
Serum Corticosterone and Insulin Resistance as Early Biomarkers in the hAPP23 Overexpressing Mouse Model of Alzheimer’s Disease

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
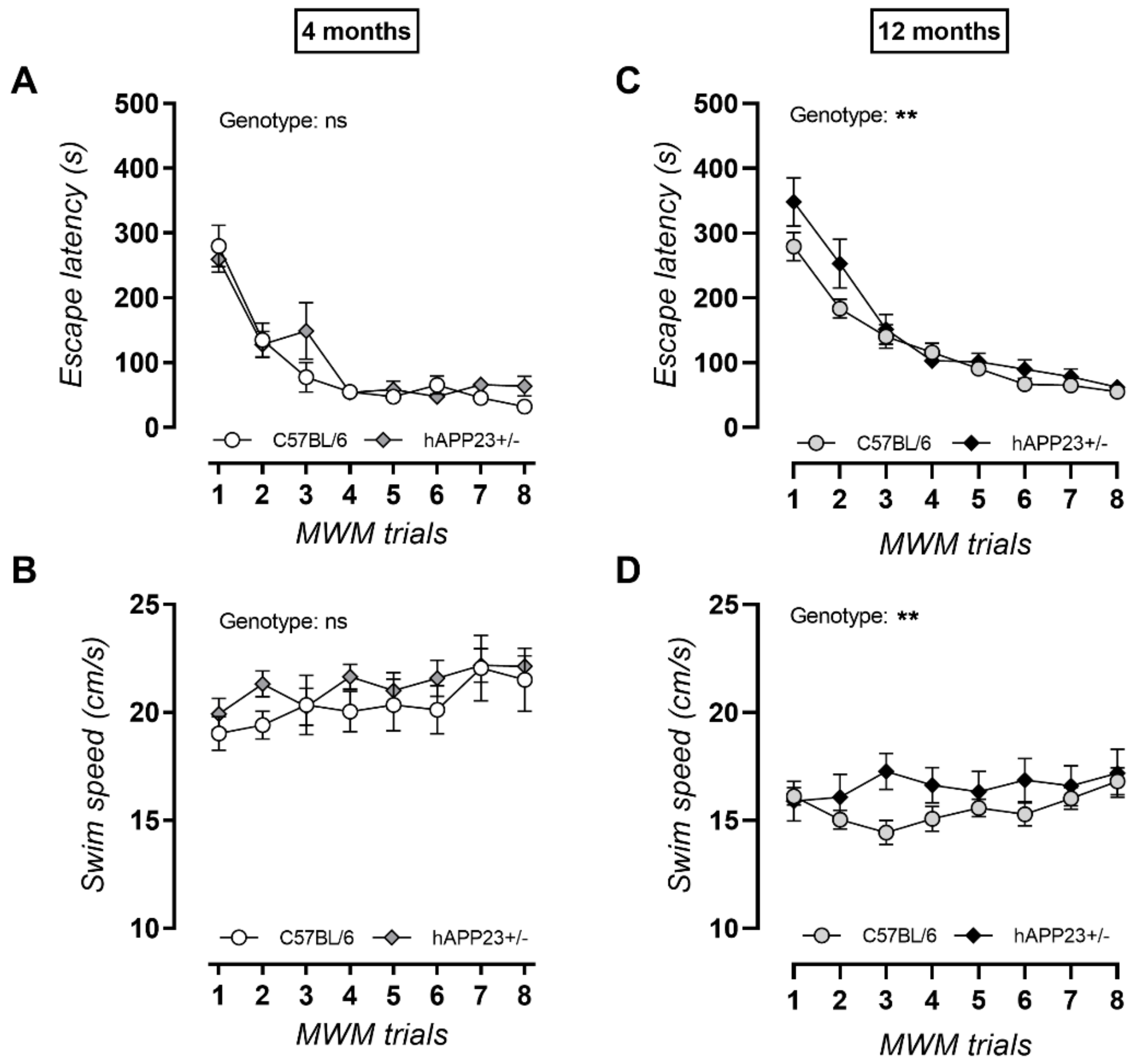
2.1. Progressive Cognitive Decline with Increasing Age for hAPP23+/− Mice
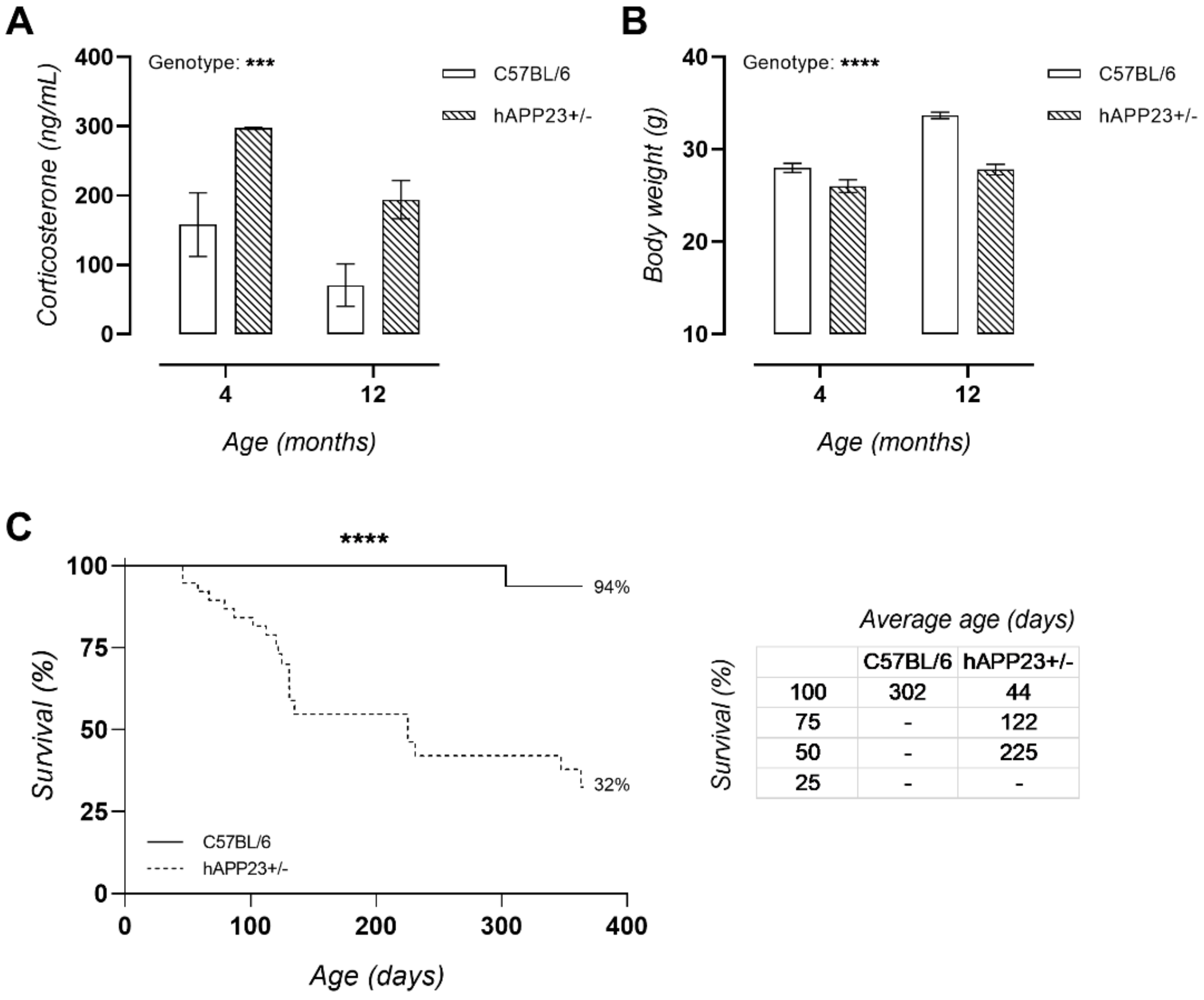
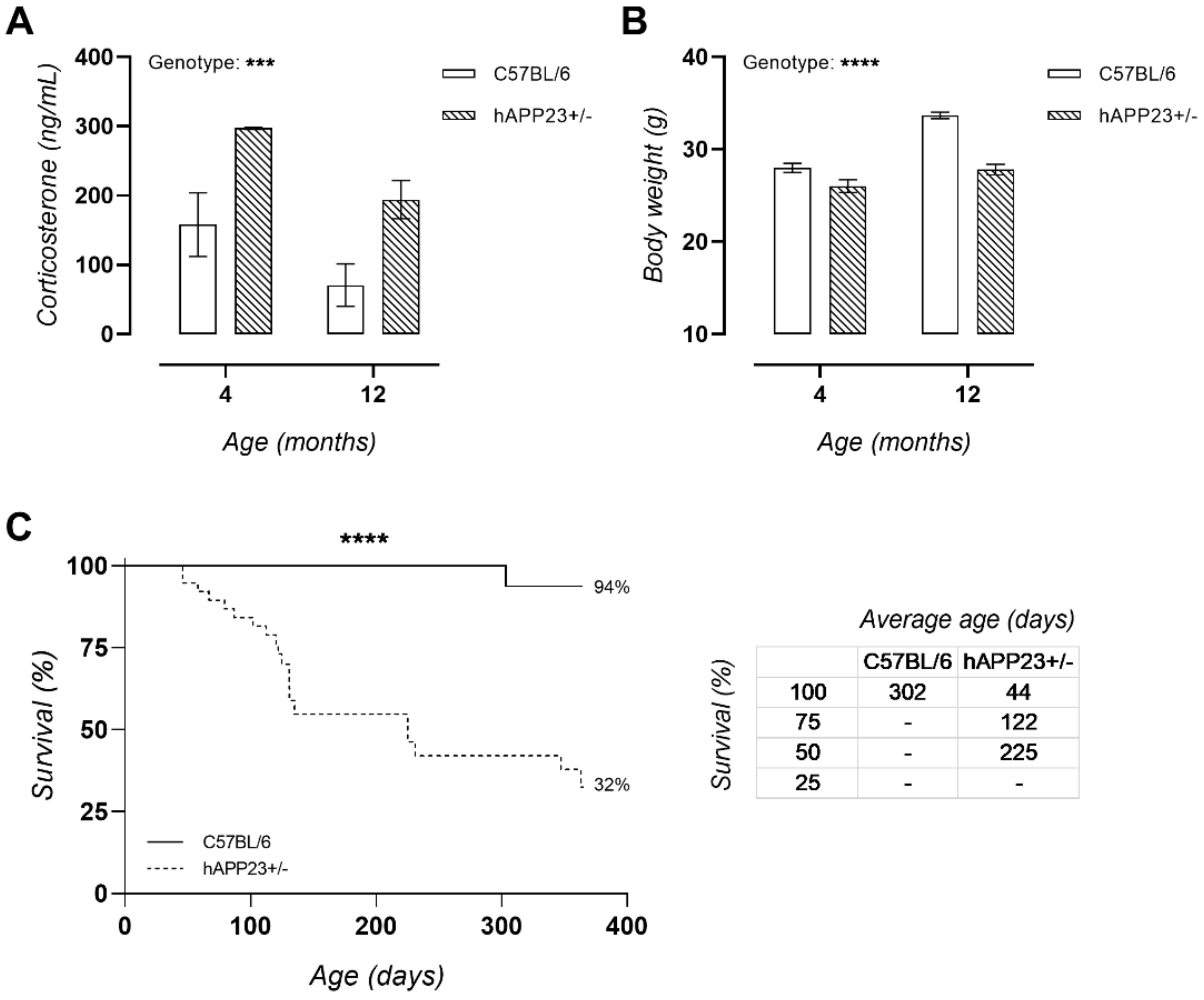
2.2. Chronically Elevated Serum Corticosterone Levels in hAPP23+/− Mice
2.3. hAPP23+/− Display Decreased Urinary β-Hydroxybutyrate Levels Compared to C57BL/6 Mice
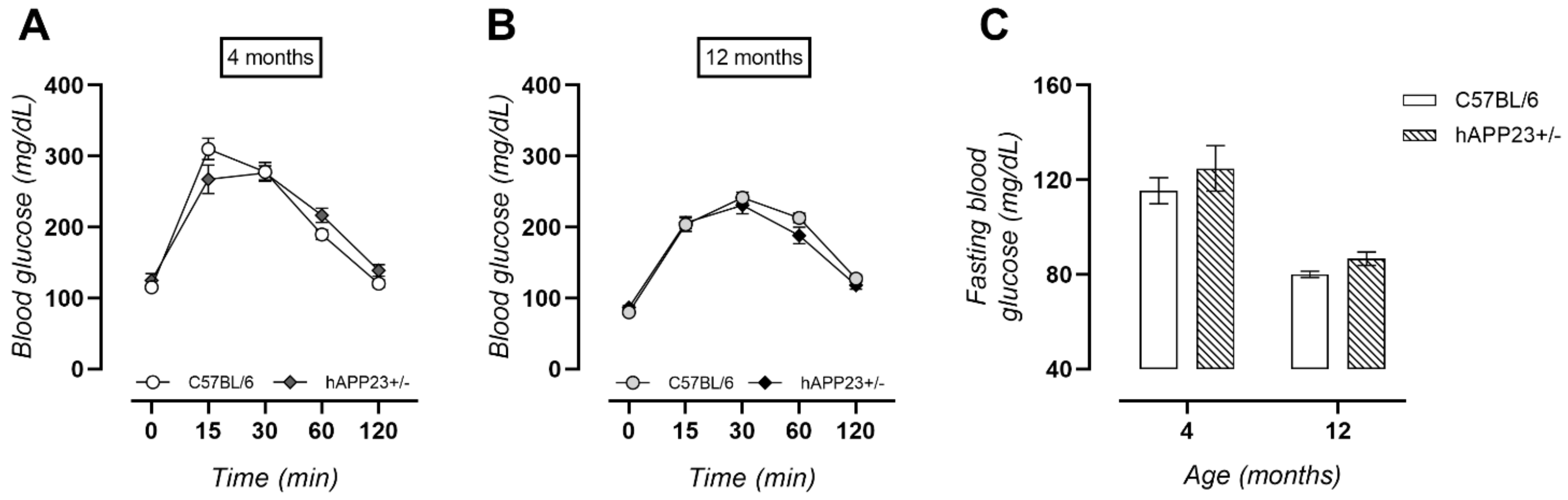
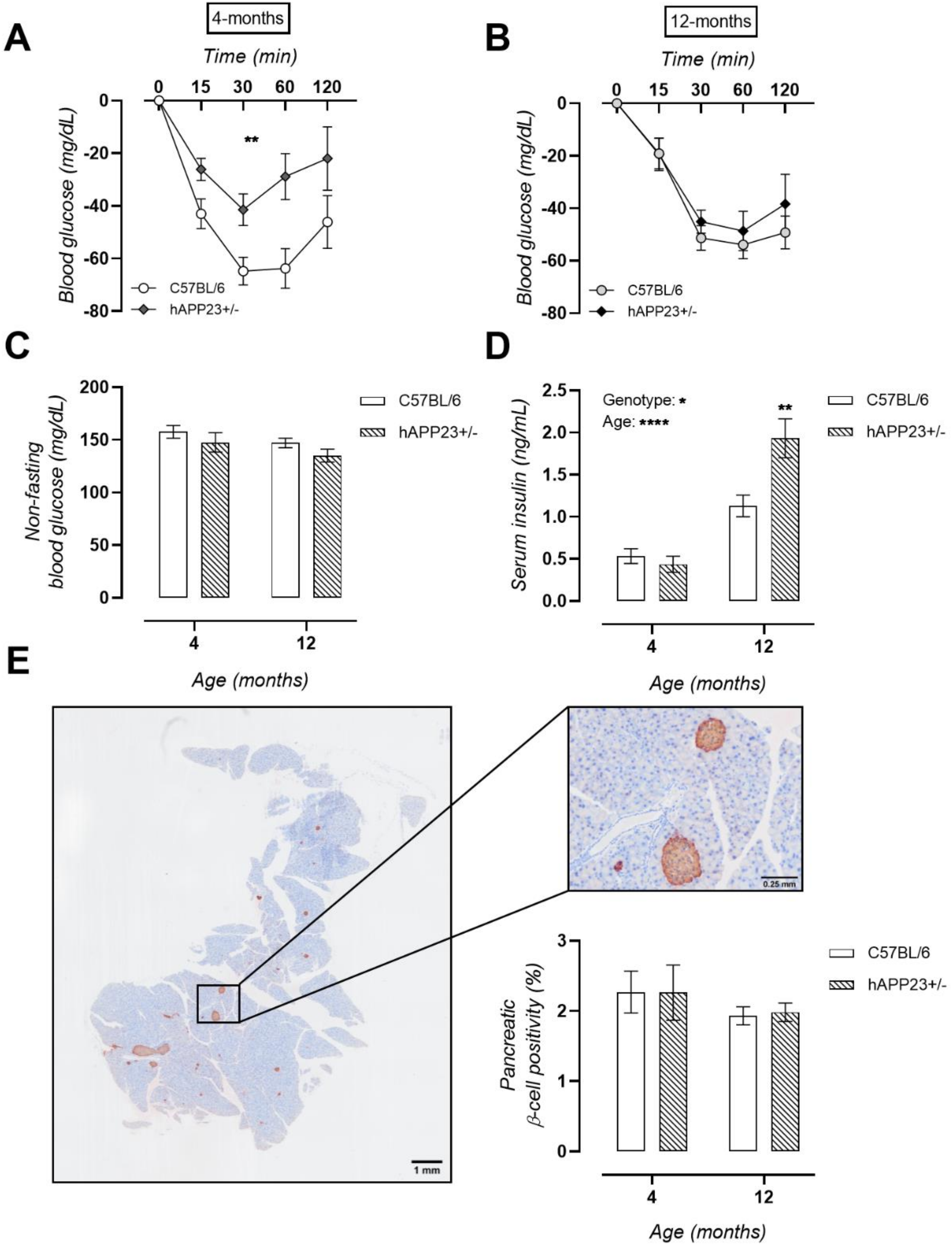
2.4. Unaltered Glucose Tolerance in hAPP23+/− Mice
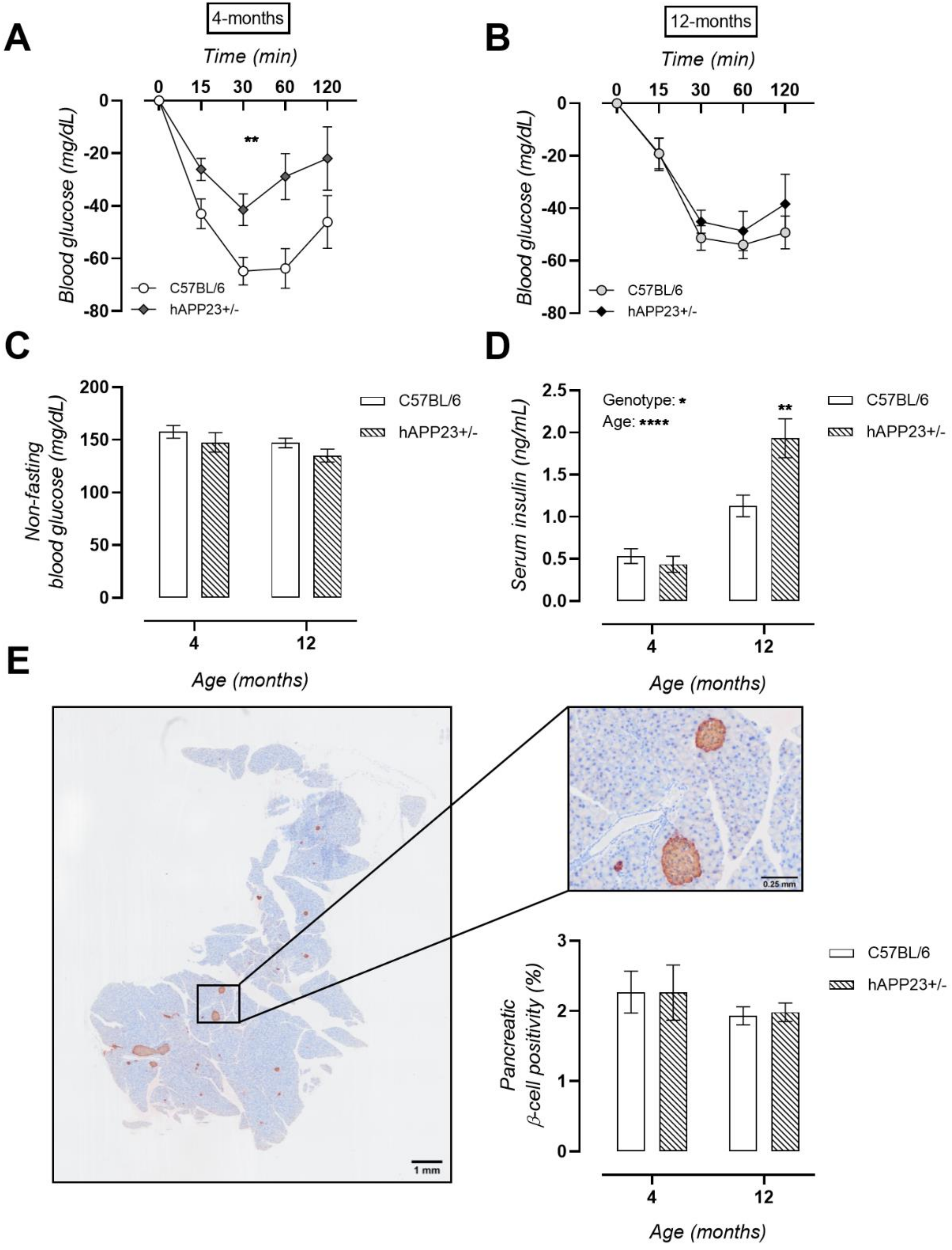
2.5. Pre-Symptomatic Insulin Resistance Evolves into Hyperinsulinaemia in hAPP23+/− Mice
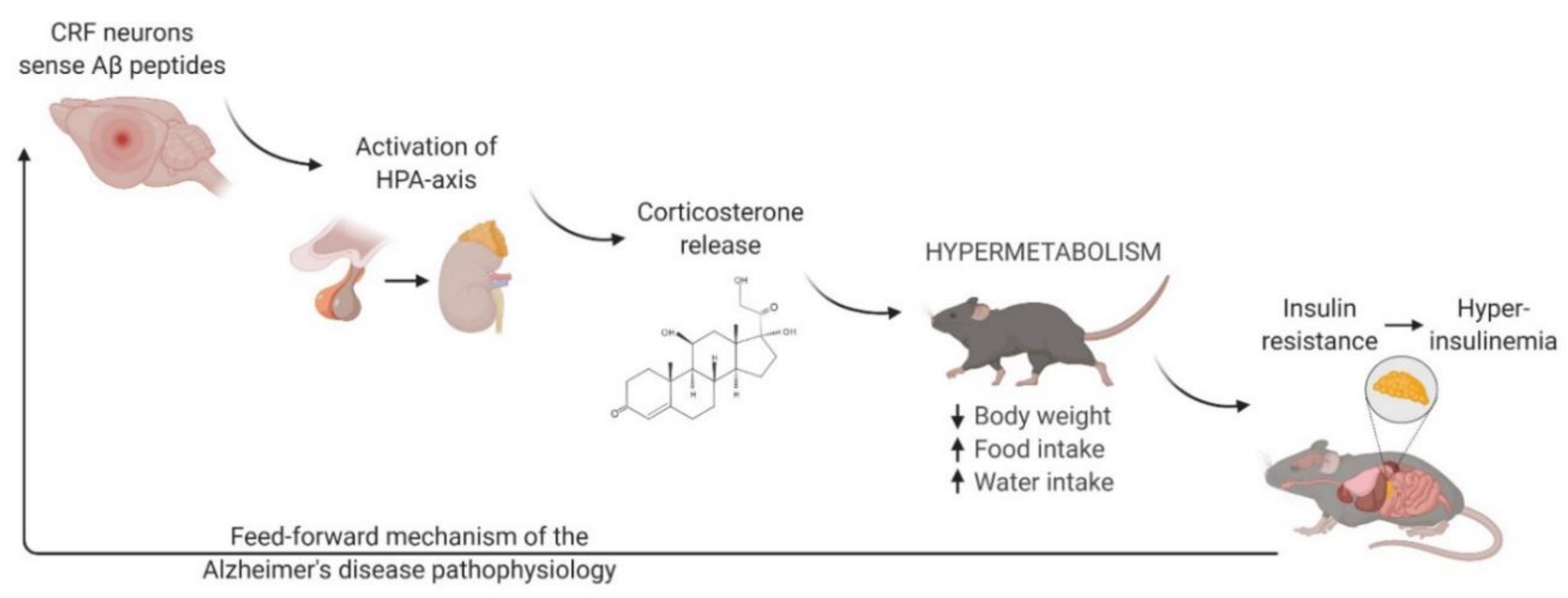
3. Discussion
4. Materials and Methods
4.1. Animal and Tissue Collection
4.2. Spatial Learning and Memory
4.3. Glucose and Insulin Tolerance Test (GTT, ITT)
4.4. Histological Analysis
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Perini, G.; Rodriguez-Vieitez, E.; Kadir, A.; Sala, A.; Savitcheva, I.; Nordberg, A. Clinical impact of 18 F-FDG-PET among memory clinic patients with uncertain diagnosis. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 612–622. [Google Scholar] [CrossRef]
- Yamanakkanavar, N.; Choi, J.Y.; Lee, B. MRI segmentation and classification of human brain using deep learning for diagnosis of alzheimer’s disease: A survey. Sensors 2020, 20, 3243. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. Biomarkers for Alzheimer’s disease: Current status and prospects for the future. J. Intern. Med. 2018, 284, 643–663. [Google Scholar] [PubMed] [Green Version]
