [83]. Various Tet-controlled lines have been created that allow for more control over the timing and location of transgene expression, but have the added complication of requiring an extra transgene [84,85,86]. All of these promoters are selected for ease of use or particular benefits, but because none of them are the endogenous promoter, the natural expression pattern of APP is not replicated in any of the models.
4.1.4. Murine APP Knock in Models
In an attempt to overcome the limitations of APP TG models, a small number of knock in (KI) App models have been created with targeted gene editing. Inserting selected mutations in the endogenous genes should mean expression is quantitatively, spatially, and temporally appropriate. Mouse App was ‘humanised’ in these models by converting the codons for the three amino acids that differ between human and mice in the Aβ coding portion of mApp. This allows murine BACE1 to cleave mAPP at the human equivalent position [87,88,89,90]. These mice did not develop overt phenotypes such as memory deficits, synaptic loss, and/or plaque pathology. These phenotypes only became evident after the insertion of multiple APP mutations (combinations of Swedish, London, Dutch, Iberian, and Artic) [91], and usually only after breeding to homozygosity in concert with homozygous FAD PSEN1 mutations [90,92].
The necessity of including multiple mutations to induce human equivalent disease confounds the use of these models, but they have helped differentiate between phenotypes due to the TG process, and those that represent the disease in a mouse. Consistent phenotypes observed in TG and KI models include plaque formation, changes to glial cells and astrocytes, and lowered rates of hippocampal neurogenesis, although some artifacts such as transgene calpain activation have been noted [93,94,95]. The presence of cognitive impairment appears to vary more between KI models than TG models. The KI models with cognitive impairment have plaque pathology prior to memory impairment, unlike the commonly used TG mice models [89,96]. Memory impairment following plaque formation is the order of events seen in patients [97], so KI models do appear to more faithfully replicate symptom clusters. Despite this, the higher variability of phenotypes in KI models, along with their milder symptom profile, means that transgenic models are still widely used.
4.1.5. Murine PSEN1 Knock in Models
Murine Psen1 (mPsen1) does not require ‘humanising’ like the mApp and (mPsen1) models made with targeted gene editing by introducing FAD mutations, which show similar phenotypes to TG hPSEN1 mouse lines [98,99]. Whether they are created by transgenesis or targeted gene editing, in the absence of hAPP or humanised mApp, all modelled PSEN1 mutations only increased the level of murine Aβ1–42 in mice, had little or no effect on murine Aβ1–40 levels, and did not result in AD equivalent symptoms [65,100,101,102]. For this reason, more recently generated KI mouse models carrying a PSEN1 mutation usually incorporate a transgene overexpressing mutant hAPP. The resulting animals have a more acute phenotype than APP mutations alone [103,104,105].
4.1.6. Construct Validity of MAPT Mouse Models
Compared to hTAU with six isoforms (named 4R2N, 4R1N, 4R0N, 3R2N, 3R1N, and 3R0N) [106,107,108] murine TAU (mTAU) only has three of the human equivalent isoforms (4R0N, 4R1N, 4R2N). There is also variability in TAU protein conservation. Some regions of mTAU tau are very similar to hTAU, while other regions differ greatly. There are species-specific differences in the presence of different isoforms during development, and spatially across the brain [108,109]. In TG models, the presence of endogenous mouse Mapt (mMapt) can alter the splicing ratios of introduced hMAPT [110,111].
As stated above, unlike in humans, tangles do not form naturally with age in the mouse brain. Indeed, it appears that replacing the mMapt gene with the human equivalent, and in some lines with a FTD mutation, is necessary to create a TAU dysfunction phenotype in mice [112]. The inclusion of FTD mutations to ensure a tangle phenotype in murine models is a major issue for construct validity. There are probably better models of frontotemporal dementia and other tauopathies than AD, even though they have provided insights about TAU toxicity [56,57]. Unexpected non-disease associated deficits have been found in some models, for example, the commonly used JNPL3 line (P103L mutation in MAPT) has motor impairments and develops eye irritations [54,113]. Further the Tau P301S line develops severe paraparesis at 5–6 months [114]. However severe motor impairment is not usually observed in AD until late in the disease course [115].
4.1.7. Predictive Validity of Murine Models
Almost no mouse model of AD has shown predictive validity in human clinical trials to date, despite many therapeutic agents ‘curing’ a mouse of AD symptoms (for reviews, see [112,113]). Those that have been successful were based on the cholinergic system or NMDA receptors and only provide temporary symptomatic relief. While symptomatic relief is important, the predicted increase in the prevalence of AD means that finding a method to prevent or cure the disease is now becoming an urgent priority.
In addition to drug failures, there is the issue of differences in drug metabolism between species; something well tolerated in mice may not be so in humans [116,117]. Many clinical trials have failed to make it to later stages due to adverse side effects, which were not present in mice. For example, immunisation of mice with Aβ1–42 (named AN1792 in the clinical trial) was able to lower the volume of plaque material in the brain and preserve cognitive function. Unfortunately, this approach failed to show benefits in clinical trials and 6% of the immunised patients developed meningoencephalitis [118,119]. The adverse effects were thought to be due to a T-cell response in humans against the large Aβ1–42 fragment. Subsequent immunisation trials with smaller epitopes that were beneficial in mice including the drugs Bapineuzumab [120] and Solanezumab [121] showed a similar lack of efficacy and/or adverse side effects [122,123,124].
To date, well over 200 compounds have failed to affect the disease course [10], and this appears to have led to some controversial decisions. Recently, the drug aducanumab (sold as Aduhelm) was approved by the FDA through an accelerated approval pathway, on the condition that follow-up trials are performed to determine efficacy. This drug showed mixed results in clinical trials, with a benefit seen at the highest dose, but only in one of the two trials. Given that 35% of patients developed brain swelling (cerebral adema) and 19% brain bleeds (intracerebral haemorrhage), there are serious safety considerations [125]. It is clear that models of AD with higher predictive validity are desperately needed.
4.1.8. Murine Model Summary
In summary, while successive generations of mouse models come closer to attaining the desired symptom clusters, this has created a trade-off between face and construct validity. The drive to replicated AD’s defining features of both plaques and tangles in a model is understandable. However, the inclusion of multiple mutations, with some from a different condition altogether, brings the construct validity of these models into question. Do they represent the disease process or a derived phenocopy? Discovering the mechanism by which amyloid pathology triggers TAU dysfunction would be invaluable for understanding the disease. Unfortunately, it appears that the mouse is too genetically and physiologically dissimilar to be able to capture this transition, even with genetic modification. The lack of translatability of treatments developed using these models is suggestive that they may not adequately represent AD. Work to understand the mechanistic nature of various FAD and MAPT mutations continues, and many of the aforementioned models are still utilized. Mouse models of LOAD variants have also been made including APOE and TREM2 [126,127,128]. It is likely that mechanistic work via mouse models will continue as more LOAD disease related genetic or environmental risk factors are discovered. However, over the last ten years, there have been repeated calls for new models of AD that can show predictive validity, causing researchers to look outside mice. Figure 3 summarises the desired qualities of an AD model, showing how improved construct validity could lead to higher translatability in clinical trials.
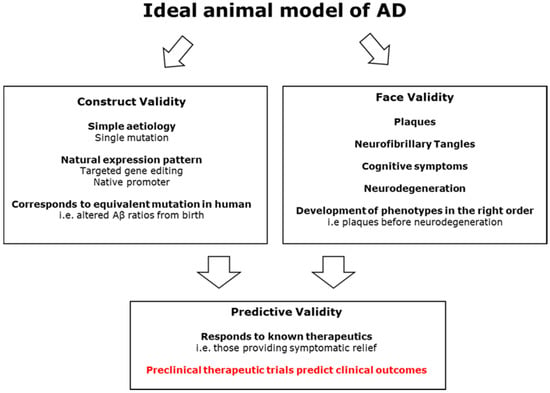
Figure 3.
A diagram showing how a model of AD with high face and construct validity is likely to improve predictive validity and lead to effective therapies.
4.2. Rat Models of AD
Rats are an attractive model system because they are genetically and physiologically more similar to humans than mice. They display more complex behaviour, and numerous assessment methods have been developed for mood and cognition for this species [47,129,130,131]. The first rat models of AD were based on knowledge from mouse models, designed to express hAPP with FAD mutations such as the UKUR25 line (with the Swedish and Indiana APP mutations, with the M146L PSEN1 mutation [132]) or the McGill-R-Thy1-APP line (with the Swedish and Indiana hAPP mutations, expressed under the murine Thy1.2 promoter [133,134]). Interestingly, these models failed to develop the plaques seen in mice, but did accumulate intracellular Aβ and developed memory deficits seen in the equivalent mouse models. Rat models that exhibited plaque pathology were finally created in the mid 2000s, sometimes with differing phenotypes from their murine genetic equivalents. The rat TgF344-AD line carries both Swedish hAPP and PSEN1 ΔE9 mutations, driven by the same murine PrP promoter [135]. This line develops both plaque and tangle-like pathology with loss of neurons. Interestingly the tangle-like structures appear despite the non-inclusion of a hMAPT transgene, even though tangles are not naturally seen in aged rats. This may be because unlike mTau, rat Tau (rTau) is spliced to create all six human equivalent isoforms [136,137]. Rat models expressing hMAPT with FTD mutations have also been developed, some of which exhibit tangles, while all show increased phospho-TAU in the brain and develop cognitive symptoms [138,139,140]. The development of Tau pathology, along with their larger brain and more complex behaviours, may confer on these models the potential to improve our understanding of AD.
5. Large Animal Models in AD Research
In pursuit of more translatable results across the medical sciences, researchers are more frequently turning to large animal models, particularly those that are evolutionarily closer to humans and have longer lifespans, thus making them better suited to recapitulate complex human diseases, especially late onset disorders [141,142,143]. Massively overexpressing transgenes speeds up the development of a phenotype, but also leads to acute inflammatory processes not present in human AD. Small mammals also have a smooth (lissencephalic) brain, while most larger mammals including humans have a more complex convoluted (gyrencephalic) brain. This makes larger mammals ideal for studying neurological disorders.
Many large animals naturally develop plaques and/or tangles as they age, and these features may be the norm in larger animals. They have been found in many primate species, and across a range of large herbivorous and carnivorous animals (summarised in Table 2). This propensity to develop plaques appears to be due, at least in part, to conservation of the amyloid beta peptide sequence in most mammals [144]. Of note is that in some large animals (e.g., dogs, sheep), only one of either plaques or tangles were originally identified, but later research revealed both [145,146]. They are typically found in aged animals, and vary widely in density between individuals of a species, so it is entirely possible that both hallmarks of AD will eventually be found in most large animals. Age related neurodegeneration has not been extensively studied in most species (Table 2), however, many are known to develop cognitive decline with age. Cognitive decline has rarely been studied in detail outside of humans, dogs, cats, and some primates.
Table 2.
A summary of the large animals in which plaque, tangle pathology, or neurodegeneration with older age has been identified.
