
Early Development of the GABAergic System and the Associated Risks of Neonatal Anesthesia


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Function and Development of the GABAergic System
2.1. GABAergic Inhibition and Neurovascular Coupling
2.2. Structure-Specific GABAergic System Function
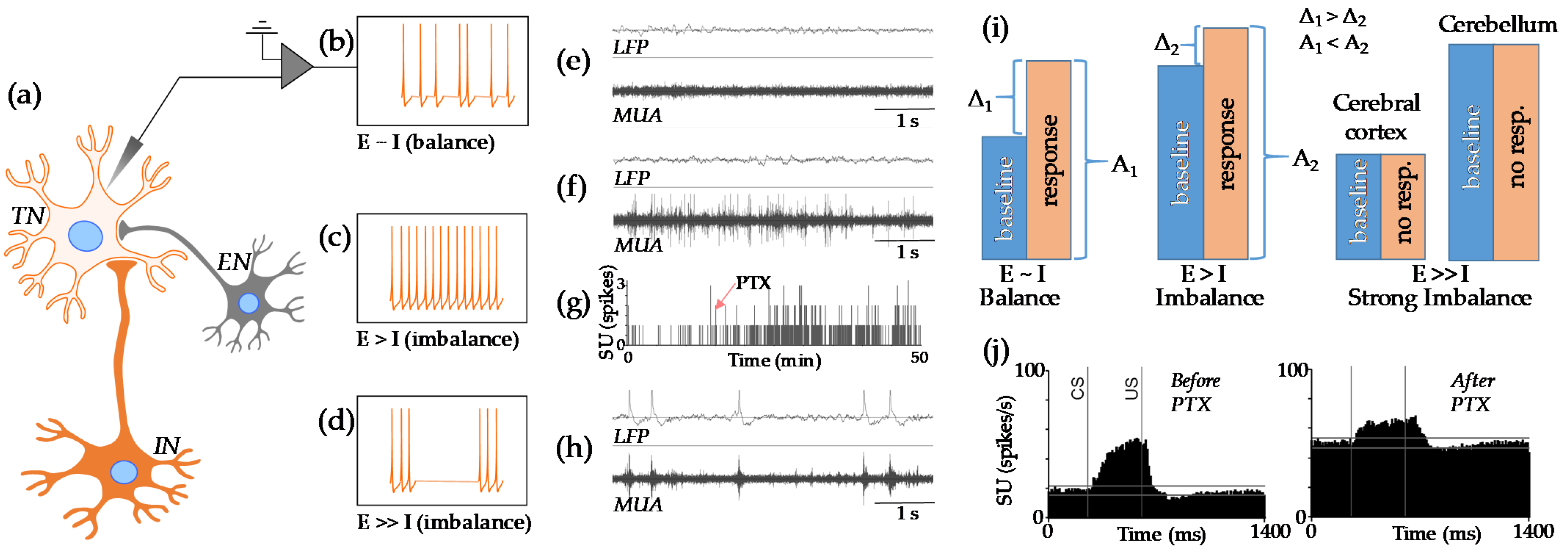
2.3. Physiology of the E-I Balance
2.4. GABA Signaling Development
2.5. Structure-Specific GABAergic Development
3. Consequences of Neonatal Anesthesia
3.1. Neuroapoptosis in GABAergic Structures
3.2. Short-Term Neurotoxicity
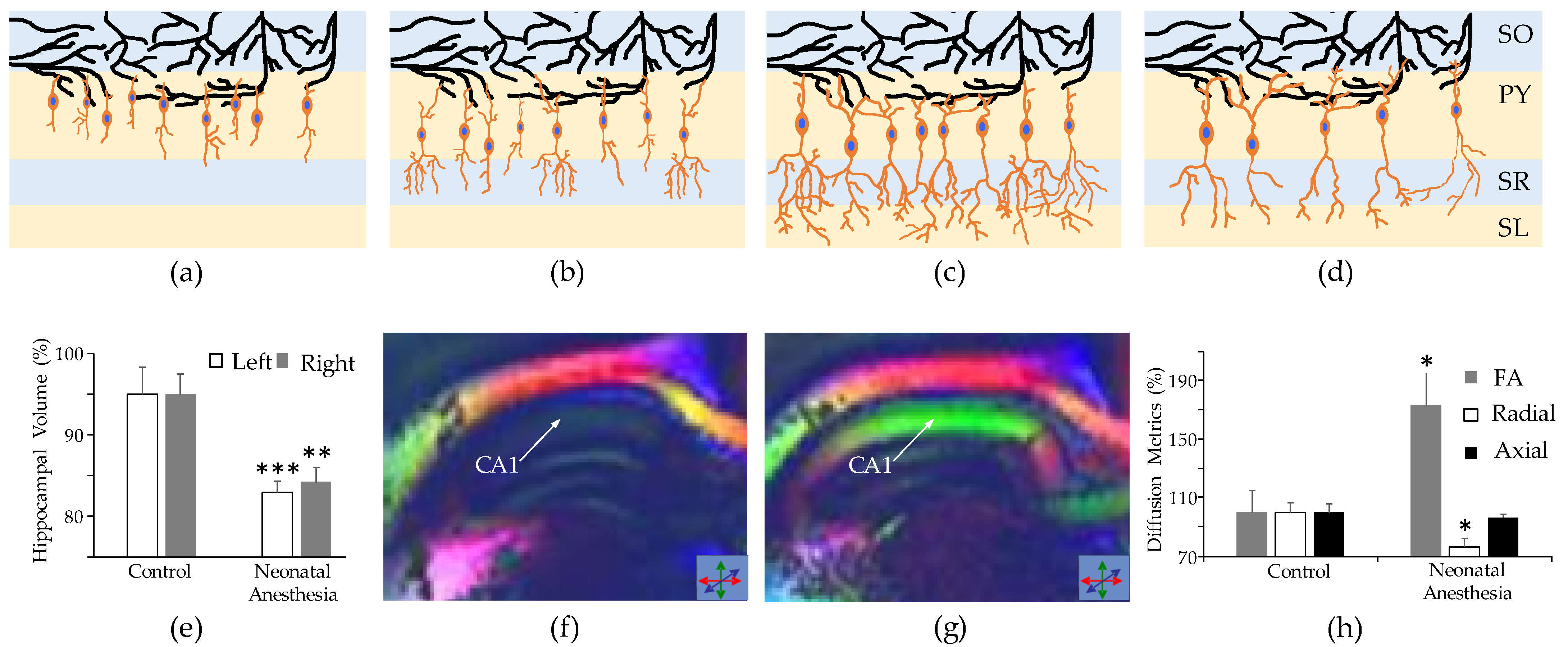
3.3. Long-Term Neurovascular Coupling Deficiency
3.4. E-I Imbalance after Neonatal Anesthesia
4. Methodological Outlook
4.1. Neuroimaging Techniques
4.2. Neuroimaging Alternatives
5. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Shi, Y.; Hu, D.; Rodgers, E.L.; Katusic, S.K.; Gleich, S.J.; Hanson, A.C.; Schroeder, D.R.; Flick, R.P.; Warner, D.O. Epidemiology of general anesthesia prior to age 3 in a population-based birth cohort. Paediatr. Anaesth. 2018, 28, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Wilder, R.T.; Flick, R.P.; Sprung, J.; Katusic, S.K.; Barbaresi, W.J.; Mickelson, C.; Gleich, S.J.; Schroeder, D.R.; Weaver, A.L.; Warner, D.O. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesth. 2009, 110, 796–804. [Google Scholar] [CrossRef]
- Chemaly, M.; El-Rajab, M.A.; Ziade, F.M.; Naja, Z.M. Effect of one anesthetic exposure on long-term behavioral changes in children. J. Clin. Anesth. 2014, 26, 551–556. [Google Scholar] [CrossRef]
- Crosby, G.; Davis, P.J. General anesthesia in infancy is associated with learning disabilities-or not. Anesth. Analg. 2013, 117, 1270–1272. [Google Scholar] [CrossRef] [PubMed]
- Sun, L. Early childhood general anaesthesia exposure and neurocognitive development. Br. J. Anaesth. 2010, 105 (Suppl. 1), i61–i68. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Zhang, J.; Wei, L.; Yu, S.P. Neurodevelopmental implications of the general anesthesia in neonate and infants. Exp. Neurol. 2015, 272, 50–60. [Google Scholar] [CrossRef]
- Olsen, E.A.; Brambrink, A.M. Anesthetic neurotoxicity in the newborn and infant. Curr. Opin. Anaesthesiol. 2013, 26, 535–542. [Google Scholar] [CrossRef]
- Bong, C.L.; Allen, J.C.; Kim, J.T. The effects of exposure to general anesthesia in infancy on academic performance at age 12. Anesth. Analg. 2013, 117, 1419–1428. [Google Scholar] [CrossRef]
- Maloney, S.E.; Yuede, C.M.; Creeley, C.E.; Williams, S.L.; Huffman, J.N.; Taylor, G.T.; Noguchi, K.N.; Wozniak, D.F. Repeated neonatal isoflurane exposures in the mouse induce apoptotic degenerative changes in the brain and relatively mild long-term behavioral deficits. Sci. Rep. 2019, 9, 2779. [Google Scholar] [CrossRef]
- Creeley, C.; Dikranian, K.; Dissen, G.; Martin, L.; Olney, J.; Brambrink, A. Propofol-induced apoptosis of neurones and oligodendrocytes in fetal and neonatal rhesus macaque brain. Br. J. Anaesth. 2013, 110 (Suppl. 1), i29–i38. [Google Scholar] [CrossRef]
- Kodama, M.; Satoh, Y.; Otsubo, Y.; Araki, Y.; Yonamine, R.; Masui, K.; Kazama, T. Neonatal desflurane exposure induces more robust neuroapoptosis than do isoflurane and sevoflurane and impairs working memory. J. Am. Soc. Anesthesiol. 2011, 115, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.K.; Johnson, S.A.; Dissen, G.A.; Martin, L.D.; Manzella, F.M.; Schenning, K.J.; Olney, J.W.; Brambrink, A.M. Isoflurane exposure for three hours triggers apoptotic cell death in neonatal macaque brain. Br. J. Anaesth. 2017, 119, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Satomoto, M.; Satoh, Y.; Terui, K.; Miyao, H.; Takishima, K.; Ito, M.; Imaki, J. Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. J. Am. Soc. Anesthesiol. 2009, 110, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Brambrink, A.M.; Evers, A.S.; Avidan, M.S.; Farber, N.B.; Smith, D.J.; Zhang, X.; Dissen, G.A.; Creeley, C.E.; Olney, J.W. Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. J. Am. Soc. Anesthesiol. 2010, 112, 834–841. [Google Scholar] [CrossRef]
- Aksenov, D.P.; Miller, M.J.; Dixon, C.J.; Drobyshevsky, A. Impact of anesthesia exposure in early development on learning and sensory functions. Dev. Psychobiol. 2020, 62, 559–572. [Google Scholar] [CrossRef]
- Amrock, L.G.; Starner, M.L.; Murphy, K.L.; Baxter, M.G. Long-term effects of single or multiple neonatal sevoflurane exposures on rat hippocampal ultrastructure. Anesthesiology 2015, 122, 87–95. [Google Scholar] [CrossRef]
- McCann, M.E.; de Graaff, J.C.; Dorris, L.; Disma, N.; Withington, D.; Bell, G.; Grobler, A.; Stargatt, R.; Hunt, R.W.; Sheppard, S.J.; et al. Neurodevelopmental outcome at 5 years of age after general anaesthesia or awake-regional anaesthesia in infancy (GAS): An international, multicentre, randomised, controlled equivalence trial. Lancet 2019, 393, 664–677. [Google Scholar] [CrossRef]
- Davidson, A.J.; Disma, N.; de Graaff, J.C.; Withington, D.E.; Dorris, L.; Bell, G.; Stargatt, R.; Bellinger, D.C.; Schuster, T.; Arnup, S.J.; et al. Neurodevelopmental outcome at 2 years of age after general anaesthesia and awake-regional anaesthesia in infancy (GAS): An international multicentre, randomised controlled trial. Lancet 2016, 387, 239–250. [Google Scholar] [CrossRef]