- Cummings, J. Lessons learned from Alzheimer disease: Clinical trials with negative outcomes. Clin. Transl. Sci. 2018, 11, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Aisen, P.; Barton, R.; Bork, J.; Doody, R.; Dwyer, J.; Egan, J.; Feldman, H.; Lappin, D.; Truyen, L. Re-engineering Alzheimer clinical trials: Global Alzheimer’s Platform network. J. Prev. Alzheimer’s Dis. 2016, 3, 114. [Google Scholar]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Cong, W.-N.; Ji, S.; Rothman, S.; Maudsley, S.; Martin, B. Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr. Alzheimer Res. 2012, 9, 5–17. [Google Scholar] [CrossRef]
- Cheng, G.; Huang, C.; Deng, H.; Wang, H. Diabetes as a risk factor for dementia and mild cognitive impairment: A meta-analysis of longitudinal studies. Intern. Med. J. 2012, 42, 484–491. [Google Scholar] [CrossRef]
- Ikram, M.A.; Brusselle, G.G.; Murad, S.D.; van Duijn, C.M.; Franco, O.H.; Goedegebure, A.; Klaver, C.C.; Nijsten, T.E.; Peeters, R.P.; Stricker, B.H. The Rotterdam Study: 2018 update on objectives, design and main results. Eur. J. Epidemiol. 2017, 32, 807–850. [Google Scholar] [PubMed] [Green Version]
- Razay, G.; Vreugdenhil, A.; Wilcock, G. Obesity, abdominal obesity and Alzheimer disease. Dement. Geriatr. Cogn. Disord. 2006, 22, 173–176. [Google Scholar] [CrossRef]
- Strachan, M. RD Lawrence Lecture 2010. The brain as a target organ in Type 2 diabetes: Exploring the links with cognitive impairment and dementia. Diabet. Med. 2011, 28, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G. Alzheimer’s disease and insulin resistance: Translating basic science into clinical applications. J. Clin. Investig. 2013, 123, 531–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neergaard, J.S.; Dragsbæk, K.; Christiansen, C.; Nielsen, H.B.; Brix, S.; Karsdal, M.A.; Henriksen, K. Metabolic syndrome, insulin resistance, and cognitive dysfunction: Does your metabolic profile affect your brain? Diabetes 2017, 66, 1957–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Van Giau, V. Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3165. [Google Scholar] [CrossRef] [PubMed]
- Nisar, O.; Pervez, H.; Mandalia, B.; Waqas, M.; Sra, H.K. Type 3 Diabetes Mellitus: A Link Between Alzheimer’s Disease and Type 2 Diabetes Mellitus. Cureus 2020, 12, 11. [Google Scholar]