Humans are fairly unique in outliving our reproductive lifespan, which no doubt contributes to the presence of AD and other dementias [191]. However, if other animals with medium to long lifespans can develop the hallmarks of AD, it is likely that the development can be accelerated with mutations from FAD. Large animals have long enough lifespans that introducing a single FAD mutation via KI methods will likely accelerate the disease in line with natural human forms of AD. This would remove the need for artificial promoters to massively overexpress transgenes to generate a robust phenotype within the 1–2 year lifespan of a mouse. If a large animal can develop all of the hallmarks of AD with the introduction of a single FAD mutation, this will represent a major step forward for construct validity. It would also be the first model to fully recapitulate a form of AD from its underlying cause.
5.1. Primate Models of AD
Being our closest relatives, primates show great promise for accurately representing human disease. Evidence of age related plaques, tangles, or both have been found across a range of primate species (see Table 2). Like humans, the pathological hallmark are only seen in some individuals, and is more likely with age, suggesting biological or environmental triggers that make certain individuals more prone to these precursors of dementia [147]. The great apes (bonobo, chimpanzee, orang-utan, and gorilla) have the highest genetic similarity to humans. Ethical concerns combined with slow reproduction make these animals relatively impractical models of AD [192,193,194]. Efforts have instead focussed on smaller primates already utilised to model human behaviour and disease.
Macaques are a promising species for modelling AD with a lifespan of 30 to 40 years, reaching old age around 20–25 years [183,195,196]. Elements of AD have been identified in multiple macaque species (Table 2). In particular, the rhesus monkey is a relevant model based on extensive data collected on the ageing process in these animals, and the similarity of their plaque morphology and staging to human [158,197,198].
Two smaller monkey species, the mouse lemur and the common marmoset, have also been considered for AD modelling because of their small size and lifespan. The mouse lemur has high rates of cognitive decline associated with plaque formation and neurodegeneration including loss of cholinergic neurons in old age [165,168,199,200,201,202,203]. They live 8–14 years in captivity, but are considered elderly after five [204]. The common marmoset (Callithrix jacchus) with a lifespan of 7–17 years, is another smaller primate that has shown promise as a monkey model of AD [168,169,205,206]. Perhaps surprisingly, most primates studied so far have shown a higher level of Aβ1–40 in plaques than Aβ1-42, whereas the common marmoset has a higher level of Aβ1-42, similar to human [207].
There is some debate about the usefulness of monkeys as natural models of AD due to the time taken to reach old age and the sporadic nature of naturally occurring AD. At present, monkeys are more often used in AD research for toxicology screening [208], screening of brain imaging compounds [209,210, or for testing of biomarker efficacy [211]. Some ongoing research involves the seeding of amyloid beta or tau in the monkey brain to investigate the proposed spread via a prion-like mechanism [212,213]. Seeding amyloid beta substantially increases the level of amyloid in the marmoset [214], rhesus macaque, and cynomolgus macaques [215], with the latter developing tau pathology and neurodegeneration [216]. This provides a useful tool to investigate the mechanism via which amyloid dysfunction leads to tau dysfunction. TAU injected/seeded rhesus monkeys have also exhibited neurodegeneration after three months [217]. While these models have utility in showing how the disease progresses, they do not capture the underlying mechanism that initiates AD.
The introduction of FAD mutations could solve this problem. Very recently, genetically modified monkey models have begun to be reported. A transgenic cynomolgus monkey model was made by introducing hAPP with the Swedish, Arctic, and Iberian mutations under a CAG promoter. Plasma Aβ1–40 levels were double that of wild type monkeys at birth, while Aβ1–42 levels were increased 50-fold, increasing the ratio of Aβ1–42 to Aβ1–40 approximately 20-fold [218]. As this is a TG rather than a KI model, the expression of mutant APP is not tissue specific, which may complicate interpretation. Nonetheless, it is a promising development. Around the same time, a KI marmoset model of AD was reported in bioRxiv, carrying the PSEN1 delta E9 mutation [219]. The ratio of Aβ1–42 to Aβ1–40 production in fibroblasts was double that of the controls in the juvenile monkeys, indicating an early pathological change. Both of these potential monkey models are still juveniles, so time will be needed to see what phenotypes arise. However, these are the first reports on the genetically modified monkey model of AD, and they represent exciting developments for the field.
5.2. Larger Non-Primate Mammalian Models
Larger mammals outside the primate group present a compromise between the limits of small animal models and the difficulties of working with primates. While primate models have great potential, they are also very expensive and most laboratories do not have the required facilities to house and maintain large numbers for experimental trials. The two most commonly suggested groups are larger companion animals and farm animals. They have a lifespan that is typically 10–15 years and have the advantage of a larger body and more human-like brain than a mouse.
5.2.1. Larger Companion Animals
The domestic dog has been suggested a number of times as a suitable model of AD, both in natural and genetically modified form [220,221,222,223]. Aged dogs develop a dementia-like syndrome called canine cognitive dysfunction (CCD), which has been suggested to be the canine counterpart to AD [180,224,225]. The presence of CCD symptoms has been in more than a quarter of dogs in the 11–12 age range, and nearly 70% in dogs 15–16 [226]. Dogs are the only animal outside humans where cognitive impairment in older age has been reasonably well characterised, and at least three standardised tests exist for assessing CCD [227,228,229]. Dogs naturally develop plaque pathology and cerebral amyloid angiopathy (CAA) [179,230], although it is as still unclear whether this correlates with the symptoms of cognitive decline [178,181,231]. Tau dysfunction and tangles have been reported and associated with cognitive decline [179,182,183]. Aged dogs have occasionally been used as a natural model of AD for therapeutic testing, with aged beagles being used to test the effects of the statin drug Atorvastatin on physiological and behavioural measures of cognitive decline. The research revealed a positive effect on a number of markers for enhanced cognitive function such as biliverdin reductase-A, heme oxygenase-1, and nitric oxide synthase in the brain as well as being significantly correlated with lower discrimination learning error scores [232,233,234]. An immunotherapy effective in mice was once trialled in dogs, with positive results [235].
Domestic cats also develop plaques, tangles, and brain atrophy along with cognitive decline with age [184,236,237]. Research into age-related cognitive dysfunction in cats is not as well developed as that of dogs, but there is increasing interest in this area [185,238,239,240].
5.2.2. Farm Animals
Mammalian farm animals have significant advantages over the aforementioned models in terms of cost and maintenance. Well refined animal husbandry and accelerated reproductive methods means that farm animals can be kept in large numbers at low cost. They can be kept in large groups outdoors, enabling low stress, and natural behaviour. In particular cows, pigs, goats and sheep have been suggested as AD models [144,145,173]. TAU pathology including tangles develop in aged sheep and goats [173,174,175], and plaques have recently been identified in sheep [145]. Plaque and tangle like pathology has also been seen after traumatic brain injury (TBI) in pigs [173]. Pigs and sheep are already in use as models of neurodegenerative disorders, so these will be covered here.
5.2.3. Pigs
With a high degree of genetic similarity, brain structure, and weight, pigs have been selected as a model system for a number of human disorders so there is a growing body of resources for working with this species (see reviews [241,242,243,244]).
Two TG pig models of AD has been reported using minipigs. The first reported model carries a hAPP transgene with the Swedish mutation driven by the human BDGFβ promoter, resulting in high levels of brain-specific expression [245]. A subsequent publication reported that the animals did not have memory deficits [246]. The most recent report identified the altered activity of APP and TAU in astrocytes derived from embryonic stem cells isolated from these TG pigs [247]. No behavioural phenotype has yet been reported.
The second minipig model was reported in 2016. This model carries three copies of a transgene expressing the 695 variant of hAPP with the Swedish mutation, and a human PSEN1 transgene with the M146L mutation. Both transgenes were expressed in the brain with normal processing of their protein products. Intraneuronal accumulation of Aβ1–42 was detected in two pigs: one at 10 months, and one at 18 months [248]. This may represent the early stages of AD, so it will be interesting to see if a more overt pathology is reported in future. Given the rapid development in targeted gene editing technology since these models were made, it should be possible to generate pig models utilising KI methods in the future to enhance construct validity.
5.2.4. Sheep
As above-mentioned, sheep appear to develop plaques and tangles as a natural part of the ageing process [145]. There are many anecdotal reports of age-related cognitive decline in sheep, although this has not been researched and the rate of naturally occurring AD-like dementia is unknown. However, Batten’s disease, a neurodegenerative disease of childhood, has been extensively investigated in sheep due to four models based on naturally occurring genetic mutations [249,250,251,252].
Our lab group has successfully made a transgenic model of the neurodegenerative disorder Huntington’s disease (HD) in sheep [253]. These sheep show early pathological markers of HD, and were used to show urea dysfunction in HD, opening up new avenues of research that are ongoing [254,255]. This line is also being used to test potential therapies [256]. Due to the existence of these sheep models, a number of genetic, physiological and behavioural tools are becoming available. Importantly, the sheep genome has recently been published and annotated [257], and thus the genome of the sheep can now be precisely manipulated for human disease research. In addition, JIVET (juvenile in vitro embryo transfer) technology developed specifically for sheep means that genetically modified ewe lambs can produce viable oocytes at six weeks of age [253]. These oocytes can be fertilized in vitro and implanted into adult recipient ewes, drastically shortening generation times. This can potentially reduce the total time from the implantation of the edited founder embryo, to its offspring being born, to less than one year. This can result in a flock large enough for research use.
Despite their reputation, sheep are reasonably intelligent, and have face recognition systems comparable to humans [258]. This higher cognitive ability makes sheep readily trainable for tests of cognitive function [259]. As in pigs, brain activity in sheep can be monitored longitudinally using EEG [260] and MRI [261]. Our lab group has shown that wild-type sheep amyloid is processed in the same manner as humans, with comparable levels of the disease-related Aβ1–40 and Aβ1–42 forms in cerebrospinal fluid (CSF). Similar levels of CSF total-Tau have been found, suggesting that the CSF profile of sheep could be an indicator of disease state.
6. Conclusions
Understandably, in a condition defined by pathological features, considerable effort has been expended on deriving models that represent the definition of the disease. This, combined with the time imperative to develop models presenting with plaques and tangles, has resulted in murine models that are a phenocopy of end stage disease but potentially do not represent the natural disease process in humans. Unarguably, the murine models of AD have greatly added to our mechanistic understanding of AD, but they have potentially reached the limits of their utility. There have been widespread calls for more valid models of AD over the last several years, and this has prompted a number of lab groups to turn to large animals. There are a number of potentially suitable large mammals, which can be broadly split into non-human primates and larger companion or farm animals. Primates’ close evolutionary relationship to humans means that discoveries based on them should have high translatability. However the cost and practicalities including breeding and housing sufficient numbers considerably limits their utility, especially for pharmaceutical testing. Larger companion or farm animal models represent a trade-off between primates with their high similarity to humans, and rodents with their ease of use in a laboratory setting. There is considerable potential, especially in farm animals, to produce models that have both high face and construct validity. Regardless, it is clear that to produce an effective treatment for AD, and to avoid the expensive failures at late stage human testing, new preclinical pharmaceutical testing models are required.