- Sun, L.S.; Li, G.; Miller, T.L.; Salorio, C.; Byrne, M.W.; Bellinger, D.C.; Ing, C.; Park, R.; Radcliffe, J.; Hays, S.R.; et al. Association Between a Single General Anesthesia Exposure Before Age 36 Months and Neurocognitive Outcomes in Later Childhood. JAMA 2016, 315, 2312–2320. [Google Scholar] [CrossRef]
- Warner, D.O.; Zaccariello, M.J.; Katusic, S.K.; Schroeder, D.R.; Hanson, A.C.; Schulte, P.J.; Buenvenida, S.L.; Gleich, S.J.; Wilder, R.T.; Sprung, J.; et al. Neuropsychological and Behavioral Outcomes after Exposure of Young Children to Procedures Requiring General Anesthesia: The Mayo Anesthesia Safety in Kids (MASK) Study. Anesthesiology 2018, 129, 89–105. [Google Scholar] [CrossRef]
- Grabowski, J.; Goldin, A.; Arthur, L.G.; Beres, A.L.; Guner, Y.S.; Hu, Y.Y.; Kawaguchi, A.L.; Kelley-Quon, L.I.; McAteer, J.P.; Miniati, D.; et al. The effects of early anesthesia on neurodevelopment: A systematic review. J. Pediatr. Surg. 2021, 56, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.P.; Yang, T.S.; Chung, C.H.; Chien, W.C.; Wong, C.S. Early childhood general anesthesia exposure associated with later developmental delay: A national population-based cohort study. PLoS ONE 2020, 15, e0238289. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, D.P.; Dmitriev, A.V.; Miller, M.J.; Wyrwicz, A.M.; Linsenmeier, R.A. Brain tissue oxygen regulation in awake and anesthetized neonates. Neuropharmacology 2018, 135, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shan, Y.; Tang, Z.; Gao, L.; Liu, H. Neuroprotective effects of dexmedetomidine against isoflurane-induced neuronal injury via glutamate regulation in neonatal rats. Drug Des. Dev. Ther. 2019, 13, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Manzella, F.M.; Joksimovic, S.M.; Orfila, J.E.; Fine, B.R.; Dietz, R.M.; Sampath, D.; Fiedler, H.K.; Tesic, V.; Atluri, N.; Raol, Y.H.; et al. Neonatal Ketamine Alters High-Frequency Oscillations and Synaptic Plasticity in the Subiculum But Does not Affect Sleep Macrostructure in Adolescent Rats. Front. Syst. Neurosci. 2020, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Istaphanous, G.K.; Ward, C.G.; Nan, X.; Hughes, E.A.; McCann, J.C.; McAuliffe, J.J.; Danzer, S.C.; Loepke, A.W. Characterization and quantification of isoflurane-induced developmental apoptotic cell death in mouse cerebral cortex. Anesth. Analg. 2013, 116, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Farrant, M.; Kaila, K. The cellular, molecular and ionic basis of GABA(A) receptor signalling. Prog Brain Res 2007, 160, 59–87. [Google Scholar]
- Tremblay, R.; Lee, S.; Rudy, B. GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 2016, 91, 260–292. [Google Scholar] [CrossRef]
- Gulyas, A.I.; Megias, M.; Emri, Z.; Freund, T.F. Total number and ratio of excitatory and inhibitory synapses converging onto single interneurons of different types in the CA1 area of the rat hippocampus. J. Neurosci. 1999, 19, 10082–10097. [Google Scholar] [CrossRef]
- Megias, M.; Emri, Z.; Freund, T.F.; Gulyas, A.I. Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience 2001, 102, 527–540. [Google Scholar] [CrossRef]
- Bloss, E.B.; Cembrowski, M.S.; Karsh, B.; Colonell, J.; Fetter, R.D.; Spruston, N. Structured Dendritic Inhibition Supports Branch-Selective Integration in CA1 Pyramidal Cells. Neuron 2016, 89, 1016–1030. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, D.P. Normal Development of Local Neurovascular Interactions and the Diagnostic Value of Resting State Functional MRI in Neurovascular Deficiency Based on the Example of Neonatal Anesthesia Exposure. Front. Neurol. 2021, 12, 664706. [Google Scholar] [CrossRef]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Nippert, A.R.; Biesecker, K.R.; Newman, E.A. Mechanisms Mediating Functional Hyperemia in the Brain. Neuroscientist 2018, 24, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Ross, W.N. Understanding calcium waves and sparks in central neurons. Nat. Rev. Neurosci. 2012, 13, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Huang, J.M.; Hampl, V.; Nelson, D.P.; Shultz, P.J.; Weir, E.K. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1994, 91, 7583–7587. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, L.; Chow, B.W.; Gu, C. Neuronal regulation of the blood-brain barrier and neurovascular coupling. Nat. Rev. Neurosci. 2020, 21, 416–432. [Google Scholar] [CrossRef]
- Gascoigne, D.A.; Drobyshevsky, A.; Aksenov, D.P. The Contribution of Dysfunctional Chloride Channels to Neurovascular Deficiency and Neurodegeneration. Front. Pharmacol. 2021, 12, 2741. [Google Scholar] [CrossRef]
- Kocharyan, A.; Fernandes, P.; Tong, X.K.; Vaucher, E.; Hamel, E. Specific subtypes of cortical GABA interneurons contribute to the neurovascular coupling response to basal forebrain stimulation. J. Cereb. Blood Flow Metab. 2008, 28, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, D.P.; Li, L.; Miller, M.J.; Wyrwicz, A.M. Role of the inhibitory system in shaping the BOLD fMRI response. Neuroimage 2019, 201, 116034. [Google Scholar] [CrossRef]