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, L.-Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [Green Version]
- Luchsinger, J.A.; Tang, M.-X.; Shea, S.; Mayeux, R. Hyperinsulinemia and risk of Alzheimer disease. Neurology 2004, 63, 1187–1192. [Google Scholar] [CrossRef]
- Willette, A.A.; Johnson, S.C.; Birdsill, A.C.; Sager, M.A.; Christian, B.; Baker, L.D.; Craft, S.; Oh, J.; Statz, E.; Hermann, B.P. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimer’s Dement. 2015, 11, 504–510.e1. [Google Scholar] [CrossRef] [Green Version]
- Geer, E.B.; Islam, J.; Buettner, C. Mechanisms of glucocorticoid-induced insulin resistance: Focus on adipose tissue function and lipid metabolism. Endocrinol. Metab. Clin. 2014, 43, 75–102. [Google Scholar] [CrossRef] [Green Version]
- Kamba, A.; Daimon, M.; Murakami, H.; Otaka, H.; Matsuki, K.; Sato, E.; Tanabe, J.; Takayasu, S.; Matsuhashi, Y.; Yanagimachi, M. Association between higher serum cortisol levels and decreased insulin secretion in a general population. PLoS ONE 2016, 11, e0166077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priyadarshini, E.; Anuradha, C.V. Glucocorticoid antagonism reduces insulin resistance and associated lipid abnormalities in high-fructose-fed mice. Can. J. Diabetes 2017, 41, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Pivonello, R.; De Leo, M.; Vitale, P.; Cozzolino, A.; Simeoli, C.; De Martino, M.C.; Lombardi, G.; Colao, A. Pathophysiology of diabetes mellitus in Cushing’s syndrome. Neuroendocrinology 2010, 92 (Suppl. 1), 77–81. [Google Scholar] [CrossRef] [PubMed]
- Van Staa, T.-P.; Leufkens, H.; Abenhaim, L.; Begaud, B.; Zhang, B.; Cooper, C. Use of oral corticosteroids in the United Kingdom. Qjm 2000, 93, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Bernini, G.; Tricò, D. Cushing’s syndrome and steroid dementia. Recent Pat. Endocr. Metab. Immune Drug Discov. 2016, 10, 50–55. [Google Scholar] [CrossRef]