Author Contributions
Conceptualisation, N.E.M. and R.G.S.; Writing—original draft preparation, N.E.M.; writing—review and editing, N.E.M., R.R.H. and R.G.S.; supervision, R.R.H. and R.G.S.; project administration, R.G.S.; funding acquisition, R.G.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Brain Research New Zealand and the University of Auckland (3715325).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors wish to thank Greg Sutherland for critically reading the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Khachaturian, Z.S. Diagnosis of Alzheimer’s disease. Arch. Neurol. 1985, 42, 1097–1105. [Google Scholar] [CrossRef]
- Hebert, L.E.; Beckett, L.A.; Scherr, P.A.; Evans, D.A. Annual incidence of Alzheimer disease in the United States projected to the years 2000 through 2050. Alzheimer Dis. Assoc. Disord. 2001, 15, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Sloane, P.D.; Zimmerman, S.; Suchindran, C.; Reed, P.; Wang, L.; Boustani, M.; Sudha, S. The public health impact of Alzheimer’s disease, 2000–2050: Potential implication of treatment advances. Annu. Rev. Public Health 2002, 23, 213–231. [Google Scholar] [CrossRef]
- Fox, P.J.; Kohatsu, N.; Max, W.; Arnsberger, P. Estimating the costs of caring for people with Alzheimer disease in California: 2000–2040. J. Public Health Policy 2001, 22, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Katzman, R.; Fox, P. The World-Wide Impact of Dementia. Projections of Prevalance and Costs. In Epidemiology of Alzheimer’s Disease: From Gene to Prevention; Springer: Berlin, Germany, 1999; pp. 1–17. [Google Scholar]
- Tagarelli, A.; Piro, A.; Tagarelli, G.; Lagonia, P.; Quattrone, A. Alois Alzheimer: A hundred years after the discovery of the eponymous disorder. Int. J. Biomed. Sci. 2006, 2, 196. [Google Scholar]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef]
- Kidd, M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature 1963, 197, 192–193. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2021. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12179. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Feldman, H.H.; Scheltens, P. The “rights” of precision drug development for Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef]
- Masters, C.L.; Beyreuther, K. Pathways to the discovery of the Aβ amyloid of Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 155–161. [Google Scholar] [CrossRef]
- Miller, D.L.; Papayannopoulos, I.A.; Styles, J.; Bobin, S.A.; Lin, Y.Y.; Biemann, K.; Iqbal, K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’ s disease. Arch. Biochem. Biophys. 1993, 301, 41–52. [Google Scholar] [CrossRef]
- Nunan, J.; Small, D.H. Regulation of APP cleavage by α-, β-and γ-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid [beta]-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101. [Google Scholar] [CrossRef]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. Neuromol. Med. 2010, 12, 1–12. [Google Scholar] [CrossRef]
- Reid, S.J.; van Roon-Mom, W.M.; Wood, P.C.; Rees, M.I.; Owen, M.J.; Faull, R.L.; Dragunow, M.; Snell, R.G. TBP, a polyglutamine tract containing protein, accumulates in Alzheimer’s disease. Mol. Brain Res. 2004, 125, 120–128. [Google Scholar] [CrossRef]
- Alonso, A.d.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Hyman, B.T.; Betensky, R.A. Neurofibrillary tangle stage and the rate of progression of Alzheimer symptoms: Modeling using an autopsy cohort and application to clinical trial design. JAMA Neurol. 2017, 74, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Isla, T.; Hollister, R.; West, H.; Mui, S.; Growdon, J.H.; Petersen, R.C.; Parisi, J.E.; Hyman, B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1997, 41, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184. [Google Scholar] [CrossRef]
- Hardy, J. Alzheimer’s disease: The amyloid cascade hypothesis: An update and reappraisal. J. Alzheimer’s Dis. 2006, 9, 151–153. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Campion, D.; Dumanchin, C.; Hannequin, D.; Dubois, B.; Belliard, S.; Puel, M.; Thomas-Anterion, C.; Michon, A.; Martin, C.; Charbonnier, F. Early-onset autosomal dominant Alzheimer disease: Prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 1999, 65, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Berchtold, N.C.; Cotman, C.W. Evolution in the conceptualization of dementia and Alzheimer’s disease: Greco-Roman period to the 1960s. Neurobiol. Aging 1998, 19, 173–189. [Google Scholar] [CrossRef]
- Takasugi, N.; Tomita, T.; Hayashi, I.; Tsuruoka, M. The role of presenilin cofactors in the gamma-secretase complex. Nature 2003, 422, 438. [Google Scholar] [CrossRef]
- Wu, L.; Rosa-Neto, P.; Hsiung, G.-Y.R.; Sadovnick, A.D.; Masellis, M.; Black, S.E.; Jia, J.; Gauthier, S. Early-onset familial Alzheimer’s disease (EOFAD). Can. J. Neurol. Sci. 2012, 39, 436–445. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Bird, T.D. Genetic factors in Alzheimer’s disease: A review of recent advances. Ann. Neurol. 1996, 40, 829–840. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Bellenguez, C.; Küçükali, F.; Jansen, I.; Andrade, V.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Grenier-Boley, B.; Campos-Martin, R.; Holmans, P.A. New insights on the genetic etiology of Alzheimer’s and related dementia. MedRxiv 2020. [Google Scholar] [CrossRef]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Coon, K.D.; Myers, A.J.; Craig, D.W.; Webster, J.A.; Pearson, J.V.; Lince, D.H.; Zismann, V.L.; Beach, T.G.; Leung, D.; Bryden, L. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J. Clin. Psychiatry 2007, 68, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Justice, M.J.; Dhillon, P. Using the Mouse to Model Human Disease: Increasing Validity and Reproducibility; The Company of Biologists Ltd.: Cambridge, UK, 2016. [Google Scholar]
- Toledano, A.; Alvarez, M.I. Lesion-Induced Vertebrate Models of Alzheimer Dementia. Neuromethods 2011, 48, 295–345. [Google Scholar] [CrossRef]
- Beach, T.G.; Potter, P.E.; Kuo, Y.M.; Emmerling, M.R.; Durham, R.A.; Webster, S.D.; Walker, D.G.; Sue, L.I.; Scott, S.; Layne, K.J.; et al. Cholinergic deafferentation of the rabbit cortex: A new animal model of A beta deposition. Neurosci. Lett. 2000, 283, 9–12. [Google Scholar] [CrossRef]
- Wenk, G.L. A Primate Model of Alzheimers-Disease. Behav. Brain Res. 1993, 57, 117–122. [Google Scholar] [CrossRef]
- Ridley, R.M.; Murray, T.K.; Johnson, J.A.; Baker, H.F. Learning Impairment Following Lesion of the Basal Nucleus of Meynert in the Marmoset—Modification by Cholinergic Drugs. Brain Res. 1986, 376, 108–116. [Google Scholar] [CrossRef]
- Coyle, J.T.; Price, D.L.; Delong, M.R. Alzheimers-Disease—A Disorder of Cortical Cholinergic Innervation. Science 1983, 219, 1184–1190. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delong, M.R. Alzheimers-Disease and Senile Dementia—Loss of Neurons in the Basal Forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- McGonigle, P. Animal models of CNS disorders. Biochem. Pharm. 2014, 87, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Borthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C. Correlative memory deficits, A beta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 98. [Google Scholar] [CrossRef]
- Chishti, M.A.; Yang, D.-S.; Janus, C.; Phinney, A.L.; Horne, P.; Pearson, J.; Strome, R.; Zuker, N.; Loukides, J.; French, J. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 2001, 276, 21562–21570. [Google Scholar] [CrossRef]
- Holcomb, L.; Gordon, M.N.; McGowan, E.; Yu, X.; Benkovic, S.; Jantzen, P.; Wright, K.; Saad, I.; Mueller, R.; Morgan, D. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 1998, 4, 97–100. [Google Scholar] [CrossRef]
- McGowan, E.; Pickford, F.; Kim, J.; Onstead, L.; Eriksen, J.; Yu, C.; Skipper, L.; Murphy, M.P.; Beard, J.; Das, P. Aβ42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 2005, 47, 191–199. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Murphy, M.P.; Baker, M.; Yu, X. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, M.; Kotilinek, L.; Forster, C.; Paulson, J.; McGowan, E.; SantaCruz, K.; Guimaraes, A.; Yue, M.; Lewis, J.; Carlson, G.; et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J. Neurosci. 2005, 25, 10637–10647. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef]
- Lewis, J.; Dickson, D.W.; Lin, W.-L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.-H.; Sahara, N.; Skipper, L.; Yager, D. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef]
- Irizarry, M.C.; Soriano, F.; McNamara, M.; Page, K.J.; Schenk, D.; Games, D.; Hyman, B.T. Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J. Neurosci. 1997, 17, 7053–7059. [Google Scholar] [CrossRef] [PubMed]
- King, D.L.; Arendash, G.W. Behavioral characterization of the Tg2576 transgenic model of Alzheimer’s disease through 19 months. Physiol. Behav. 2002, 75, 627–642. [Google Scholar] [CrossRef]
- Westerman, M.A.; Cooper-Blacketer, D.; Mariash, A.; Kotilinek, L.; Kawarabayashi, T.; Younkin, L.H.; Carlson, G.A.; Younkin, S.G.; Ashe, K.H. The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2002, 22, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Kotilinek, L.A.; Bacskai, B.; Westerman, M.; Kawarabayashi, T.; Younkin, L.; Hyman, B.T.; Younkin, S.; Ashe, K.H. Reversible memory loss in a mouse transgenic model of Alzheimer’s disease. J. Neurosci. 2002, 22, 6331–6335. [Google Scholar] [CrossRef]