- Anenberg, E.; Chan, A.W.; Xie, Y.; LeDue, J.M.; Murphy, T.H. Optogenetic stimulation of GABA neurons can decrease local neuronal activity while increasing cortical blood flow. J. Cereb. Blood Flow Metab. 2015, 35, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Saillet, S.; Quilichini, P.P.; Ghestem, A.; Giusiano, B.; Ivanov, A.I.; Hitziger, S.; Vanzetta, I.; Bernard, C.; Benar, C.G. Interneurons contribute to the hemodynamic/metabolic response to epileptiform discharges. J. Neurophysiol. 2016, 115, 1157–1169. [Google Scholar] [CrossRef]
- Vaucher, E.; Tong, X.K.; Cholet, N.; Lantin, S.; Hamel, E. GABA neurons provide a rich input to microvessels but not nitric oxide neurons in the rat cerebral cortex: A means for direct regulation of local cerebral blood flow. J. Comp. Neurol. 2000, 421, 161–171. [Google Scholar] [CrossRef]
- Cauli, B.; Tong, X.K.; Rancillac, A.; Serluca, N.; Lambolez, B.; Rossier, J.; Hamel, E. Cortical GABA interneurons in neurovascular coupling: Relays for subcortical vasoactive pathways. J. Neurosci. 2004, 24, 8940–8949. [Google Scholar] [CrossRef]
- Rubenstein, J.L. Annual Research Review: Development of the cerebral cortex: Implications for neurodevelopmental disorders. J. Child Psychol. Psychiatry 2011, 52, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, K.; Steinecke, A.; Bolz, J. GABA through the ages: Regulation of cortical function and plasticity by inhibitory interneurons. Neural. Plast. 2012, 2012, 892784. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Kastner, S. Thalamic functions in distributed cognitive control. Nat. Neurosci. 2017, 20, 1669–1679. [Google Scholar] [CrossRef]
- Jager, P.; Ye, Z.; Yu, X.; Zagoraiou, L.; Prekop, H.T.; Partanen, J.; Jessell, T.M.; Wisden, W.; Brickley, S.G.; Delogu, A. Tectal-derived interneurons contribute to phasic and tonic inhibition in the visual thalamus. Nat. Commun. 2016, 7, 13579. [Google Scholar] [CrossRef]
- Kim, U.; Sanchez-Vives, M.V.; McCormick, D.A. Functional dynamics of GABAergic inhibition in the thalamus. Science 1997, 278, 130–134. [Google Scholar] [CrossRef]
- Pelkey, K.A.; Chittajallu, R.; Craig, M.T.; Tricoire, L.; Wester, J.C.; McBain, C.J. Hippocampal GABAergic Inhibitory Interneurons. Physiol. Rev. 2017, 97, 1619–1747. [Google Scholar] [CrossRef]
- Jimenez-Balado, J.; Eich, T.S. GABAergic dysfunction, neural network hyperactivity and memory impairments in human aging and Alzheimer’s disease. Semin. Cell Dev. Biol. 2021, 116, 146–159. [Google Scholar] [CrossRef]
- Fasano, C.; Rocchetti, J.; Pietrajtis, K.; Zander, J.F.; Manseau, F.; Sakae, D.Y.; Marcus-Sells, M.; Ramet, L.; Morel, L.J.; Carrel, D.; et al. Regulation of the Hippocampal Network by VGLUT3-Positive CCK- GABAergic Basket Cells. Front. Cell Neurosci. 2017, 11, 140. [Google Scholar] [CrossRef]
- Kullmann, D.M.; Lamsa, K.P. Long-term synaptic plasticity in hippocampal interneurons. Nat. Rev. Neurosci. 2007, 8, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, T.W.; Correia, M.M.; Ferreira, C.S.; Prescot, A.P.; Anderson, M.C. Hippocampal GABA enables inhibitory control over unwanted thoughts. Nat. Commun. 2017, 8, 1311. [Google Scholar] [CrossRef]
- Toyoda, H.; Li, X.Y.; Wu, L.J.; Zhao, M.G.; Descalzi, G.; Chen, T.; Koga, K.; Zhuo, M. Interplay of amygdala and cingulate plasticity in emotional fear. Neural. Plast. 2011, 2011, 813749. [Google Scholar] [CrossRef]
- Koehl, M.; Abrous, D.N. A new chapter in the field of memory: Adult hippocampal neurogenesis. Eur. J. Neurosci. 2011, 33, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Takayama, C. Formation of GABAergic synapses in the cerebellum. Cerebellum 2005, 4, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Nairn, J.G.; Bedi, K.S.; Mayhew, T.M.; Campbell, L.F. On the number of Purkinje cells in the human cerebellum: Unbiased estimates obtained by using the “fractionator”. J. Comp. Neurol. 1989, 290, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Middleton, F.A.; Strick, P.L. The cerebellum: An overview. Trends. Neurosci. 1998, 21, 367–369. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, G.; Lewis, D.A. GABA neurons and the mechanisms of network oscillations: Implications for understanding cortical dysfunction in schizophrenia. Schizophr. Bull. 2008, 34, 944–961. [Google Scholar] [CrossRef] [PubMed]
- Reimbayev, R.; Daley, K.; Belykh, I. When two wrongs make a right: Synchronized neuronal bursting from combined electrical and inhibitory coupling. Philos. Trans. A Math. Phys. Eng. Sci. 2017, 375, 2096. [Google Scholar] [CrossRef]
- Cobb, S.R.; Buhl, E.H.; Halasy, K.; Paulsen, O.; Somogyi, P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature 1995, 378, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.I.; Tao, H.W.; Holt, C.E.; Harris, W.A.; Poo, M. A critical window for cooperation and competition among developing retinotectal synapses. Nature 1998, 395, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Bi, G.Q.; Poo, M.M. Synaptic modifications in cultured hippocampal neurons: Dependence on spike timing, synaptic strength, and postsynaptic cell type. J. Neurosci. 1998, 18, 10464–10472. [Google Scholar] [CrossRef] [PubMed]