- Ennis, G.E.; An, Y.; Resnick, S.M.; Ferrucci, L.; O’Brien, R.J.; Moffat, S.D. Long-term cortisol measures predict Alzheimer disease risk. Neurology 2017, 88, 371–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guldiken, S.; Guldiken, B. Subclinical Cushing’s syndrome is a potential cause of metabolic dementia and rapidly progressive Alzheimer-type dementia. Med. Hypotheses 2008, 71, 703–705. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-López, C.; Molina, M.G.; Salio, A.M. Rapidly Progressing Dementia as a Form of Presentation of Cushing Syndrome. Neurologia (Barcelona, Spain) 2019, 34, 339–340. [Google Scholar] [CrossRef]
- Zhang, S.-Q.; Cao, L.-L.; Liang, Y.-Y.; Wang, P. The molecular mechanism of chronic high-dose corticosterone-induced aggravation of cognitive impairment in APP/PS1 Transgenic Mice. Front. Mol. Neurosci. 2020, 13. [Google Scholar] [CrossRef]
- Maugard, M.; Doux, C.; Bonvento, G. A new statistical method to analyze Morris Water Maze data using Dirichlet distribution. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Snellman, A.; López-Picón, F.R.; Rokka, J.; Salmona, M.; Forloni, G.; Scheinin, M.; Solin, O.; Rinne, J.O.; Haaparanta-Solin, M. Longitudinal amyloid imaging in mouse brain with 11C-PIB: Comparison of APP23, Tg2576, and APPswe-PS1dE9 mouse models of Alzheimer disease. J. Nucl. Med. 2013, 54, 1434–1441. [Google Scholar] [CrossRef] [Green Version]
- Kelly, P.; Bondolfi, L.; Hunziker, D.; Schlecht, H.-P.; Carver, K.; Maguire, E.; Abramowski, D.; Wiederhold, K.-H.; Sturchler-Pierrat, C.; Jucker, M. Progressive age-related impairment of cognitive behavior in APP23 transgenic mice. Neurobiol. Aging 2003, 24, 365–378. [Google Scholar] [CrossRef]