- Janus, C.; Phinney, A.L.; Chishti, M.A.; Westaway, D. New developments in animal models of Alzheimer’s disease. Curr. Neurol. Neurosci. Rep. 2001, 1, 451–457. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.-M.; Kim, G.; Seekins, S.; Yager, D. Familial Alzheimer’s disease–linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef]
- Citron, M.; Westaway, D.; Xia, W.; Carlson, G.; Diehl, T.; Levesque, G.; Johnson-Wood, K.; Lee, M.; Seubert, P.; Davis, A.; et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat. Med. 1997, 3, 67–72. [Google Scholar] [CrossRef]
- Duff, K.; Eckman, C.; Zehr, C.; Yu, X.; Prada, C.-M.; Perez-Tur, J.; Hutton, M.; Buee, L.; Harigaya, Y.; Yager, D. Increased amyloid-β42 (43) in brains of mice expressing mutant presenilin 1. Nature 1996, 383, 710–713. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Ratovitski, T.; Van Lare, J.; Lee, M.K.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Price, D.L.; Sisodia, S.S. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 1997, 19, 939–945. [Google Scholar] [CrossRef]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 196.e29–196.e40. [Google Scholar] [CrossRef]
- Chen, F.; David, D.; Ferrari, A.; Gotz, J. Posttranslational modifications of tau—Role in human tauopathies and modeling in transgenic animals. Curr. Drug Targets 2004, 5, 503–515. [Google Scholar] [CrossRef]
- Rademakers, R.; Cruts, M.; van Broeckhoven, C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum. Mutat. 2004, 24, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; Deture, M.; Ramsden, M.; McGowan, E. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef]
- Eriksen, J.L.; Janus, C.G. Plaques, tangles, and memory loss in mouse models of neurodegeneration. Behav. Genet. 2007, 37, 79–100. [Google Scholar] [CrossRef]
- Yamada, T.; Sasaki, H.; Furuya, H.; Miyata, T.; Goto, I.; Sakaki, Y. Complementary DNA for the mouse homolog of the human amyloid beta protein precursor. Biochem. Biophys. Res. Commun. 1987, 149, 665–671. [Google Scholar] [CrossRef]
- Liu, K.; Doms, R.W.; Lee, V.M.-Y. Glu11 site cleavage and N-terminally truncated Aβ production upon BACE overexpression. Biochemistry 2002, 41, 3128–3136. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Wang, Y.; McCarthy, D.; Wen, H.; Borchelt, D.R.; Price, D.L.; Wong, P.C. BACE1 is the major [beta]-secretase for generation of A [beta] peptides by neurons. Nat. Neurosci. 2001, 4, 233. [Google Scholar] [CrossRef]
- Kawasumi, M.; Chiba, T.; Yamada, M.; Miyamae-Kaneko, M.; Matsuoka, M.; Nakahara, J.; Tomita, T.; Iwatsubo, T.; Kato, S.; Aiso, S.; et al. Targeted introduction of V642I mutation in amyloid precursor protein gene causes functional abnormality resembling early stage of Alzheimer’s disease in aged mice. Eur. J. Neurosci. 2004, 19, 2826–2838. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Shiojiri, S.; Takahashi, Y.; Kitaguchi, N.; Ito, H.; Kameyama, M.; Kimura, J.; Nakamura, S.; Ueda, K. Tissue-specific expression of three types of beta-protein precursor mRNA: Enhancement of protease inhibitor-harboring types in Alzheimer’s disease brain. Biochem. Biophys. Res. Commun. 1989, 165, 1406–1414. [Google Scholar] [CrossRef]
- Tanaka, S.; Liu, L.; Kimura, J.; Shiojiri, S.; Takahashi, Y.; Kitaguchi, N.; Nakamura, S.; Ueda, K. Age-related changes in the proportion of amyloid precursor protein mRNAs in Alzheimer’s disease and other neurological disorders. Brain Res. Mol. Brain Res. 1992, 15, 303–310. [Google Scholar] [CrossRef]
- Moir, R.D.; Lynch, T.; Bush, A.I.; Whyte, S.; Henry, A.; Portbury, S.; Multhaup, G.; Small, D.H.; Tanzi, R.E.; Beyreuther, K.; et al. Relative increase in Alzheimer’s disease of soluble forms of cerebral Abeta amyloid protein precursor containing the Kunitz protease inhibitory domain. J. Biol. Chem. 1998, 273, 5013–5019. [Google Scholar] [CrossRef]
- Higgins, L.S.; Catalano, R.; Quon, D.; Cordell, B. Transgenic mice expressing human beta-APP751, but not mice expressing beta-APP695, display early Alzheimer’s disease-like histopathology. Ann. N. Y. Acad. Sci. 1993, 695, 224–227. [Google Scholar] [CrossRef]
- Sasahara, M.; Fries, J.W.; Raines, E.W.; Gown, A.M.; Westrum, L.E.; Frosch, M.P.; Bonthron, D.T.; Ross, R.; Collins, T. PDGF B-chain in neurons of the central nervous system, posterior pituitary, and in a transgenic model. Cell 1991, 64, 217–227. [Google Scholar] [CrossRef]
- Caroni, P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J. Neurosci. Methods 1997, 71, 3–9. [Google Scholar] [CrossRef]
- Asante, E.A.; Gowland, I.; Linehan, J.M.; Mahal, S.P.; Collinge, J. Expression pattern of a mini human PrP gene promoter in transgenic mice. Neurobiol. Dis. 2002, 10, 1–7. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.-H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Slunt, H.H.; Gonzales, V.; Savonenko, A.V.; Wen, J.C.; Jenkins, N.A.; Copeland, N.G.; Younkin, L.H.; Lester, H.A.; Younkin, S.G. Persistent amyloidosis following suppression of Aβ production in a transgenic model of Alzheimer disease. PLoS Med. 2005, 2, e355. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Johnson-Venkatesh, E.M.; Zhang, H.; Parent, J.M.; Sutton, M.A.; Umemori, H. Multiple forms of activity-dependent competition refine hippocampal circuits in vivo. Neuron 2011, 70, 1128–1142. [Google Scholar] [CrossRef]
- Liu, P.; Paulson, J.B.; Forster, C.L.; Shapiro, S.L.; Ashe, K.H.; Zahs, K.R. Characterization of a novel mouse model of Alzheimer’s disease—Amyloid pathology and unique β-Amyloid oligomer profile. PLoS ONE 2015, 10, e126317. [Google Scholar] [CrossRef] [PubMed]
- Reaume, A.G.; Howland, D.S.; Trusko, S.P.; Savage, M.J.; Lang, D.M.; Greenberg, B.D.; Siman, R.; Scott, R.W. Enhanced amyloidogenic processing of the β-amyloid precursor protein in gene-targeted mice bearing the Swedish familial Alzheimer’s disease mutations and a “humanized” Aβ sequence. J. Biol. Chem. 1996, 271, 23380–23388. [Google Scholar] [CrossRef]
- Li, H.; Guo, Q.; Inoue, T.; Polito, V.A.; Tabuchi, K.; Hammer, R.E.; Pautler, R.G.; Taffet, G.E.; Zheng, H. Vascular and parenchymal amyloid pathology in an Alzheimer disease knock-in mouse model: Interplay with cerebral blood flow. Mol. Neurodegener 2014, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef]
- Masuda, A.; Kobayashi, Y.; Kogo, N.; Saito, T.; Saido, T.C.; Itohara, S. Cognitive deficits in single App knock-in mouse models. Neurobiol. Learn. Mem. 2016, 135, 73–82. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener 2017, 12, 89. [Google Scholar] [CrossRef]
- Anantharaman, M.; Tangpong, J.; Keller, J.N.; Murphy, M.P.; Markesbery, W.R.; Kiningham, K.K.; Clair, D.K.S. β-Amyloid mediated nitration of manganese superoxide dismutase: Implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am. J. Pathol. 2006, 168, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; McNeil, E.; Dressler, L.; Siman, R. Long-lasting impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer’s disease. Exp. Neurol. 2007, 204, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Matsuba, Y.; Yamazaki, N.; Hashimoto, S.; Saido, T.C. Calpain activation in Alzheimer’s model mice is an artifact of APP and presenilin overexpression. J. Neurosci. 2016, 36, 9933–9936. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Ishii, A.; Kamano, N.; Watamura, N.; Saito, T.; Ohshima, T.; Yokosuka, M.; Saido, T.C. Endoplasmic reticulum stress responses in mouse models of Alzheimer’s disease: Overexpression paradigm versus knockin paradigm. J. Biol. Chem. 2018, 293, 3118–3125. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Dickson, D.W.; Crystal, H.A.; Mattiace, L.A.; Masur, D.M.; Blau, A.D.; Davies, P.; Yen, S.-H.; Aronson, M.K. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol. Aging 1992, 13, 179–189. [Google Scholar] [CrossRef]
- Nakano, Y.; Kondoh, G.; Kudo, T.; Imaizumi, K.; Kato, M.; Miyazaki, J.i.; Tohyama, M.; Takeda, J.; Takeda, M. Accumulation of murine amyloidβ42 in a gene-dosage-dependent manner in PS1 ‘knock-in’mice. Eur. J. Neurosci. 1999, 11, 2577–2581. [Google Scholar] [CrossRef]
- Flood, D.G.; Reaume, A.G.; Dorfman, K.S.; Lin, Y.-G.; Lang, D.M.; Trusko, S.P.; Savage, M.J.; Annaert, W.G.; De Strooper, B.; Siman, R. FAD mutant PS-1 gene-targeted mice: Increased Aβ42 and Aβ deposition without APP overproduction. Neurobiol. Aging 2002, 23, 335–348. [Google Scholar] [CrossRef]
- Holcomb, L.A.; Gordon, M.N.; Jantzen, P.; Hsiao, K.; Duff, K.; Morgan, D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: Lack of association with amyloid deposits. Behav. Genet. 1999, 29, 177–185. [Google Scholar] [CrossRef]
- Huang, X.; Yee, B.; Nag, S.; Chan, S.; Tang, F. Behavioral and neurochemical characterization of transgenic mice carrying the human presenilin-1 gene with or without the leucine-to-proline mutation at codon 235. Exp. Neurol. 2003, 183, 673–681. [Google Scholar] [CrossRef]
- Janus, C.; D’Amelio, S.; Amitay, O.; Chishti, M.; Strome, R.; Fraser, P.; Carlson, G.; Roder, J.; George–Hyslop, P.S.; Westaway, D. Spatial learning in transgenic mice expressing human presenilin 1 (PS1) transgenes. Neurobiol. Aging 2000, 21, 541–549. [Google Scholar] [CrossRef]