- Debanne, D.; Gahwiler, B.H.; Thompson, S.M. Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J. Physiol. 1998, 507, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Jutras, M.J.; Buffalo, E.A. Synchronous neural activity and memory formation. Curr. Opin. Neurobiol. 2010, 20, 150–155. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M.; Caba, M.; Komisaruk, B.R.; Beyer, C. Modulation by estrogen and progesterone of the effect of muscimol on nociception in the spinal cord. Pharmacol. Biochem. Behav. 1990, 37, 123–128. [Google Scholar] [CrossRef]
- Biggio, G.; Casu, M.; Corda, M.G.; Vernaleone, F.; Gessa, G.L. Effect of muscimol, a GABA-mimetic agent, on dopamine metabolism in the mouse brain. Life Sci. 1977, 21, 525–531. [Google Scholar] [CrossRef]
- Helmstetter, F.J.; Bellgowan, P.S. Effects of muscimol applied to the basolateral amygdala on acquisition and expression of contextual fear conditioning in rats. Behav. Neurosci. 1994, 108, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, D.; Serdyukova, N.; Irwin, K.; Bracha, V. GABA neurotransmission in the cerebellar interposed nuclei: Involvement in classically conditioned eyeblinks and neuronal activity. J. Neurophysiol. 2004, 91, 719–727. [Google Scholar] [CrossRef][Green Version]
- Rubenstein, J.L.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Sohal, V.S.; Rubenstein, J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Staley, K. Molecular mechanisms of epilepsy. Nat. Neurosci. 2015, 18, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Obata, K.; Oide, M.; Tanaka, H. Excitatory and inhibitory actions of GABA and glycine on embryonic chick spinal neurons in culture. Brain Res. 1978, 144, 179–184. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Cherubini, E.; Corradetti, R.; Gaiarsa, J.L. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 1989, 416, 303–325. [Google Scholar] [CrossRef] [PubMed]
- Leinekugel, X.; Medina, I.; Khalilov, I.; Ben-Ari, Y.; Khazipov, R. Ca2+ oscillations mediated by the synergistic excitatory actions of GABA(A) and NMDA receptors in the neonatal hippocampus. Neuron 1997, 18, 243–255. [Google Scholar] [CrossRef]
- Hollrigel, G.S.; Ross, S.T.; Soltesz, I. Temporal patterns and depolarizing actions of spontaneous GABAA receptor activation in granule cells of the early postnatal dentate gyrus. J. Neurophysiol. 1998, 80, 2340–2351. [Google Scholar] [CrossRef]
- Owens, D.F.; Boyce, L.H.; Davis, M.B.; Kriegstein, A.R. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J. Neurosci. 1996, 16, 6414–6423. [Google Scholar] [CrossRef]
- Dammerman, R.S.; Flint, A.C.; Noctor, S.; Kriegstein, A.R. An excitatory GABAergic plexus in developing neocortical layer 1. J. Neurophysiol. 2000, 84, 428–434. [Google Scholar] [CrossRef]
- Gao, X.B.; van den Pol, A.N. GABA, not glutamate, a primary transmitter driving action potentials in developing hypothalamic neurons. J. Neurophysiol. 2001, 85, 425–434. [Google Scholar] [CrossRef][Green Version]
- Eilers, J.; Plant, T.D.; Marandi, N.; Konnerth, A. GABA-mediated Ca2+ signalling in developing rat cerebellar Purkinje neurones. J. Physiol. 2001, 536, 429–437. [Google Scholar] [CrossRef]
- Serafini, R.; Valeyev, A.Y.; Barker, J.L.; Poulter, M.O. Depolarizing GABA-activated Cl- channels in embryonic rat spinal and olfactory bulb cells. J. Physiol. 1995, 488, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Reichling, D.B.; Kyrozis, A.; Wang, J.; MacDermott, A.B. Mechanisms of GABA and glycine depolarization-induced calcium transients in rat dorsal horn neurons. J. Physiol. 1994, 476, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. Excitatory actions of gaba during development: The nature of the nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [CrossRef]
- Spitzer, N.C. How GABA generates depolarization. J. Physiol. 2010, 588, 757–758. [Google Scholar] [CrossRef]
- Fukuda, A.; Tanaka, M.; Yamada, Y.; Muramatsu, K.; Shimano, Y.; Nishino, H. Simultaneous optical imaging of intracellular Cl− in neurons in different layers of rat neocortical slices: Advantages and limitations. Neurosci. Res. 1998, 32, 363–371. [Google Scholar] [CrossRef]
- Kuner, T.; Augustine, G.J. A genetically encoded ratiometric indicator for chloride: Capturing chloride transients in cultured hippocampal neurons. Neuron 2000, 27, 447–459. [Google Scholar] [CrossRef]
- Bockhorst, K.H.; Narayana, P.A.; Liu, R.; Ahobila-Vijjula, P.; Ramu, J.; Kamel, M.; Wosik, J.; Bockhorst, T.; Hahn, K.; Hasan, K.M.; et al. Early postnatal development of rat brain: In vivo diffusion tensor imaging. J. Neurosci. Res. 2008, 86, 1520–1528. [Google Scholar] [CrossRef]
- Downes, N.; Mullins, P. The development of myelin in the brain of the juvenile rat. Toxicol. Pathol. 2014, 42, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Murakami, J.W.; Weinberger, E.; Shaw, D.W. Normal myelination of the pediatric brain imaged with fluid-attenuated inversion-recovery (FLAIR) MR imaging. AJNR Am. J. Neuroradiol. 1999, 20, 1406–1411. [Google Scholar] [PubMed]
- Kriegstein, A.; Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef]
- Catalani, A.; Sabbatini, M.; Consoli, C.; Cinque, C.; Tomassoni, D.; Azmitia, E.; Angelucci, L.; Amenta, F. Glial fibrillary acidic protein immunoreactive astrocytes in developing rat hippocampus. Mech. Ageing Dev. 2002, 123, 481–490. [Google Scholar] [CrossRef]