- Van Dam, D.; d’Hooge, R.; Staufenbiel, M.; Van Ginneken, C.; Van Meir, F.; De Deyn, P.P. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur. J. Neurosci. 2003, 17, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Vloeberghs, E.; Van Dam, D.; Franck, F.; Serroyen, J.; Geert, M.; Staufenbiel, M.; De Deyn, P.P. Altered ingestive behavior, weight changes, and intact olfactory sense in an APP overexpression model. Behav. Neurosci. 2008, 122, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holcomb, L.; Gordon, M.N.; McGowan, E.; Yu, X.; Benkovic, S.; Jantzen, P.; Wright, K.; Saad, I.; Mueller, R.; Morgan, D. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 1998, 4, 97–100. [Google Scholar] [CrossRef]
- Touma, C.; Ambrée, O.; Görtz, N.; Keyvani, K.; Lewejohann, L.; Palme, R.; Paulus, W.; Schwarze-Eicker, K.; Sachser, N. Age-and sex-dependent development of adrenocortical hyperactivity in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2004, 25, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Venturelli, M.; Cè, E.; Limonta, E.; Muti, E.; Scarsini, R.; Brasioli, A.; Schena, F.; Esposito, F. Possible Predictors of Involuntary Weight Loss in Patients with Alzheimer?s Disease. PLoS ONE 2016, 11, e0157384. [Google Scholar] [CrossRef]
- Csernansky, J.G.; Dong, H.; Fagan, A.M.; Wang, L.; Xiong, C.; Holtzman, D.M.; Morris, J.C. Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am. J. Psychiatry 2006, 163, 2164–2169. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Ricobaraza, A.; Frechilla, D.; Franco, R.; Pérez-Mediavilla, A.; Garcia-Osta, A. Chronic mild stress accelerates the onset and progression of the Alzheimer’s disease phenotype in Tg2576 mice. J. Alzheimer’s Dis. 2012, 28, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Ouanes, S.; Popp, J. High cortisol and the risk of dementia and Alzheimer’s disease: A review of the literature. Front. Aging Neurosci. 2019, 11, 43. [Google Scholar] [CrossRef]
- Wilson, R.; Evans, D.; Bienias, J.; De Leon, C.M.; Schneider, J.; Bennett, D. Proneness to psychological distress is associated with risk of Alzheimer’s disease. Neurology 2003, 61, 1479–1485. [Google Scholar] [CrossRef]
