
Nicotine Neurotoxicity Involves Low Wnt1 Signaling in Spinal Locomotor Networks of the Postnatal Rodent Spinal Cord



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
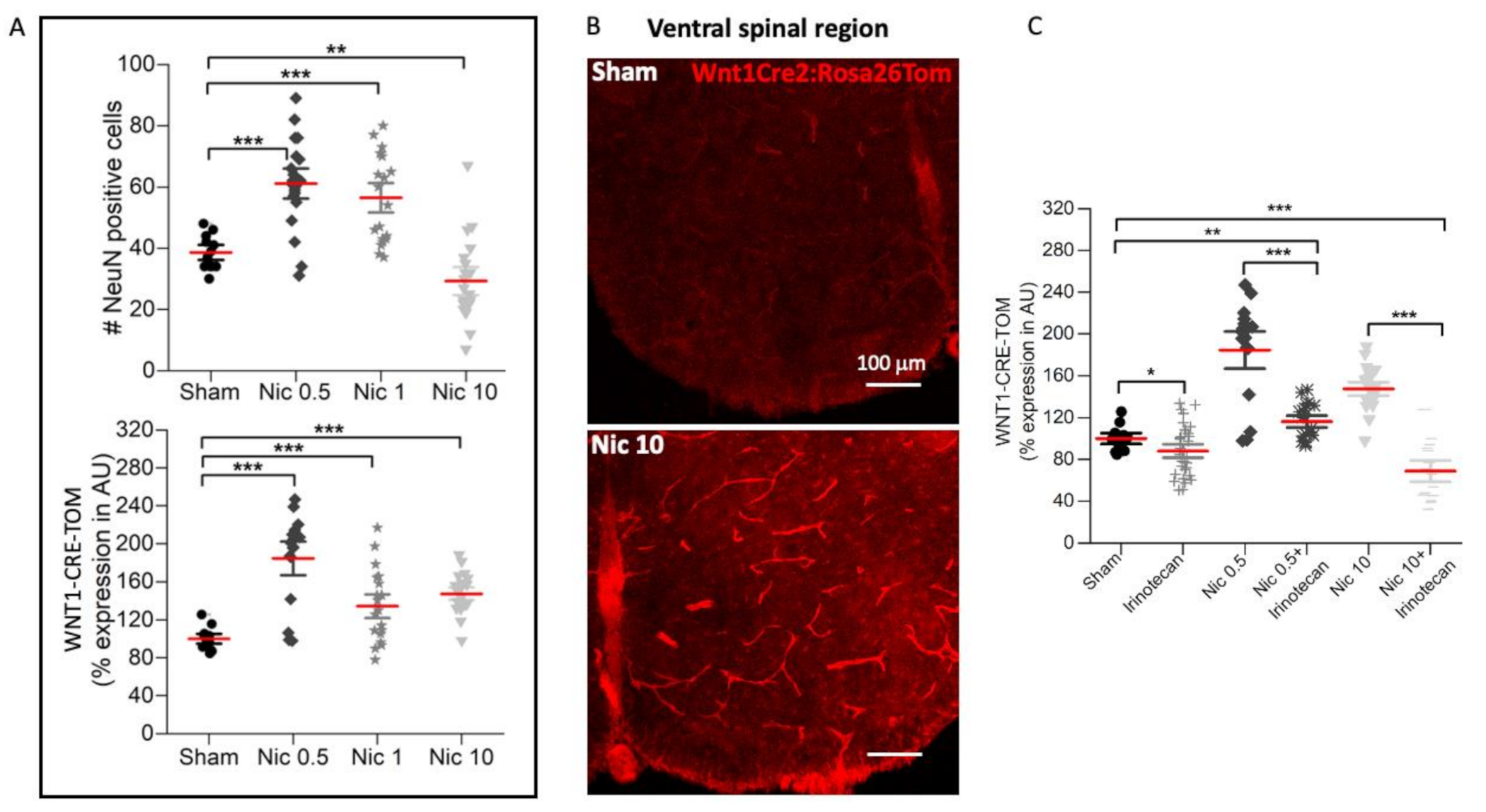