- Dineley, K.T.; Xia, X.; Bui, D.; Sweatt, J.D.; Zheng, H. Accelerated plaque accumulation, associative learning deficits, and up-regulation of α7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J. Biol. Chem. 2002, 277, 22768–22780. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: Evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef]
- Casas, C.; Sergeant, N.; Itier, J.-M.; Blanchard, V.; Wirths, O.; van der Kolk, N.; Vingtdeux, V.; van de Steeg, E.; Ret, G.; Canton, T. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Aβ 42 accumulation in a novel Alzheimer transgenic model. Am. J. Pathol. 2004, 165, 1289–1300. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple Isoforms of Human Microtubule-Associated Protein-Tau—Sequences and Localization in Neurofibrillary Tangles of Alzheimers-Disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Hampel, H.; Blennow, K.; Shaw, L.M.; Hoessler, Y.C.; Zetterberg, H.; Trojanowski, J.Q. Total and phosphorylated tau protein as biological markers of Alzheimer’s disease. Exp. Gerontol. 2010, 45, 30–40. [Google Scholar] [CrossRef] [PubMed]
- McMillan, P.; Korvatska, E.; Poorkaj, P.; Evstafjeva, Z.; Robinson, L.; Greenup, L.; Leverenz, J.; Schellenberg, G.D.; D’Souza, I. Tau Isoform Regulation Is Region- and Cell-Specific in Mouse Brain. J. Comp. Neurol. 2008, 511, 788–803. [Google Scholar] [CrossRef]
- Liu, C.; Götz, J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS ONE 2013, 8, e84849. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.-Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid ß-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef]
- Wegmann, S.; Maury, E.A.; Kirk, M.J.; Saqran, L.; Roe, A.; DeVos, S.L.; Nicholls, S.; Fan, Z.; Takeda, S.; Cagsal-Getkin, O. Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J. 2015, 34, 3028–3041. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, J.J.; Kinney, J.W.; Cummings, J.L. Animal systems in the development of treatments for Alzheimer’s disease: Challenges, methods, and implications. Neurobiol. Aging 2013, 34, 169–183. [Google Scholar] [CrossRef]
- Mullane, K.; Williams, M. Alzheimer’s therapeutics: Continued clinical failures question the validity of the amyloid hypothesis-but what lies beyond? Biochem. Pharm. 2013, 85, 289–305. [Google Scholar] [CrossRef]
- Allen, B.; Ingram, E.; Takao, M.; Smith, M.J.; Jakes, R.; Virdee, K.; Yoshida, H.; Holzer, M.; Craxton, M.; Emson, P.C. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J. Neurosci. 2002, 22, 9340–9351. [Google Scholar] [CrossRef]
- Förstl, H.; Kurz, A. Clinical features of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Perel, P.; Roberts, I.; Sena, E.; Wheble, P.; Briscoe, C.; Sandercock, P.; Macleod, M.; Mignini, L.E.; Jayaram, P.; Khan, K.S. Comparison of treatment effects between animal experiments and clinical trials: Systematic review. Br. Med. J. 2007, 334, 197–200. [Google Scholar] [CrossRef]
- Ioannidis, J.P.A. Extrapolating from Animals to Humans. Sci. Transl. Med. 2012, 4, 151ps15. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Boche, D.; Wilkinson, D.; Yadegarfar, G.; Hopkins, V.; Bayer, A.; Jones, R.W.; Bullock, R.; Love, S.; Neal, J.W.; et al. Long-term effects of A beta(42) immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet 2008, 372, 216–223. [Google Scholar] [CrossRef]
- Gilman, S.; Koller, M.; Black, R.S.; Jenkins, L.; Griffith, S.G.; Fox, N.C.; Eisner, L.; Kirby, L.; Rovira, M.B.; Forette, F.; et al. Clinical effects of A beta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.P.; Johnson-Wood, K.; et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Dodart, J.C.; Bales, K.R.; Gannon, K.S.; Greene, S.J.; DeMattos, R.B.; Mathis, C.; DeLong, C.A.; Wu, S.; Wu, X.; Holtzman, D.M.; et al. Immunization reverses memory deficits without reducing brain A beta burden in Alzheimer’s disease model. Nat. Neurosci. 2002, 5, 452–457. [Google Scholar] [CrossRef]
- Salloway, S.P.; Black, R.; Sperling, R.; Fox, N.; Gilman, S.; Schenk, D.; Grundman, M. A Phase 2 Multiple Ascending Dose Trial of Bapineuzumab in Mild to Moderate Alzheimer Disease Reply. Neurology 2010, 74, 2026–2027. [Google Scholar]
- Farlow, M.; Amold, S.E.; van Dyck, C.H.; Aisen, P.S.; Snider, B.J.; Porsteinsson, A.P.; Friedrich, S.; Dean, R.A.; Gonzales, C.; Sethuraman, G.; et al. Safety and biomarker effects of Solanezumab in patients with Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 261–271. [Google Scholar] [CrossRef]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.Y.; Aisen, P.S.; et al. Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Abyadeh, M.; Gupta, V.; Gupta, V.; Chitranshi, N.; Wu, Y.; Amirkhani, A.; Meyfour, A.; Sheriff, S.; Shen, T.; Dhiman, K. Comparative Analysis of Aducanumab, Zagotenemab and Pioglitazone as Targeted Treatment Strategies for Alzheimer’s Disease. Aging Dis. 2022, 12, 1964–1976. [Google Scholar]
- Holtzman, D.M.; Bales, K.R.; Tenkova, T.; Fagan, A.M.; Parsadanian, M.; Sartorius, L.J.; Mackey, B.; Olney, J.; McKeel, D.; Wozniak, D. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2892–2897. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.R.; Hirsch, A.M.; Broihier, M.L.; Miller, C.M.; Neilson, L.E.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci. 2017, 37, 637–647. [Google Scholar] [CrossRef]
- Lewandowski, C.T.; Weng, J.M.; LaDu, M.J. Alzheimer’s disease pathology in APOE transgenic mouse models: The Who, What, When, Where, Why, and How. Neurobiol. Dis. 2020, 139, 104811. [Google Scholar] [CrossRef]
- Do Carmo, S.; Cuello, A.C. Modeling Alzheimer’s disease in transgenic rats. Mol. Neurodegener 2013, 8, 37. [Google Scholar] [CrossRef]
- Lin, J.H. Species similarities and differences in pharmacokinetics. Drug Metab. Dispos. 1995, 23, 1008–1021. [Google Scholar]
- Jacob, H.J.; Kwitek, A.E. Rat genetics: Attachign physiology and pharmacology to the genome. Nat. Rev. Genet. 2002, 3, 33–42. [Google Scholar] [CrossRef]
- Echeverria, V.; Ducatenzeiler, A.; Alhonen, L.; Janne, J.; Grant, S.M.; Wandosell, F.; Muro, A.; Baralle, F.; Li, H.S.; Duff, K.; et al. Rat transgenic models with a phenotype of intracellular A beta accumulation in hippocampus and cortex. J. Alzheimer’s Dis. 2004, 6, 209–219. [Google Scholar] [CrossRef]
- Leon, W.C.; Canneva, F.; Partridge, V.; Allard, S.; Ferretti, M.T.; DeWilde, A.; Vercauteren, F.; Atifeh, R.; Ducatenzeiler, A.; Klein, W. A novel transgenic rat model with a full Alzheimer’s-like amyloid pathology displays pre-plaque intracellular amyloid-β-associated cognitive impairment. J. Alzheimer’s Dis. 2010, 20, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Flood, D.G.; Lin, Y.-G.; Lang, D.M.; Trusko, S.P.; Hirsch, J.D.; Savage, M.J.; Scott, R.W.; Howland, D.S. A transgenic rat model of Alzheimer’s disease with extracellular Aβ deposition. Neurobiol. Aging 2009, 30, 1078–1090. [Google Scholar] [CrossRef]
- Cohen, R.M.; Rezai-Zadeh, K.; Weitz, T.M.; Rentsendorj, A.; Gate, D.; Spivak, I.; Bholat, Y.; Vasilevko, V.; Glabe, C.G.; Breunig, J.J.; et al. A Transgenic Alzheimer Rat with Plaques, Tau Pathology, Behavioral Impairment, Oligomeric A beta, and Frank Neuronal Loss. J. Neurosci. 2013, 33, 6245–6256. [Google Scholar] [CrossRef]
- Hanes, J.; Zilka, N.; Bartkova, M.; Caletkova, M.; Dobrota, D.; Novak, M. Rat tau proteome consists of six tau isoforms: Implication for animal models of human tauopathies. J. Neurochem. 2009, 108, 1167–1176. [Google Scholar] [CrossRef]
- Kosik, K.S.; Orecchio, L.D.; Bakalis, S.; Neve, R.L. Developmentally regulated expression of specific tau sequences. Neuron 1989, 2, 1389–1397. [Google Scholar] [CrossRef]
- Filipcik, P.; Zilka, N.; Bugos, O.; Kucerak, J.; Koson, P.; Novak, P.; Novak, M. First transgenic rat model developing progressive cortical neurofibrillary tangles. Neurobiol. Aging 2012, 33, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Koson, P.; Zilka, N.; Kovac, A.; Kovacech, B.; Korenova, M.; Filipcik, P.; Novak, M. Truncated tau expression levels determine life span of a rat model of tauopathy without causing neuronal loss or correlating with terminal neurofibrillary tangle load. Eur. J. Neurosci. 2008, 28, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Hrnkova, M.; Zilka, N.; Minichova, Z.; Koson, P.; Novak, M. Neurodegeneration caused by expression of human truncated tau leads to progressive neurobehavioural impairment in transgenic rats. Brain Res. 2007, 1130, 206–213. [Google Scholar] [CrossRef]
- Aigner, B.; Renner, S.; Kessler, B.; Klymiuk, N.; Kurome, M.; Wunsch, A.; Wolf, E. Transgenic pigs as models for translational biomedical research. J. Mol. Med. 2010, 88, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.L.; Lin, L.; Bolund, L.; Jensen, T.G.; Sorensen, C.B. Genetically modified pigs for biomedical research. J. Inherit. Metab. Dis. 2012, 35, 695–713. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, J.C.; Bawden, C.S.; Rudiger, S.R.; McLaughlan, C.J.; Reid, S.J.; Waldvogel, H.J.; MacDonald, M.E.; Gusella, J.F.; Walker, S.K.; Kelly, J.M. An ovine transgenic Huntington’s disease model. Hum. Mol. Genet. 2010, ddq063. [Google Scholar] [CrossRef]
- Johnstone, E.; Chaney, M.; Norris, F.; Pascual, R.; Little, S. Conservation of the sequence of the Alzheimer’s disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Mol. Brain Res. 1991, 10, 299–305. [Google Scholar] [CrossRef]