- Baloch, S.; Verma, R.; Huang, H.; Khurd, P.; Clark, S.; Yarowsky, P.; Abel, T.; Mori, S.; Davatzikos, C. Quantification of brain maturation and growth patterns in C57BL/6J mice via computational neuroanatomy of diffusion tensor images. Cereb. Cortex 2009, 19, 675–687. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Quantitative growth and development of human brain. Arch. Dis. Child. 1973, 48, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef]
- Micheva, K.D.; Beaulieu, C. Quantitative aspects of synaptogenesis in the rat barrel field cortex with special reference to GABA circuitry. J. Comp. Neurol. 1996, 373, 340–354. [Google Scholar] [CrossRef]
- Keshavan, M.S.; Diwadkar, V.A.; DeBellis, M.; Dick, E.; Kotwal, R.; Rosenberg, D.R.; Sweeney, J.A.; Minshew, N.; Pettegrew, J.W. Development of the corpus callosum in childhood, adolescence and early adulthood. Life Sci. 2002, 70, 1909–1922. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Broadbelt, K.G.; Haynes, R.L.; Folkerth, R.D.; Borenstein, N.S.; Belliveau, R.A.; Trachtenberg, F.L.; Volpe, J.J.; Kinney, H.C. Late development of the GABAergic system in the human cerebral cortex and white matter. J. Neuropathol. Exp. Neurol. 2011, 70, 841–858. [Google Scholar] [CrossRef] [PubMed]
- Tyzio, R.; Represa, A.; Jorquera, I.; Ben-Ari, Y.; Gozlan, H.; Aniksztejn, L. The establishment of GABAergic and glutamatergic synapses on CA1 pyramidal neurons is sequential and correlates with the development of the apical dendrite. J. Neurosci. 1999, 19, 10372–10382. [Google Scholar] [CrossRef]
- Khazipov, R.; Esclapez, M.; Caillard, O.; Bernard, C.; Khalilov, I.; Tyzio, R.; Hirsch, J.; Dzhala, V.; Berger, B.; Ben-Ari, Y. Early development of neuronal activity in the primate hippocampus in utero. J. Neurosci. 2001, 21, 9770–9781. [Google Scholar] [CrossRef] [PubMed]
- Gubellini, P.; Ben-Ari, Y.; Gaiarsa, J.L. Activity- and age-dependent GABAergic synaptic plasticity in the developing rat hippocampus. Eur. J. Neurosci. 2001, 14, 1937–1946. [Google Scholar] [CrossRef] [PubMed]
- Kilb, W. Development of the GABAergic system from birth to adolescence. Neuroscientist 2012, 18, 613–630. [Google Scholar] [CrossRef] [PubMed]
- Premachandran, H.; Zhao, M.; Arruda-Carvalho, M. Sex Differences in the Development of the Rodent Corticolimbic System. Front. Neurosci. 2020, 14, 583477. [Google Scholar] [CrossRef]
- Bosman, C.A.; Lansink, C.S.; Pennartz, C.M. Functions of gamma-band synchronization in cognition: From single circuits to functional diversity across cortical and subcortical systems. Eur. J. Neurosci. 2014, 39, 1982–1999. [Google Scholar] [CrossRef] [PubMed]
- Leinekugel, X.; Khazipov, R.; Cannon, R.; Hirase, H.; Ben-Ari, Y.; Buzsaki, G. Correlated bursts of activity in the neonatal hippocampus in vivo. Science 2002, 296, 2049–2052. [Google Scholar] [CrossRef]
- Doischer, D.; Hosp, J.A.; Yanagawa, Y.; Obata, K.; Jonas, P.; Vida, I.; Bartos, M. Postnatal differentiation of basket cells from slow to fast signaling devices. J. Neurosci. 2008, 28, 12956–12968. [Google Scholar] [CrossRef]
- Okaty, B.W.; Miller, M.N.; Sugino, K.; Hempel, C.M.; Nelson, S.B. Transcriptional and electrophysiological maturation of neocortical fast-spiking GABAergic interneurons. J. Neurosci. 2009, 29, 7040–7052. [Google Scholar] [CrossRef]
- Kobayashi, M.; Hamada, T.; Kogo, M.; Yanagawa, Y.; Obata, K.; Kang, Y. Developmental profile of GABAA-mediated synaptic transmission in pyramidal cells of the somatosensory cortex. Eur. J. Neurosci. 2008, 28, 849–861. [Google Scholar] [CrossRef]
- Fan, S.J.; Sun, A.B.; Liu, L. Epigenetic modulation during hippocampal development. Biomed. Rep. 2018, 9, 463–473. [Google Scholar] [CrossRef]
- Hennou, S.; Khalilov, I.; Diabira, D.; Ben-Ari, Y.; Gozlan, H. Early sequential formation of functional GABA(A) and glutamatergic synapses on CA1 interneurons of the rat foetal hippocampus. Eur. J. Neurosci. 2002, 16, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.; Khalilov, I.; Represa, A.; Gozlan, H. Interneurons set the tune of developing networks. Trends Neurosci. 2004, 27, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, D.P.; Venkatasubramanian, P.N.; Miller, M.J.; Dixon, C.J.; Li, L.; Wyrwicz, A.M. Effects of neonatal isoflurane anesthesia exposure on learning-specific and sensory systems in adults. Sci. Rep. 2020, 10, 13832. [Google Scholar] [CrossRef]
- Letinic, K.; Zoncu, R.; Rakic, P. Origin of GABAergic neurons in the human neocortex. Nature 2002, 417, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Kelsom, C.; Lu, W. Development and specification of GABAergic cortical interneurons. Cell Biosci. 2013, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Van Eden, C.G.; Uylings, H.B. Cytoarchitectonic development of the prefrontal cortex in the rat. J. Comp. Neurol. 1985, 241, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Ganella, D.E.; Kim, J.H. Developmental rodent models of fear and anxiety: From neurobiology to pharmacology. Br. J. Pharmacol. 2014, 171, 4556–4574. [Google Scholar] [CrossRef]
- Arain, M.; Haque, M.; Johal, L.; Mathur, P.; Nel, W.; Rais, A.; Sandhu, R.; Sharma, S. Maturation of the adolescent brain. Neuropsychiatr. Dis. Treat. 2013, 9, 449–461. [Google Scholar]