2.1. Dose-Dependent Effect of Nicotine on Network Depolarization, Wnt1 Pathway, and Neuronal Numbers
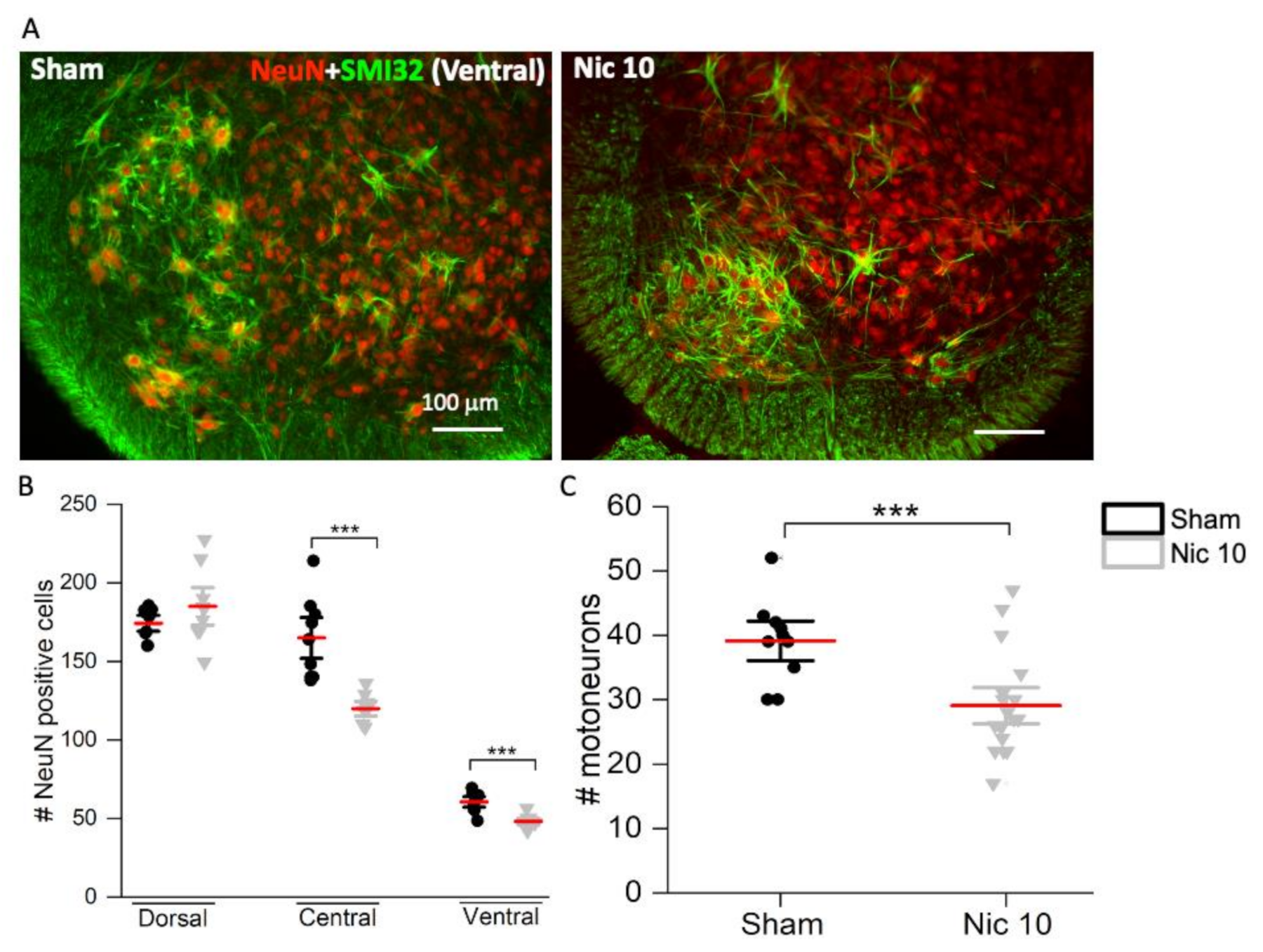
2.2. Effect of High Nicotine Levels on Rat Spinal Neurons and Motoneurons
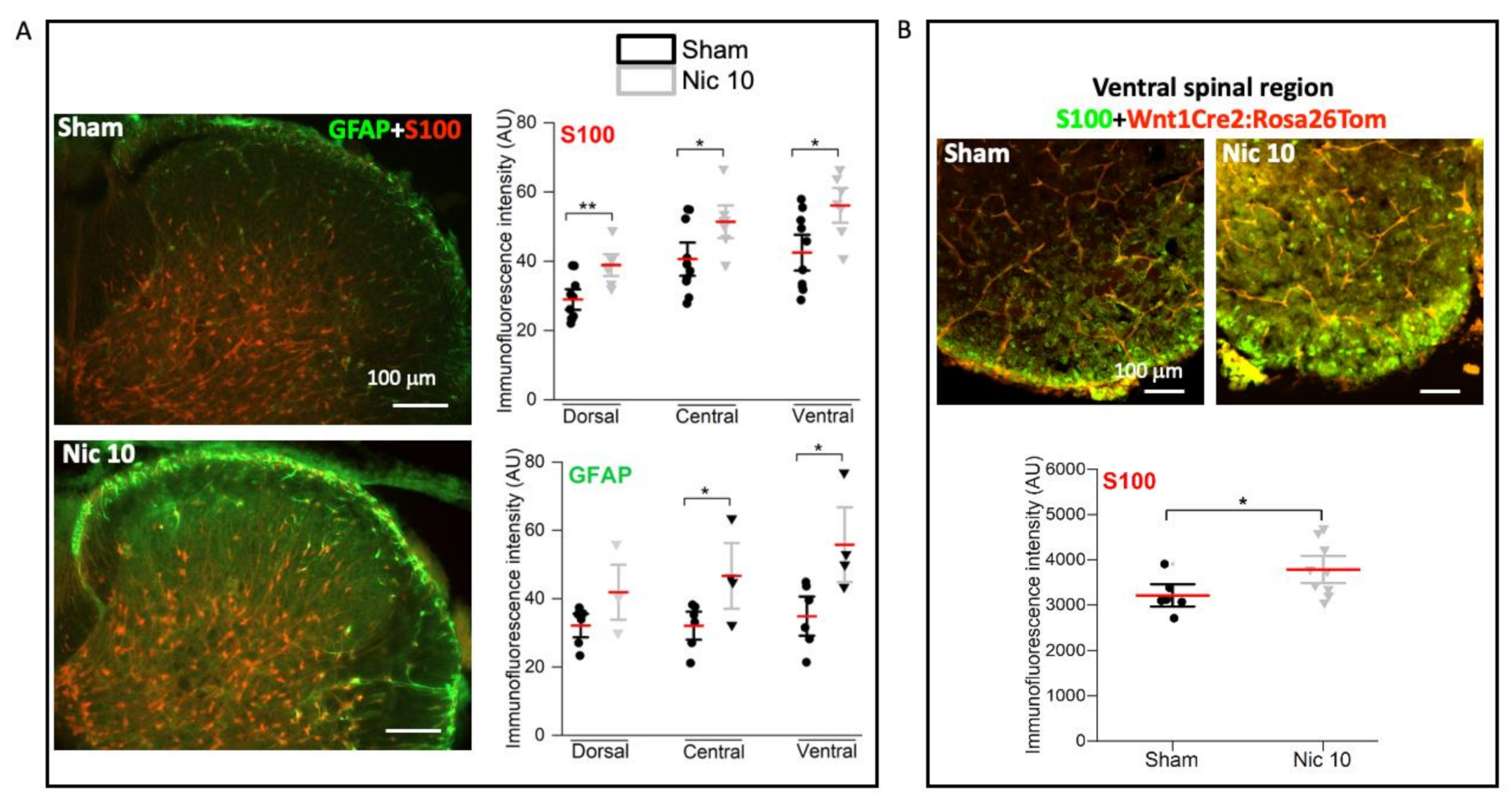