- Reid, S.J.; Mckean, N.E.; Henty, K.; Portelius, E.; Blennow, K.; Rudiger, S.R.; Bawden, C.S.; Handley, R.R.; Verma, P.J.; Faull, R.L. Alzheimer’s disease markers in the aged sheep (Ovis aries). Neurobiol. Aging 2017, 58, 112–119. [Google Scholar] [CrossRef]
- Papaioannou, N.; Tooten, P.C.; van Ederen, A.M.; Bohl, J.R.; Rofina, J.; Tsangaris, T.; Gruys, E. Immunohistochemical investigation of the brain of aged dogs. I. Detection of neurofibrillary tangles and of 4-hydroxynonenal protein, an oxidative damage product, in senile plaques. Amyloid 2001, 8, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Rosen, R.F.; Farberg, A.S.; Gearing, M.; Dooyema, J.; Long, P.M.; Anderson, D.C.; Davis-Turak, J.; Coppola, G.; Geschwind, D.H.; Pare, J.F.; et al. Tauopathy with paired helical filaments in an aged chimpanzee. J. Comp. Neurol. 2008, 509, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Gearing, M.; Rebeck, G.W.; Hyman, B.T.; Tigges, J.; Mirra, S.S. Neuropathology and apolipoprotein E profile of aged chimpanzees: Implications for Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 9382–9386. [Google Scholar] [CrossRef] [PubMed]
- Gearing, M.; Tigges, J.; Mori, H.; Mirra, S. β-Amyloid (Aβ) deposition in the brains of aged orangutans. Neurobiol. Aging 1997, 18, 139–146. [Google Scholar] [CrossRef]
- Kimura, N.; Nakamura, S.; Goto, N.; Narushima, E.; Hara, I.; Shichiri, S.; Saitou, K.; Nose, M.; Hayashi, T.; Kawamura, S.; et al. Senile plaques in an aged Western lowland gorilla. Exp. Anim. Tokyo 2001, 50, 77–81. [Google Scholar] [CrossRef]
- Perez, S.E.; Raghanti, M.A.; Hof, P.R.; Kramer, L.; Ikonomovic, M.D.; Lacor, P.N.; Erwin, J.M.; Sherwood, C.C.; Mufson, E.J. Alzheimer’s disease pathology in the neocortex and hippocampus of the western lowland gorilla (Gorilla gorilla gorilla). J. Comp. Neurol. 2013, 521, 4318–4338. [Google Scholar] [CrossRef]
- Perez, S.E.; Sherwood, C.C.; Cranfield, M.R.; Erwin, J.M.; Mudakikwa, A.; Hof, P.R.; Mufson, E.J. Early Alzheimer’s disease–type pathology in the frontal cortex of wild mountain gorillas (Gorilla beringei beringei). Neurobiol. Aging 2016, 39, 195–201. [Google Scholar] [CrossRef]
- Nakamura, S.; Kiatipattanasakul, W.; Nakayama, H.; Ono, F.; Sakakibara, I.; Yoshikawa, Y.; Goto, N.; Doi, K. Immunohistochemical characteristics of the constituents of senile plaques and amyloid angiopathy in aged cynomolgus monkeys. J. Med. Primatol. 1996, 25, 294–300. [Google Scholar] [CrossRef]
- Darusman, H.S.; Gjedde, A.; Sajuthi, D.; Schapiro, S.J.; Kalliokoski, O.; Kristianingrum, Y.P.; Handaryani, E.; Hau, J. Amyloid Beta1–42 and the Phoshorylated Tau Threonine 231 in Brains of Aged Cynomolgus Monkeys (Macaca fascicularis). Front. Aging Neurosci. 2014, 6, 313. [Google Scholar] [CrossRef]
- Nakamura, S.i.; Nakayama, H.; Goto, N.; Ono, F.; Sakakibara, I.; Yoshikawa, Y. Histopathological studies of senile plaques and cerebral amyloidosis in cynomolgus monkeys. J. Med. Primatol. 1998, 27, 244–252. [Google Scholar] [CrossRef]
- Nakamura, S.; Kimura, N.; Nishimura, M.; Torii, R.; Terao, K. Neurofibrillary tangles and senile plaques in aged cynomolgus monkeys. In Proceedings of the AFLAS and CALAS, Kyoto, Japan, 20–22 September 2008. [Google Scholar]
- Wisniewski, H.M.; Ghetti, B.; Terry, R.D. Neuritic Senile Plaques and Filamentous Changes in Aged Rhesus-Monkeys. J. Neuropathol. Exp. Neurol. 1973, 32, 566–584. [Google Scholar] [CrossRef]
- Paspalas, C.D.; Carlyle, B.C.; Leslie, S.; Preuss, T.M.; Crimins, J.L.; Huttner, A.J.; van Dyck, C.H.; Rosene, D.L.; Nairn, A.C.; Arnsten, A.F. The aged rhesus macaque manifests Braak stage III/IV Alzheimer’s-like pathology. Alzheimer’s Dement. 2018, 14, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Toledano, A.; Álvarez, M.; López-Rodríguez, A.; Toledano-Díaz, A.; Fernández-Verdecia, C. Does Alzheimer disease exist in all primates? Alzheimer pathology in non-human primates and its pathophysiological implications (II). Neurología 2014, 29, 42–55. [Google Scholar] [CrossRef]
- Latimer, C.S.; Shively, C.A.; Keene, C.D.; Jorgensen, M.J.; Andrews, R.N.; Register, T.C.; Montine, T.J.; Wilson, A.M.; Neth, B.J.; Mintz, A. A nonhuman primate model of early Alzheimer’s disease pathologic change: Implications for disease pathogenesis. Alzheimer’s Dement. 2019, 15, 93–105. [Google Scholar] [CrossRef]
- Ndung’u, M.; Hartig, W.; Wegner, F.; Mwenda, J.M.; Low, R.W.C.; Akinyemi, R.O.; Kalaria, R.N. Cerebral amyloid beta(42) deposits and microvascular pathology in ageing baboons. Neuropathol. Appl. Neurol. 2012, 38, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Schultz, C.; Dehghani, F.; Hubbard, G.B.; Thal, D.R.; Struckhoff, G.; Braak, E.; Braak, H. Filamentous tau pathology in nerve cells, astrocytes, and oligodendrocytes of aged baboons. J. Neuropathol. Exp. Neurol. 2000, 59, 39–52. [Google Scholar] [CrossRef]
- Schultz, C.; Hubbard, G.B.; Rub, U.; Braak, E.; Braak, H. Age-related progression of tau pathology in brains of baboons. Neurobiol. Aging 2000, 21, 905–912. [Google Scholar] [CrossRef]
- Lemere, C.A.; Oh, J.; Stanish, H.A.; Peng, Y.; Pepivani, I.; Fagan, A.M.; Yamaguchi, H.; Westmoreland, S.V.; Mansfield, K.G. Cerebral amyloid-beta protein accumulation with aging in cotton-top tamarins: A model of early Alzheimer’s disease? Rejuvenation Res. 2008, 11, 321–332. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Silhol, S.; Jallageas, V.; Mallet, J.; Bons, N.; Bouras, C.; Delaere, P. Quantitative analysis of tau protein-immunoreactive accumulations and β amyloid protein deposits in the cerebral cortex of the mouse lemur, Microcebus murinus. Acta Neuropathol. 1997, 94, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Kraska, A.; Dorieux, O.; Picq, J.-L.; Petit, F.; Bourrin, E.; Chenu, E.; Volk, A.; Perret, M.; Hantraye, P.; Mestre-Frances, N. Age-associated cerebral atrophy in mouse lemur primates. Neurobiol. Aging 2011, 32, 894–906. [Google Scholar] [CrossRef]
- Mestre, N.; Bons, N. Age-related cytological changes and neuronal loss in basal forebrain cholinergic neurons in Microcebus murinus (Lemurian primate). Neurodegeneration 1993, 2, 25–32. [Google Scholar]
- Geula, C.; Nagykery, N.; Wu, C.-K. Amyloid-β deposits in the cerebral cortex of the aged common marmoset (Callithrix jacchus): Incidence and chemical composition. Acta Neuropathol. 2002, 103, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Callejas, J.D.; Fuchs, E.; Perez-Cruz, C. Evidence of tau hyperphosphorylation and dystrophic microglia in the common marmoset. Front. Aging Neurosci. 2016, 8, 315. [Google Scholar] [CrossRef] [PubMed]
- Elfenbein, H.A.; Rosen, R.F.; Stephens, S.L.; Switzer, R.C.; Smith, Y.; Pare, J.; Mehta, P.D.; Warzok, R.; Walker, L.C. Cerebral beta-amyloid angiopathy in aged squirrel monkeys. Histol. Histopathol. 2007, 22, 155–167. [Google Scholar]
- Walker, L.; Masters, C.; Beyreuther, K.; Price, D. Amyloid in the brains of aged squirrel monkeys. Acta Neuropathol. 1990, 80, 381–387. [Google Scholar] [CrossRef]
- Smith, D.; Chen, X.; Nonaka, M.; Trojanowski, J.; Lee, V.-Y.; Saatman, K.; Leoni, M.; Xu, B.; Wolf, J.; Meaney, D. Accumulation of amyloid β and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J. Neuropathol. Exp. Neurol. 1999, 58, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E.; Strothjohann, M. Abnormally phosphorylated tau protein related to the formation of neurofibrillary tangles and neuropil threads in the cerebral cortex of sheep and goat. Neurosci. Lett. 1994, 171, 1–4. [Google Scholar] [CrossRef]
- Nelson, P.; Saper, C. Ultrastructure of neurofibrillary tangles in the cerebral cortex of sheep. Neurobiol. Aging 1995, 16, 315–323. [Google Scholar] [CrossRef]
- Nelson, P.T.; Greenberg, S.G.; Saper, C.B. Neurofibrillary tangles in the cerebral cortex of sheep. Neurosci. Lett. 1994, 170, 187–190. [Google Scholar] [CrossRef]
- Nakamura, S.-I.; Nakayama, H.; Uetsuka, K.; Sasaki, N.; Uchida, K.; Goto, N. Senile plaques in an aged two-humped (Bactrian) camel (Camelus bactrianus). Acta Neuropathol. 1995, 90, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Härtig, W.; Klein, C.; Brauer, K.; Schüppel, K.-F.; Arendt, T.; Brückner, G.; Bigl, V. Abnormally phosphorylated protein tau in the cortex of aged individuals of various mammalian orders. Acta Neuropathol. 2000, 100, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Colle, M.-A.; Hauw, J.-J.; Crespeau, F.; Uchihara, T.; Akiyama, H.; Checler, F.; Pageat, P.; Duykaerts, C. Vascular and parenchymal Aβ deposition in the aging dog: Correlation with behavior. Neurobiol. Aging 2000, 21, 695–704. [Google Scholar] [CrossRef]
- Yu, C.H.; Song, G.S.; Yhee, J.Y.; Kim, J.H.; Im, K.S.; Nho, W.G.; Lee, J.H.; Sur, J.H. Histopathological and Immunohistochemical Comparison of the Brain of Human Patients with Alzheimer’s Disease and the Brain of Aged Dogs with Cognitive Dysfunction. J. Comp. Pathol. 2011, 145, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Abey, A.; Davies, D.; Goldsbury, C.; Buckland, M.; Valenzuela, M.; Duncan, T. Distribution of tau hyperphosphorylation in canine dementia resembles early Alzheimer’s disease and other tauopathies. Brain Pathol. 2021, 31, 144–162. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, M.; Chambers, J.K.; Uchida, K.; Nakayama, H. The Relation between canine cognitive dysfunction and age-related brain lesions. J. Vet. Med Sci. 2016, 78, 991–1006. [Google Scholar] [CrossRef]