- Jager, P.; Moore, G.; Calpin, P.; Durmishi, X.; Salgarella, I.; Menage, L.; Kita, Y.; Wang, Y.; Kim, D.W.; Blackshaw, S.; et al. Dual midbrain and forebrain origins of thalamic inhibitory interneurons. eLife 2021, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Mojsilovic, J.; Zecevic, N. Early development of the human thalamus: Golgi and Nissl study. Early Hum. Dev. 1991, 27, 119–144. [Google Scholar] [CrossRef]
- Nakagawa, Y. Development of the thalamus: From early patterning to regulation of cortical functions. Wiley Interdiscip. Rev. Dev. Biol. 2019, 8, e345. [Google Scholar] [CrossRef] [PubMed]
- Alcauter, S.; Lin, W.; Smith, J.K.; Short, S.J.; Goldman, B.D.; Reznick, J.S.; Gilmore, J.H.; Gao, W. Development of thalamocortical connectivity during infancy and its cognitive correlations. J. Neurosci. 2014, 34, 9067–9075. [Google Scholar] [CrossRef]
- Hou, G.; Smith, A.G.; Zhang, Z.W. Lack of Intrinsic GABAergic Connections in the Thalamic Reticular Nucleus of the Mouse. J. Neurosci. 2016, 36, 7246–7252. [Google Scholar] [CrossRef] [PubMed]
- Leto, K.; Bartolini, A.; Yanagawa, Y.; Obata, K.; Magrassi, L.; Schilling, K.; Rossi, F. Laminar fate and phenotype specification of cerebellar GABAergic interneurons. J. Neurosci. 2009, 29, 7079–7091. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Tang, C.; Ren, J.; Zhang, C.; Dong, L.; Zhu, Z. Effect of multiple neonatal sevoflurane exposures on hippocampal apolipoprotein E levels and learning and memory abilities. Pediatr. Neonatol. 2018, 59, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.W.; Shu, Y.; Li, M.; Guo, X.; Pac-Soo, C.; Maze, M.; Ma, D. The glutaminergic, GABAergic, dopaminergic but not cholinergic neurons are susceptible to anaesthesia-induced cell death in the rat developing brain. Neuroscience 2011, 174, 64–70. [Google Scholar] [CrossRef]
- Deng, M.; Hofacer, R.D.; Jiang, C.; Joseph, B.; Hughes, E.A.; Jia, B.; Danzer, S.C.; Loepke, A.W. Brain regional vulnerability to anaesthesia-induced neuroapoptosis shifts with age at exposure and extends into adulthood for some regions. Br. J. Anaesth. 2014, 113, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Yu, D.; Li, X.; Huang, J.; Jing, S.; Bao, X.; Yang, T.; Fan, X. Propofol Exposure in Early Life Induced Developmental Impairments in the Mouse Cerebellum. Front. Cell Neurosci. 2017, 11, 373. [Google Scholar] [CrossRef] [PubMed]
- Woodward, T.J.; Timic Stamenic, T.; Todorovic, S.M. Neonatal general anesthesia causes lasting alterations in excitatory and inhibitory synaptic transmission in the ventrobasal thalamus of adolescent female rats. Neurobiol. Dis. 2019, 127, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bie, B.; Naguib, M. Epigenetic Manipulation of Brain-derived Neurotrophic Factor Improves Memory Deficiency Induced by Neonatal Anesthesia in Rats. Anesthesiology 2016, 124, 624–640. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liang, G.; Hawkins, B.J.; Madesh, M.; Pierwola, A.; Wei, H. Inhalational anesthetics induce cell damage by disruption of intracellular calcium homeostasis with different potencies. Anesthesiology 2008, 109, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Ramanantsoa, N.; Fleiss, B.; Bouslama, M.; Matrot, B.; Schwendimann, L.; Cohen-Salmon, C.; Gressens, P.; Gallego, J. Bench to Cribside: The Path for Developing a Neuroprotectant. Transl. Stroke Res. 2013, 4, 258–277. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Yu, S.P. Ionic regulation of cell volume changes and cell death after ischemic stroke. Transl. Stroke Res. 2014, 5, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Macri, M.A.; D’Alessandro, N.; Di Giulio, C.; Di Iorio, P.; Di Luzio, S.; Giuliani, P.; Esposito, E.; Pokorski, M. Region-specific effects on brain metabolites of hypoxia and hyperoxia overlaid on cerebral ischemia in young and old rats: A quantitative proton magnetic resonance spectroscopy study. J. Biomed. Sci. 2010, 17, 14. [Google Scholar] [CrossRef]
- Aligny, C.; Le Roux, C.W.; Dourmap, N.; Ramdani, Y.; Do-Rego, J.-C.; Jegou, S.; Leroux, P.; Leroux-Nicollet, I.; Marret, S.; Gonzalez, B.J. Ketamine alters cortical integration of GABAergic interneurons and induces long-term sex-dependent impairments in transgenic Gad67-GFP mice. Cell Death Dis. 2014, 5, e1311. [Google Scholar] [CrossRef]
- Chung, W.; Ryu, M.J.; Heo, J.Y.; Lee, S.; Yoon, S.; Park, H.; Park, S.; Kim, Y.; Kim, Y.H.; Yoon, S.H.; et al. Sevoflurane Exposure during the Critical Period Affects Synaptic Transmission and Mitochondrial Respiration but Not Long-term Behavior in Mice. Anesthesiology 2017, 126, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, O.H.; Gulvezan, T.; Symmes, B.; Quillinan, N.; Jevtovic-Todorovic, V. Sex differences in neurodevelopmental abnormalities caused by early-life anaesthesia exposure: A narrative review. Br. J. Anaesth. 2020, 124, e81–e91. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, D.P.; Miller, M.J.; Li, L.; Wyrwicz, A.M. Eyeblink classical conditioning and BOLD fMRI of anesthesia-induced changes in the developing brain. Physiol. Behav. 2016, 167, 10–15. [Google Scholar] [CrossRef]