2.3. Upregulation of S100 and GFAP by Nicotine 10 μM
2.4. Impaired Monosynaptic Transmission and Fictive Locomotion Induced by Nic 10
2.5. Effect of Nicotine (10 μM) on Dorsal–Dorsal Root Potential
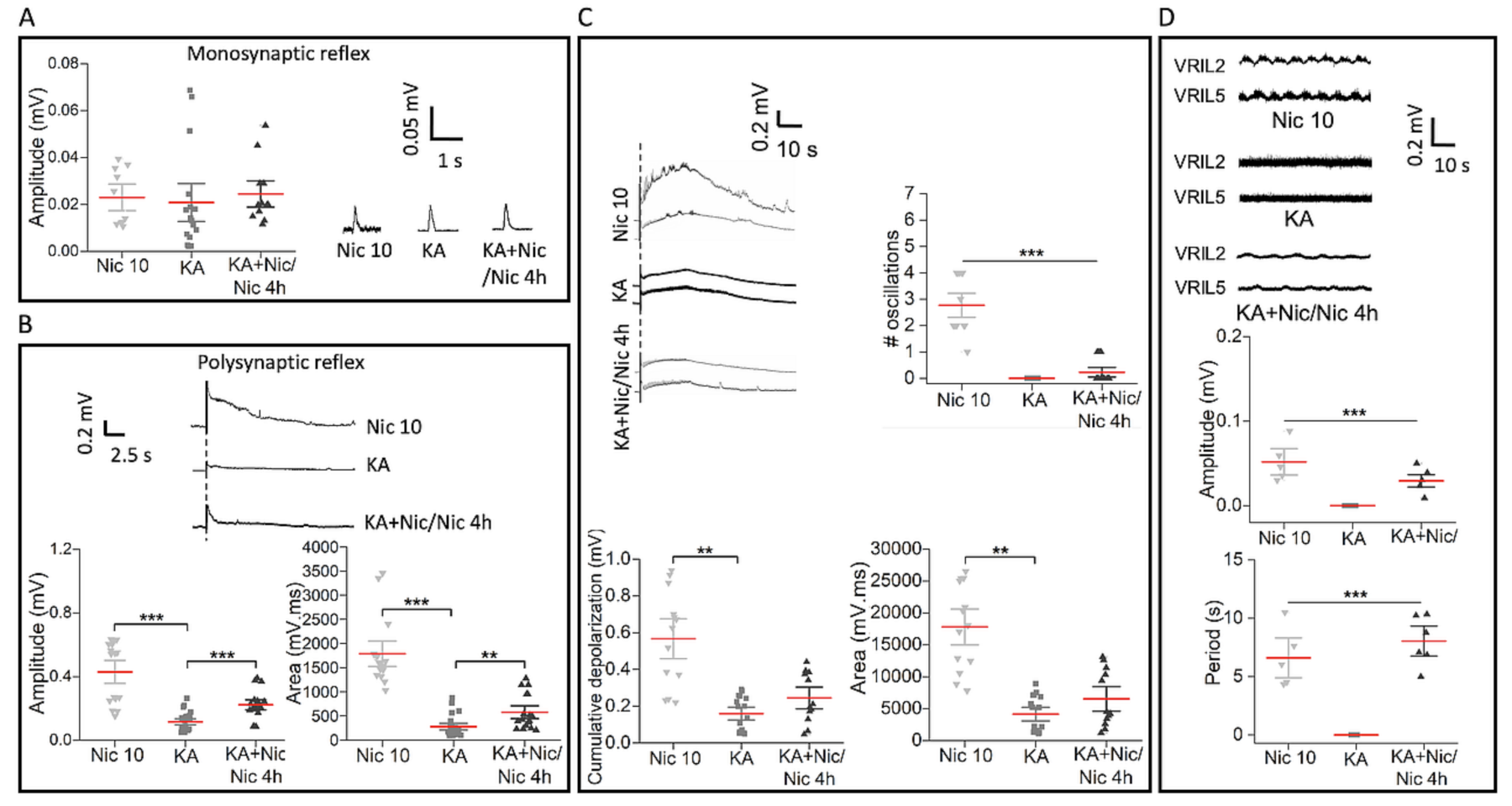