- Schmidt, F.; Boltze, J.; Jäger, C.; Hofmann, S.; Willems, N.; Seeger, J.; Härtig, W.; Stolzing, A. Detection and quantification of β-amyloid, pyroglutamyl Aβ, and tau in aged canines. J. Neuropathol. Exp. Neurol. 2015, 74, 912–923. [Google Scholar] [CrossRef]
- Smolek, T.; Madari, A.; Farbakova, J.; Kandrac, O.; Jadhav, S.; Cente, M.; Brezovakova, V.; Novak, M.; Zilka, N. Tau hyperphosphorylation in synaptosomes and neuroinflammation are associated with canine cognitive impairment. J. Comp. Neurol. 2016, 524, 874–895. [Google Scholar] [CrossRef]
- Fiock, K.L.; Smith, J.D.; Crary, J.F.; Hefti, M.M. β-amyloid and tau pathology in the aging feline brain. J. Comp. Neurol. 2020, 528, 112–117. [Google Scholar] [CrossRef]
- Sordo Sordo, L. Neuropathology, Diagnosis, and Potential Treatment of Feline Cognitive Dysfunction Syndrome and Its Similarities to Alzheimer’s Disease; University of Edinburgh: Edinburgh, UK, 2021. [Google Scholar]
- Chambers, J.K.; Uchida, K.; Harada, T.; Tsuboi, M.; Sato, M.; Kubo, M.; Kawaguchi, H.; Miyoshi, N.; Tsujimoto, H.; Nakayama, H. Neurofibrillary tangles and the deposition of a beta amyloid peptide with a novel N-terminal epitope in the brains of wild Tsushima leopard cats. PLoS ONE 2012, 7, e46452. [Google Scholar] [CrossRef]
- Cork, L.C.; Powers, R.E.; Selkoe, D.J.; Davies, P.; Geyer, J.J.; Price, D.L. Neurofibrillary tangles and senile plaques in aged bears. J. Neuropathol. Exp. Neurol. 1988, 47, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Yoshino, T.; Yamaguchi, R.; Tateyama, S.; Kimoto, Y.; Nakayama, H.; Goto, N. Senile plaques and other senile changes in the brain of an aged American black bear. Vet. Pathol. 1995, 32, 412–414. [Google Scholar] [CrossRef]
- Roertgen, K.E.; Parisi, J.E.; Clark, H.B.; Barnes, D.L.; O’Brien, T.D.; Johnson, K.H. Aβ-associated cerebral angiopathy and senile plaques with neurofibrillary tangles and cerebral hemorrhage in an aged wolverine (Gulo gulo). Neurobiol. Aging 1996, 17, 243–247. [Google Scholar] [CrossRef]
- Takaichi, Y.; Chambers, J.K.; Takahashi, K.; Soeda, Y.; Koike, R.; Katsumata, E.; Kita, C.; Matsuda, F.; Haritani, M.; Takashima, A. Amyloid β and tau pathology in brains of aged pinniped species (sea lion, seal, and walrus). Acta Neuropathol. Commun. 2021, 9, 1–15. [Google Scholar] [CrossRef]
- Finch, C.E. Evolution of the human lifespan and diseases of aging: Roles of infection, inflammation, and nutrition. Proc. Natl. Acad. Sci. USA 2010, 107, 1718–1724. [Google Scholar] [CrossRef]
- Knight, A. The beginning of the end for chimpanzee experiments? Philos. Ethics Humanit. Med. 2008, 3, 1–14. [Google Scholar] [CrossRef]
- Shumaker, R.W.; Wich, S.A.; Perkins, L. Reproductive life history traits of female orangutans (Pongo spp.). In Primate Reproductive Aging; Karger Publishers: Basel, Switzerland, 2008; Volume 36, pp. 147–161. [Google Scholar]
- Nishida, T.; Corp, N.; Hamai, M.; Hasegawa, T.; Hiraiwa-Hasegawa, M.; Hosaka, K.; Hunt, K.D.; Itoh, N.; Kawanaka, K.; Matsumoto-Oda, A. Demography, female life history, and reproductive profiles among the chimpanzees of Mahale. Am. J. Primatol. Off. J. Am. Soc. Primatol. 2003, 59, 99–121. [Google Scholar] [CrossRef] [PubMed]
- Li, H.W.; Zhang, L.; Qin, C. Current state of research on non-human primate models of Alzheimer’s disease. Anim. Models Exp. Med. 2019, 2, 227–238. [Google Scholar] [CrossRef]
- Uno, H. Age-related pathology and biosenescent markers in captive rhesus macaques. Age 1997, 20, 1–13. [Google Scholar] [CrossRef]
- Souder, D.C.; Dreischmeier, I.A.; Smith, A.B.; Wright, S.; Martin, S.A.; Sagar, M.A.K.; Eliceiri, K.W.; Salamat, S.M.; Bendlin, B.B.; Colman, R.J. Rhesus monkeys as a translational model for late-onset Alzheimer’s disease. Aging Cell 2021, 20, e13374. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Picq, J.-L. Aging affects executive functions and memory in mouse lemur primates. Exp. Gerontol. 2007, 42, 223–232. [Google Scholar] [CrossRef]
- Mestre-Francés, N.; Trouche, S.G.; Fontes, P.; Lautier, C.; Devau, G.; Lasbleiz, C.; Dhenain, M.; Verdier, J.-M. Old Gray Mouse Lemur Behavior, Cognition, and Neuropathology. In Conn’s Handbook of Models for Human Aging; Elsevier: Amsterdam, The Netherlands, 2018; pp. 287–300. [Google Scholar]
- Bons, N.; Mestre, N.; Petter, A. Senile plaques and neurofibrillary changes in the brain of an aged lemurian primate, Microcebus murinus. Neurobiol. Aging 1992, 13, 99–105. [Google Scholar] [CrossRef]
- Bons, N.; Mestre, N.; Ritchie, K.; Petter, A.; Podlisny, M.; Selkoe, D. Identification of Amyloid-Beta Protein in the Brain of the Small, Short-Lived Lemurian Primate Microcebus-Murinus. Neurobiol. Aging 1994, 15, 215–220. [Google Scholar] [CrossRef]
- Bons, N.; Rieger, F.; Prudhomme, D.; Fisher, A.; Krause, K.H. Microcebus murinus: A useful primate model for human cerebral aging and Alzheimer’s disease? Genes Brain Behav. 2006, 5, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Dhenain, M.; Chenu, E.; Hisley, C.K.; Aujard, F.; Volk, A. Regional atrophy in the brain of lissencephalic mouse lemur primates: Measurement by automatic histogram-based segmentation of MR images. Magn. Reson. Med. Off. J. Int. Soc. Magn. Reson. Med. 2003, 50, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Okano, H.; Hikishima, K.; Iriki, A.; Sasaki, E. The common marmoset as a novel animal model system for biomedical and neuroscience research applications. In Seminars in Fetal and Neonatal Medicine; Elsevier: Amsterdam, The Netherlands, 2012; pp. 336–340. [Google Scholar]
- King, A. The search for better animal models of Alzheimer’s disease. Nature 2018, 559, S13. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.; Ridley, R.; Baker, H.; Maclean, C.; Honer, W.; Francis, P. Chronic elevation of amyloid precursor protein in the neocortex or hippocampus of marmosets with selective cholinergic lesions. J. Neural Transm. 2001, 108, 809–826. [Google Scholar] [CrossRef]
- Trouche, S.G.; Asuni, A.; Rouland, S.; Wisniewski, T.; Frangione, B.; Verdier, J.-M.; Sigurdsson, E.M.; Mestre-Francés, N. Antibody response and plasma Aβ1-40 levels in young Microcebus murinus primates immunized with Aβ1-42 and its derivatives. Vaccine 2009, 27, 957–964. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Seneca, N.; Cai, L.; Liow, J.-S.; Zoghbi, S.S.; Gladding, R.L.; Hong, J.; Pike, V.W.; Innis, R.B. Brain and whole-body imaging in nonhuman primates with [11C] MeS-IMPY, a candidate radioligand for β-amyloid plaques. Nuclear Med. Biol. 2007, 34, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.H.; Holland, J.P.; Stephenson, N.A.; Kassenbrock, A.; Rotstein, B.H.; Daignault, C.P.; Lewis, R.; Collier, L.; Hooker, J.M.; Vasdev, N. PET neuroimaging studies of [18F] CABS13 in a double transgenic mouse model of Alzheimer’s disease and nonhuman primates. ACS Chem. Neurosci. 2015, 6, 535–541. [Google Scholar] [CrossRef]
- Heuer, E.; Jacobs, J.; Du, R.; Wang, S.; Keifer, O.P.; Cintron, A.F.; Dooyema, J.; Meng, Y.; Zhang, X.; Walker, L.C. Amyloid-Related Imaging Abnormalities in an Aged Squirrel Monkey with Cerebral Amyloid Angiopathy. J. Alzheimer’s Dis. 2017, 57, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, J.; Watts, J.C.; Mensinger, Z.L.; Oehler, A.; Grillo, S.K.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc. Natl. Acad. Sci. USA 2012, 109, 11025–11030. [Google Scholar] [CrossRef]
- Goedert, M. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015, 349. [Google Scholar] [CrossRef]
- Ridley, R.; Baker, H.; Windle, C.; Cummings, R. Very long term studies of the seeding of β-amyloidosis in primates. J. Neural Transm. 2006, 113, 1243–1251. [Google Scholar] [CrossRef]
- Beckman, D.; Morrison, J.H. Towards developing a rhesus monkey model of early Alzheimer’s disease focusing on women’s health. Am. J. Primatol. 2021, e23289. [Google Scholar] [CrossRef]
- Forny-Germano, L.; e Silva, N.M.L.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.B. Alzheimer’s disease-like pathology induced by amyloid-β oligomers in nonhuman primates. J. Neurosci. 2014, 34, 13629–13643. [Google Scholar] [CrossRef]
- Beckman, D.; Chakrabarty, P.; Ott, S.; Dao, A.; Zhou, E.; Janssen, W.G.; Donis-Cox, K.; Muller, S.; Kordower, J.H.; Morrison, J.H. A novel tau-based rhesus monkey model of Alzheimer’s pathogenesis. Alzheimer’s Dement. 2021, 17, 933–945. [Google Scholar] [CrossRef]
- Seita, Y.; Morimura, T.; Watanabe, N.; Iwatani, C.; Tsuchiya, H.; Nakamura, S.; Suzuki, T.; Yanagisawa, D.; Tsukiyama, T.; Nakaya, M. Generation of transgenic cynomolgus monkeys overexpressing the gene for Amyloid-β precursor protein. J. Alzheimer’s Dis. 2020, 75, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Sasaguri, H.; Kumita, W.; Inoue, T.; Kurosaki, Y.; Nagata, K.; Mihira, N.; Sato, K.; Sakuma, T.; Yamamoto, T. A non-human primate model of familial Alzheimer’s disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zeiss, C.J. Utility of spontaneous animal models of Alzheimer’s disease in preclinical efficacy studies. Cell Tissue Res. 2020, 380, 273–286. [Google Scholar] [CrossRef]
- Bosch, M.N.; Pugliese, M.; Gimeno-Bayon, J.; Rodriguez, M.J.; Mahy, N. Dogs with Cognitive Dysfunction Syndrome: A Natural Model of Alzheimer’s Disease. Curr. Alzheimer Res. 2012, 9, 298–314. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Head, E. Brain aging in dogs: Parallels with human brain aging and Alzheimer’s disease. Vet. Ther. 2001, 2, 247–260. [Google Scholar] [PubMed]