- Drobyshevsky, A.; Miller, M.J.; Li, L.; Dixon, C.J.; Venkatasubramanian, P.N.; Wyrwicz, A.; Aksenov, D.P. Behavior and regional cortical BOLD signal fluctuations are altered in adult rabbits after neonatal volatile anesthetic exposure. Front. Neurosci. 2020, 14, 571486. [Google Scholar] [CrossRef]
- Doubovikov, E.D.; Aksenov, D.P. Oscillations and concentration dynamics of brain tissue oxygen in neonates and adults. J. Comput. Neurosci. 2020, 48, 21–26. [Google Scholar] [CrossRef]
- Linsenmeier, R.A.; Aksenov, D.P.; Faber, H.M.; Makar, P.; Wyrwicz, A.M. Spontaneous Fluctuations of PO2 in the Rabbit Somatosensory Cortex. Adv. Exp. Med. Biol. 2016, 876, 311–317. [Google Scholar] [PubMed]
- Unal-Cevik, I.; Kilinç, M.; Can, A.; Gürsoy-Ozdemir, Y.; Dalkara, T. Apoptotic and necrotic death mechanisms are concomitantly activated in the same cell after cerebral ischemia. Stroke 2004, 35, 2189–2194. [Google Scholar] [CrossRef] [PubMed]
- Rink, C.; Khanna, S. Significance of Brain Tissue Oxygenation and the Arachidonic Acid Cascade in Stroke. Antioxidants Redox Signal. 2011, 14, 1889–1903. [Google Scholar] [CrossRef]
- Mahakizadeh, S.; Mokhtari, T.; Navaee, F.; Poorhassan, M.; Tajik, A.; Hassanzadeh, G. Effects of chronic hypoxia on the expression of seladin-1/Tuj1 and the number of dark neurons of hippocampus. J. Chem. Neuroanat. 2020, 104, 101744. [Google Scholar] [CrossRef] [PubMed]
- Dheer, A.; Jain, V.; Kushwah, N.; Kumar, R.; Prasad, D.; Singh, S.B. Temporal and Spatial Changes in Glial Cells During Chronic Hypobaric Hypoxia: Role in Neurodegeneration. Neuroscience 2018, 383, 235–246. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Lai, U.H.; Zhu, L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive Oxygen Species Formation in the Brain at Different Oxygen Levels: The Role of Hypoxia Inducible Factors. Front. Cell Dev. Biol. 2018, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Smith, M.A.; Zhu, X.; Nunomura, A.; Castellani, R.J.; Perry, G. Oxidative stress and neurodegeneration. Ann. N. Y. Acad. Sci. 2005, 1043, 545–552. [Google Scholar] [CrossRef]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Yao, H.; Haddad, G.G. Calcium and pH homeostasis in neurons during hypoxia and ischemia. Cell Calcium 2004, 36, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Rolett, E.L.; Azzawi, A.; Liu, K.J.; Yongbi, M.N.; Swartz, H.M.; Dunn, J.F. Critical oxygen tension in rat brain: A combined (31)P-NMR and EPR oximetry study. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R9–R16. [Google Scholar] [CrossRef] [PubMed]
- Nioka, S.; Chance, B.; Smith, D.S.; Mayevsky, A.; Reilly, M.P.; Alter, C.; Asakura, T. Cerebral energy metabolism and oxygen state during hypoxia in neonate and adult dogs. Pediatr. Res. 1990, 28, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Shetty, P.K.; Galeffi, F.; Turner, D.A. Nicotinamide pre-treatment ameliorates NAD(H) hyperoxidation and improves neuronal function after severe hypoxia. Neurobiol. Dis. 2014, 62, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Tao, G.; Zhang, J.; Zhang, L.; Dong, Y.; Yu, B.; Crosby, G.; Culley, D.J.; Zhang, Y.; Xie, Z. Sevoflurane induces tau phosphorylation and glycogen synthase kinase 3β activation in young mice. Anesthesiology 2014, 121, 510–527. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Jiang, H. Effect of the inhaled anesthetics isoflurane, sevoflurane and desflurane on the neuropathogenesis of Alzheimer’s disease (Review). Mol. Med. Rep. 2015, 12, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhang, G.; Zhang, B.; Moir, R.; Xia, W.; Marcantonio, E.R.; Culley, D.J.; Crosby, G.; Tanzi, R.E.; Xie, Z. The Common Inhalational Anesthetic Sevoflurane Induces Apoptosis and Increases β-Amyloid Protein Levels. Arch. Neurol. 2009, 66, 620–631. [Google Scholar] [CrossRef]
- Bellinger, D.C.; Wypij, D.; duPlessis, A.J.; Rappaport, L.A.; Jonas, R.A.; Wernovsky, G.; Newburger, J.W. Neurodevelopmental status at eight years in children with dextro-transposition of the great arteries: The Boston Circulatory Arrest Trial. J. Thorac. Cardiovasc. Surg. 2003, 126, 1385–1396. [Google Scholar] [CrossRef]
- Huberman Samuel, M.; Meiri, G.; Dinstein, I.; Flusser, H.; Michaelovski, A.; Bashiri, A.; Menashe, I. Exposure to General Anesthesia May Contribute to the Association between Cesarean Delivery and Autism Spectrum Disorder. J. Autism Dev. Disord. 2019, 49, 3127–3135. [Google Scholar] [CrossRef] [PubMed]
- Creagh, O.; Torres, H.; Rivera, K.; Morales-Franqui, M.; Altieri-Acevedo, G.; Warner, D. Previous Exposure to Anesthesia and Autism Spectrum Disorder (ASD): A Puerto Rican Population-Based Sibling Cohort Study. Bol. Asoc. Med. Puerto Rico 2016, 108, 73–80. [Google Scholar]
- Ferguson, B.R.; Gao, W.J. PV Interneurons: Critical Regulators of E/I Balance for Prefrontal Cortex-Dependent Behavior and Psychiatric Disorders. Front. Neural Circuits 2018, 12, 37. [Google Scholar] [CrossRef]
- Aksenov, D.P.; Serdyukova, N.A.; Bloedel, J.R.; Bracha, V. Glutamate neurotransmission in the cerebellar interposed nuclei: Involvement in classically conditioned eyeblinks and neuronal activity. J. Neurophysiol. 2005, 93, 44–52. [Google Scholar] [CrossRef]
- Zhou, H.; Xie, Z.; Brambrink, A.M.; Yang, G. Behavioural impairments after exposure of neonatal mice to propofol are accompanied by reductions in neuronal activity in cortical circuitry. Br. J. Anaesth. 2021, 126, 1141–1156. [Google Scholar] [CrossRef]