- Geerlings, M.I.; Sigurdsson, S.; Eiriksdottir, G.; Garcia, M.E.; Harris, T.B.; Gudnason, V.; Launer, L.J. Salivary cortisol, brain volumes, and cognition in community-dwelling elderly without dementia. Neurology 2015, 85, 976–983. [Google Scholar] [CrossRef] [Green Version]
- Tatomir, A.; Micu, C.; Crivii, C. The impact of stress and glucocorticoids on memory. Clujul Med. 2014, 87, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapolsky, R.M. Stress, glucocorticoids, and damage to the nervous system: The current state of confusion. Stress 1996, 1, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Justice, N.J.; Huang, L.; Tian, J.-B.; Cole, A.; Pruski, M.; Hunt, A.J.; Flores, R.; Zhu, M.X.; Arenkiel, B.R.; Zheng, H. Posttraumatic stress disorder-like induction elevates β-amyloid levels, which directly activates corticotropin-releasing factor neurons to exacerbate stress responses. J. Neurosci. 2015, 35, 2612–2623. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.V.; Gordon, M.N.; Connor, K.E.; Good, R.A.; Engelman, R.W.; Mason, J.; Morgan, D.G.; Morgan, T.E.; Finch, C.E. Caloric restriction attenuates Aβ-deposition in Alzheimer transgenic models. Neurobiol. Aging 2005, 26, 995–1000. [Google Scholar] [CrossRef]
- Pedersen, W.A.; Culmsee, C.; Ziegler, D.; Herman, J.P.; Mattson, M.P. Aberrant stress response associated with severe hypoglycemia in a transgenic mouse model of Alzheimer’s disease. J. Mol. Neurosci. 1999, 13, 159–165. [Google Scholar] [CrossRef]
- Joly-Amado, A.; Serraneau, K.S.; Brownlow, M.; de Evsikova, C.M.; Speakman, J.R.; Gordon, M.N.; Morgan, D. Metabolic changes over the course of aging in a mouse model of tau deposition. Neurobiol. Aging 2016, 44, 62–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, E.M.; Verkhratsky, A.; Luckman, S.M.; Allan, S.M.; Lawrence, C.B. Hypermetabolism in a triple-transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 187–193. [Google Scholar] [CrossRef]