- Head, E. A canine model of human aging and Alzheimer’s disease. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 1384–1389. [Google Scholar] [CrossRef]
- Prpar Mihevc, S.; Majdič, G. Canine Cognitive Dysfunction and Alzheimer’s Disease–Two Facets of the Same Disease? Front. Neurosci. 2019, 13, 604. [Google Scholar] [CrossRef]
- Insua, D.; Suárez, M.-L.; Santamarina, G.; Sarasa, M.; Pesini, P. Dogs with canine counterpart of Alzheimer’s disease lose noradrenergic neurons. Neurobiol. Aging 2010, 31, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Neilson, J.C.; Hart, B.L.; Cliff, K.D.; Ruehl, W.W. Prevalence of behavioral changes associated with age-related cognitive impairment in dogs. J. Am. Vet. Med. Assoc. 2001, 218, 1787–1791. [Google Scholar] [CrossRef]
- Salvin, H.E.; McGreevy, P.D.; Sachdev, P.S.; Valenzuela, M.J. The canine cognitive dysfunction rating scale (CCDR): A data-driven and ecologically relevant assessment tool. Vet. J. 2011, 188, 331–336. [Google Scholar] [CrossRef]
- Madari, A.; Farbakova, J.; Katina, S.; Smolek, T.; Novak, P.; Weissova, T.; Novak, M.; Zilka, N. Assessment of severity and progression of canine cognitive dysfunction syndrome using the CAnine DEmentia Scale (CADES). Appl. Anim. Behav. Sci. 2015, 171, 138–145. [Google Scholar] [CrossRef]
- Osella, M.; Re, G.; Odore, R.; Girardi, C.; Badino, P.; Barbero, R.; Bergamasco, L. Canine Cognitive Dysfunction: Prevalence, Clinical Signs and Treatment with a Nutraceutical; Purdue University Press: West Lafayette, IN, USA, 2005; pp. 66–72. [Google Scholar]
- Wisniewski, H.; Johnson, A.; Raine, C.; Kay, W.; Terry, R. Senile plaques and cerebral amyloidosis in aged dogs. A histochemical and ultrastructural study. Lab. Investig. 1970, 23, 287–296. [Google Scholar] [PubMed]
- Pugliese, M.; Geloso, M.C.; Carrasco, J.L.; Mascort, J.; Michetti, F.; Mahy, N. Canine cognitive deficit correlates with diffuse plaque maturation and S100β (−) astrocytosis but not with insulin cerebrospinal fluid level. Acta Neuropathol. 2006, 111, 519. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Murphy, M.P.; Mancuso, C.; Head, E. Atorvastatin treatment in a dog preclinical model of Alzheimer’s disease leads to up-regulation of haem oxygenase-1 and is associated with reduced oxidative stress in brain. Int. J. Neuropsychopharmacol. 2012, 15, 981–987. [Google Scholar] [CrossRef]
- Barone, E.; Mancuso, C.; Di Domenico, F.; Sultana, R.; Murphy, M.P.; Head, E.; Butterfield, D.A. Biliverdin reductase-A: A novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. J. Neurochem. 2012, 120, 135–146. [Google Scholar] [CrossRef]
- Di Domenico, F.; Perluigi, M.; Barone, E. Biliverdin Reductase-A correlates with inducible nitric oxide synthasein in atorvastatin treated aged canine brain. Neural Regen. Res. 2013, 8, 1925. [Google Scholar] [PubMed]
- Bosch, M.N.; Bayon, J.G.; Rodriguez, M.J.; Pugliese, M.; Mahy, N. Rapid improvement of canine cognitive dysfunction with immunotherapy designed for Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 482–493. [Google Scholar] [CrossRef]
- Head, E.; Moffat, K.; Das, P.; Sarsoza, E.; Poon, W.W.; Landsberg, G.; Cotman, C.W.; Murphy, M.P. beta-amyloid deposition and tau phosphorylation in clinically characterized aged cats. Neurobiol. Aging 2005, 26, 749–763. [Google Scholar] [CrossRef]
- Nakamura, S.-i.; Nakayama, H.; Kiatipattanasakul, W.; Uetsuka, K.; Uchida, K.; Goto, N. Senile plaques in very aged cats. Acta Neuropathol. 1996, 91, 437–439. [Google Scholar] [CrossRef]
- Klug, J.; Snyder, J.M.; Darvas, M.; Imai, D.M.; Church, M.; Latimer, C.; Keene, C.D.; Ladiges, W. Aging pet cats develop neuropathology similar to human Alzheimer’s disease. Aging Pathobiol. Ther. 2020, 2, 120–125. [Google Scholar] [CrossRef]
- Sordo, L.; Gunn-Moore, D.A. Cognitive dysfunction in cats: Update on neuropathological and behavioural changes plus clinical management. Vet. Rec. 2021, 188, e3. [Google Scholar] [CrossRef] [PubMed]
- Gunn-Moore, D.; Moffat, K.; Christie, L.A.; Head, E. Cognitive dysfunction and the neurobiology of ageing in cats. J. Small Anim. Pract. 2007, 48, 546–553. [Google Scholar] [CrossRef]
- Perleberg, C.; Kind, A.; Schnieke, A. Genetically engineered pigs as models for human disease. Dis. Models Mech. 2018, 11, dmm030783. [Google Scholar] [CrossRef] [PubMed]
- Prather, R.S.; Lorson, M.; Ross, J.W.; Whyte, J.J.; Walters, E. Genetically engineered pig models for human diseases. Annu. Rev. Anim. Biosci. 2013, 1, 203–219. [Google Scholar] [CrossRef]
- Walters, E.M.; Agca, Y.; Ganjam, V.; Evans, T. Animal models got you puzzled? Think pig. Ann. N. Y. Acad. Sci. 2011, 1245, 63–64. [Google Scholar] [CrossRef]
- Hoffe, B.; Holahan, M.R. The use of pigs as a translational model for studying neurodegenerative diseases. Front. Physiol. 2019, 10, 838. [Google Scholar] [CrossRef]
- Kragh, P.M.; Nielsen, A.L.; Li, J.; Du, Y.; Lin, L.; Schmidt, M.; Bøgh, I.B.; Holm, I.E.; Jakobsen, J.E.; Johansen, M.G. Hemizygous minipigs produced by random gene insertion and handmade cloning express the Alzheimer’s disease-causing dominant mutation APPsw. Transgenic Res. 2009, 18, 545–558. [Google Scholar] [CrossRef]
- Søndergaard, L.V.; Ladewig, J.; Dagnæs-Hansen, F.; Herskin, M.S.; Holm, I.E. Object recognition as a measure of memory in 1–2 years old transgenic minipigs carrying the APPsw mutation for Alzheimer’s disease. Transgenic Res. 2012, 21, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Hall, V.J.; Lindblad, M.M.; Jakobsen, J.E.; Gunnarsson, A.; Schmidt, M.; Rasmussen, M.A.; Volke, D.; Zuchner, T.; Hyttel, P. Impaired APP activity and altered Tau splicing in embryonic stem cell-derived astrocytes obtained from an APPsw transgenic minipig. Dis. Models Mech. 2015, 8, 1265–1278. [Google Scholar]
- Jakobsen, J.E.; Johansen, M.G.; Schmidt, M.; Liu, Y.; Li, R.; Callesen, H.; Melnikova, M.; Habekost, M.; Matrone, C.; Bouter, Y. Expression of the Alzheimer’s disease mutations AβPP695sw and PSEN1M146I in double-transgenic göttingen minipigs. J. Alzheimer’s Dis. 2016, 53, 1617–1630. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Pearce, D.A. Large animal models for Batten disease: A review. J. Child Neurol. 2013, 28, 1123–1127. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.; Jolly, R.; Palmer, D.; Tammen, I.; Broom, M.; McKinnon, R. Neuronal ceroid lipofuscinosis in Merino sheep. Aust. Vet. J. 2002, 80, 292–297. [Google Scholar] [CrossRef]
- Jolly, R.; Arthur, D.; Kay, G.; Palmer, D. Neuronal ceroid-lipofuscinosis in Borderdale sheep. N. Z. Vet. J. 2002, 50, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Jolly, R.; Janmaat, A.; West, D.a.; Morrison, I. Ovine ceroid-lipofuscinosis: A model of Batten’s disease. Neuropathol. Appl. Neurol. 1980, 6, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.M.; Kleemann, D.O.; Walker, S.K. Enhanced efficiency in the production of offspring from 4-to 8-week-old lambs. Theriogenology 2005, 63, 1876–1890. [Google Scholar] [CrossRef]
- Reid, S.J.; Patassini, S.; Handley, R.R.; Rudiger, S.R.; McLaughlan, C.J.; Osmand, A.; Jacobsen, J.C.; Morton, A.J.; Weiss, A.; Waldvogel, H.J. Further molecular characterisation of the OVT73 transgenic sheep model of Huntington’s disease identifies cortical aggregates. J. Huntingt. Dis. 2013, 2, 279–295. [Google Scholar]
- Handley, R.R.; Reid, S.J.; Brauning, R.; Maclean, P.; Mears, E.R.; Fourie, I.; Patassini, S.; Cooper, G.J.; Rudiger, S.R.; McLaughlan, C.J. Brain urea increase is an early Huntington’s disease pathogenic event observed in a prodromal transgenic sheep model and HD cases. Proc. Natl. Acad. Sci. USA 2017, 201711243. [Google Scholar] [CrossRef] [PubMed]
- Pfister, E.L.; DiNardo, N.; Mondo, E.; Borel, F.; Conroy, F.; Fraser, C.; Gernoux, G.; Han, X.; Hu, D.; Johnson, E. Artificial miRNAs reduce human mutant Huntingtin throughout the striatum in a transgenic sheep model of Huntington’s disease. Hum. Gene Ther. 2018, 29, 663–673. [Google Scholar] [CrossRef]
- Jiang, Y.; Xie, M.; Chen, W.; Talbot, R.; Maddox, J.F.; Faraut, T.; Wu, C.; Muzny, D.M.; Li, Y.; Zhang, W.; et al. The sheep genome illuminates biology of the rumen and lipid metabolism. Science 2014, 344, 1168–1173. [Google Scholar] [CrossRef]
- Kendrick, K.M.; da Costa, A.P.; Leigh, A.E.; Hinton, M.R.; Peirce, J.W. Sheep don’t forget a face. Nature 2001, 414, 165–166. [Google Scholar] [CrossRef]
- Morton, A.J.; Avanzo, L. Executive decision-making in the domestic sheep. PLoS ONE 2011, 6, e15752. [Google Scholar] [CrossRef] [PubMed]
- Perentos, N.; Martins, A.Q.; Watson, T.C.; Bartsch, U.; Mitchell, N.L.; Palmer, D.N.; Jones, M.W.; Morton, A.J. Translational neurophysiology in sheep: Measuring sleep and neurological dysfunction in CLN5 Batten disease affected sheep. Brain 2015, 138, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Sawiak, S.J.; Perumal, S.R.; Rudiger, S.R.; Matthews, L.; Mitchell, N.L.; McLaughlan, C.J.; Bawden, C.S.; Palmer, D.N.; Kuchel, T.; Morton, A.J. Rapid and Progressive Regional Brain Atrophy in CLN6 Batten Disease Affected Sheep Measured with Longitudinal Magnetic Resonance Imaging. PLoS ONE 2015, 10, e0132331. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).