- Van der Graaf, M. In vivo magnetic resonance spectroscopy: Basic methodology and clinical applications. Eur. Biophys. J. 2009, 39, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Puts, N.A.; Edden, R.A. In vivo magnetic resonance spectroscopy of GABA: A methodological review. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 60, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Rothman, D.L.; Petroff, O.A.; Behar, K.L.; Mattson, R.H. Localized 1H NMR measurements of gamma-aminobutyric acid in human brain in vivo. Proc. Natl. Acad. Sci. USA 1993, 90, 5662–5666. [Google Scholar] [CrossRef] [PubMed]
- Edden, R.A.; Barker, P.B. Spatial effects in the detection of gamma-aminobutyric acid: Improved sensitivity at high fields using inner volume saturation. Magn. Reason. Med. 2007, 58, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Terpstra, M.; Ugurbil, K.; Gruetter, R. Direct in vivo measurement of human cerebral GABA concentration using MEGA-editing at 7 Tesla. Magn. Reason. Med. 2002, 47, 1009–1012. [Google Scholar] [CrossRef]
- Buxton, R.B. The physics of functional magnetic resonance imaging (fMRI). Rep. Prog. Phys. 2013, 76, 096601. [Google Scholar] [CrossRef]
- Aksenov, D.P.; Li, L.; Miller, M.J.; Wyrwicz, A.M. Blood oxygenation level dependent signal and neuronal adaptation to optogenetic and sensory stimulation in somatosensory cortex in awake animals. Eur. J. Neurosci. 2016, 44, 2722–2729. [Google Scholar] [CrossRef]
- Jasanoff, A. MRI contrast agents for functional molecular imaging of brain activity. Curr. Opin. Neurobiol. 2007, 17, 593–600. [Google Scholar] [CrossRef]
- Howell, A.L.; Osher, D.E.; Li, J.; Saygin, Z.M. The intrinsic neonatal hippocampal network: RsfMRI findings. J. Neurophysiol. 2020, 124, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Hüppi, P.S.; Dubois, J. Diffusion tensor imaging of brain development. Semin. Fetal Neonatal Med. 2006, 11, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.L.; Lee, J.E.; Lazar, M.; Field, A.S. Diffusion tensor imaging of the brain. Neurotherapeutics 2007, 4, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Silverman, P.M.; Cooper, C.J.; Weltman, D.I.; Zeman, R.K. Helical CT: Practical considerations and potential pitfalls. Radiographics 1995, 15, 25–36. [Google Scholar] [CrossRef]
- Lopes da Silva, F. EEG and MEG: Relevance to neuroscience. Neuron 2013, 80, 1112–1128. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, N. Cognitive neuroscience of creativity: EEG based approaches. Methods 2007, 42, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Gross, J. Magnetoencephalography in Cognitive Neuroscience: A Primer. Neuron 2019, 104, 189–204. [Google Scholar] [CrossRef] [PubMed]
- Tonnesen, J.; Pryds, A.; Larsen, E.H.; Paulson, O.B.; Hauerberg, J.; Knudsen, G.M. Laser Doppler flowmetry is valid for measurement of cerebral blood flow autoregulation lower limit in rats. Exp. Physiol. 2005, 90, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Ozbay, B.N.; Futia, G.L.; Ma, M.; Bright, V.M.; Gopinath, J.T.; Hughes, E.G.; Restrepo, D.; Gibson, E.A. Three dimensional two-photon brain imaging in freely moving mice using a miniature fiber coupled microscope with active axial-scanning. Sci. Rep. 2018, 8, 8108. [Google Scholar] [CrossRef]
- Swadlow, H.A. Fast-spike interneurons and feedforward inhibition in awake sensory neocortex. Cereb. Cortex 2003, 13, 25–32. [Google Scholar] [CrossRef]
- Aksenov, D.P.; Miller, M.J.; Dixon, C.J.; Wyrwicz, A.M. The effect of sevoflurane and isoflurane anesthesia on single unit and local field potentials. Exp. Brain Res. 2019, 237, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Yon, J.H.; Daniel-Johnson, J.; Carter, L.B.; Jevtovic-Todorovic, V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience 2005, 135, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Di Maggio, C.; Sun, L.S.; Kakavouli, A.; Byrne, M.W.; Li, G. A retrospective cohort study of the association of anesthesia and hernia repair surgery with behavioral and developmental disorders in young children. J. Neurosurg. Anesthesiol. 2009, 21, 286–291. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gascoigne, D.A.; Serdyukova, N.A.; Aksenov, D.P. Early Development of the GABAergic System and the Associated Risks of Neonatal Anesthesia. Int. J. Mol. Sci. 2021, 22, 12951. https://doi.org/10.3390/ijms222312951
Gascoigne DA, Serdyukova NA, Aksenov DP. Early Development of the GABAergic System and the Associated Risks of Neonatal Anesthesia. International Journal of Molecular Sciences. 2021; 22(23):12951. https://doi.org/10.3390/ijms222312951
Chicago/Turabian StyleGascoigne, David A., Natalya A. Serdyukova, and Daniil P. Aksenov. 2021. "Early Development of the GABAergic System and the Associated Risks of Neonatal Anesthesia" International Journal of Molecular Sciences 22, no. 23: 12951. https://doi.org/10.3390/ijms222312951
APA StyleGascoigne, D. A., Serdyukova, N. A., & Aksenov, D. P. (2021). Early Development of the GABAergic System and the Associated Risks of Neonatal Anesthesia. International Journal of Molecular Sciences, 22(23), 12951. https://doi.org/10.3390/ijms222312951