- Morais, J.B.S.; Severo, J.S.; Beserra, J.B.; de Oiveira, A.R.S.; Cruz, K.J.C.; de Sousa Melo, S.R.; do Nascimento, G.V.R.; de Macedo, G.F.S.; do Nascimento Marreiro, D. Association between cortisol, insulin resistance and zinc in obesity: A mini-review. Biol. Trace Elem. Res. 2019, 191, 323–330. [Google Scholar] [CrossRef]
- Purnell, J.Q.; Kahn, S.E.; Samuels, M.H.; Brandon, D.; Loriaux, D.L.; Brunzell, J.D. Enhanced cortisol production rates, free cortisol, and 11β-HSD-1 expression correlate with visceral fat and insulin resistance in men: Effect of weight loss. Am. J. Physiol. Endocrinol. Metab. 2009. [Google Scholar] [CrossRef]
- Malan, L.; Hamer, M.; von Känel, R.; van Wyk, R.D.; Sumner, A.E.; Nilsson, P.M.; Lambert, G.W.; Steyn, H.S.; Badenhorst, C.J.; Malan, N.T. A Stress Syndrome Prototype Reflects Type 3 Diabetes and Ischemic Stroke Risk: The SABPA Study. Biology 2021, 10, 162. [Google Scholar] [CrossRef]
- Stanley, M.; Macauley, S.L.; Holtzman, D.M. Changes in insulin and insulin signaling in Alzheimer’s disease: Cause or consequence? J. Exp. Med. 2016, 213, 1375–1385. [Google Scholar] [CrossRef]
- Baglietto-Vargas, D.; Shi, J.; Yaeger, D.M.; Ager, R.; LaFerla, F.M. Diabetes and Alzheimer’s disease crosstalk. Neurosci. Biobehav. Rev. 2016, 64, 272–287. [Google Scholar] [CrossRef]
- Lee, H.J.; Seo, H.I.; Cha, H.Y.; Yang, Y.J.; Kwon, S.H.; Yang, S.J. Diabetes and Alzheimer’s disease: Mechanisms and nutritional aspects. Clin. Nutr. Res. 2018, 7, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achanta, L.B.; Rae, C.D. β-Hydroxybutyrate in the brain: One molecule, multiple mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef]
- Odorcyk, F.; Duran-Carabali, L.; Rocha, D.; Sanches, E.; Martini, A.; Venturin, G.; Greggio, S.; da Costa, J.; Kucharski, L.; Zimmer, E. Differential glucose and beta-hydroxybutyrate metabolism confers an intrinsic neuroprotection to the immature brain in a rat model of neonatal hypoxia ischemia. Exp. Neurol. 2020, 330, 113317. [Google Scholar] [CrossRef]
- Buchanan, T.A.; Watanabe, R.M.; Xiang, A.H. Limitations in Surrogate Measures of Insulin Resistance; Oxford University Press: Oxford, UK, 2010; pp. 4874–4876. [Google Scholar]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.-H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef] [Green Version]
- Underwood, W.; Anthony, R. AVMA Guidelines for the Euthanasia of Animals: 2020 Edition. Retrieved March 2013, 30, 2020–2021. [Google Scholar]
- Van Dam, D.; Lenders, G.; De Deyn, P.P. Effect of Morris water maze diameter on visual-spatial learning in different mouse strains. Neurobiol. Learn. Mem. 2006, 85, 164–172. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hendrickx, J.O.; De Moudt, S.; Calus, E.; Martinet, W.; Guns, P.-J.D.F.; Roth, L.; De Deyn, P.P.; Van Dam, D.; De Meyer, G.R.Y. Serum Corticosterone and Insulin Resistance as Early Biomarkers in the hAPP23 Overexpressing Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 6656. https://doi.org/10.3390/ijms22136656
Hendrickx JO, De Moudt S, Calus E, Martinet W, Guns P-JDF, Roth L, De Deyn PP, Van Dam D, De Meyer GRY. Serum Corticosterone and Insulin Resistance as Early Biomarkers in the hAPP23 Overexpressing Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(13):6656. https://doi.org/10.3390/ijms22136656
Chicago/Turabian StyleHendrickx, Jhana O., Sofie De Moudt, Elke Calus, Wim Martinet, Pieter-Jan D. F. Guns, Lynn Roth, Peter P. De Deyn, Debby Van Dam, and Guido R. Y. De Meyer. 2021. "Serum Corticosterone and Insulin Resistance as Early Biomarkers in the hAPP23 Overexpressing Mouse Model of Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 13: 6656. https://doi.org/10.3390/ijms22136656
APA StyleHendrickx, J. O., De Moudt, S., Calus, E., Martinet, W., Guns, P.-J. D. F., Roth, L., De Deyn, P. P., Van Dam, D., & De Meyer, G. R. Y. (2021). Serum Corticosterone and Insulin Resistance as Early Biomarkers in the hAPP23 Overexpressing Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences, 22(13), 6656. https://doi.org/10.3390/ijms22136656