2.6. Neuronal Number, Motoneurons and Wnt1 Pathway after Excitotoxic Insult Followed by Nic 10
2.7. Poor Recovery in Reflex Responses and Fictive Locomotion
3. Discussion
3.1. An Evolving Scenario from Neuroprotection to Neurotoxicity
3.2. Nicotine, Wnt Signaling, and Neurotoxicity
3.3. Effect of High Nicotine on Kainate-Mediated Excitotoxicity
3.4. Advantages and Limitations of the Experimental Model and the Study
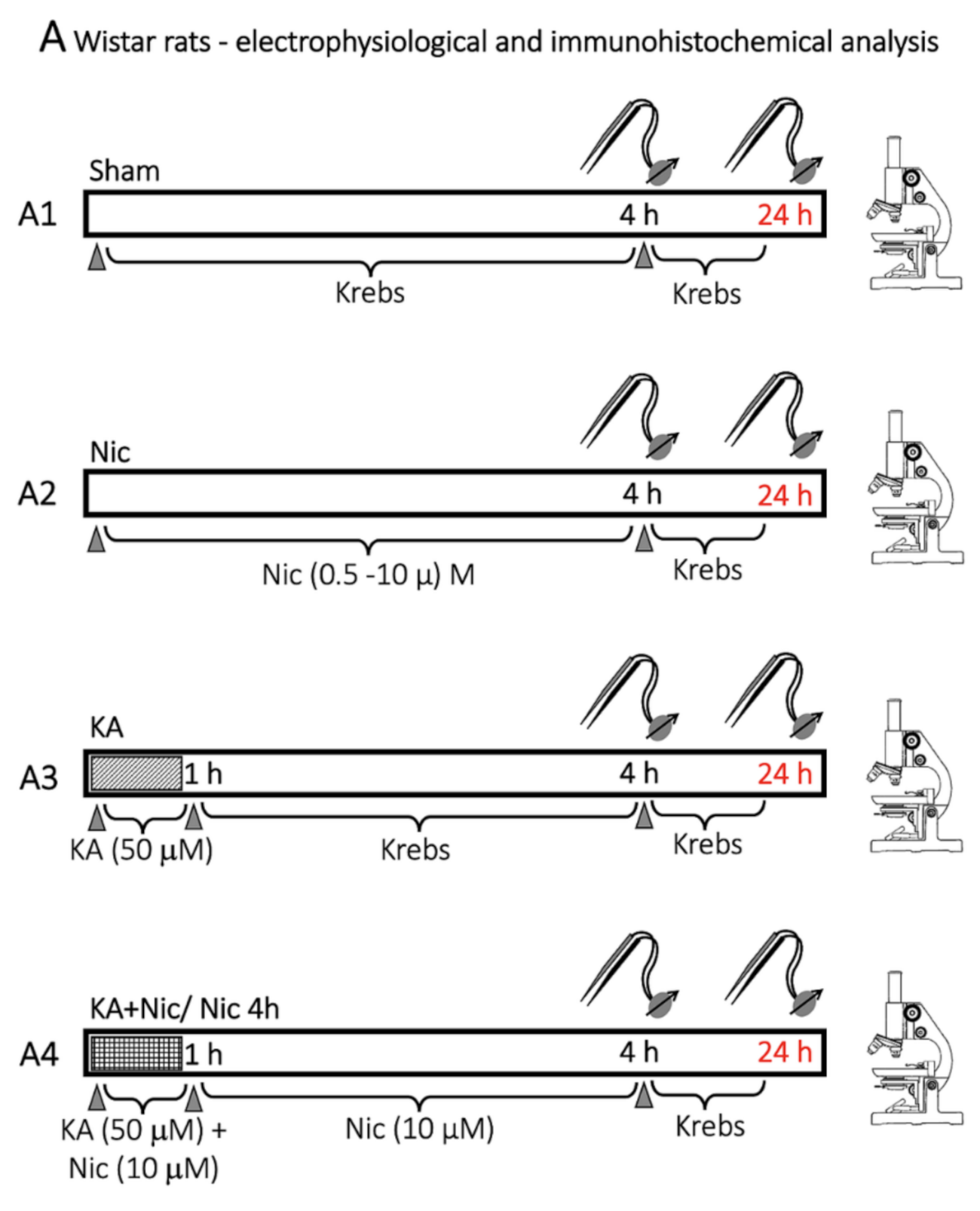
4. Materials and Methods
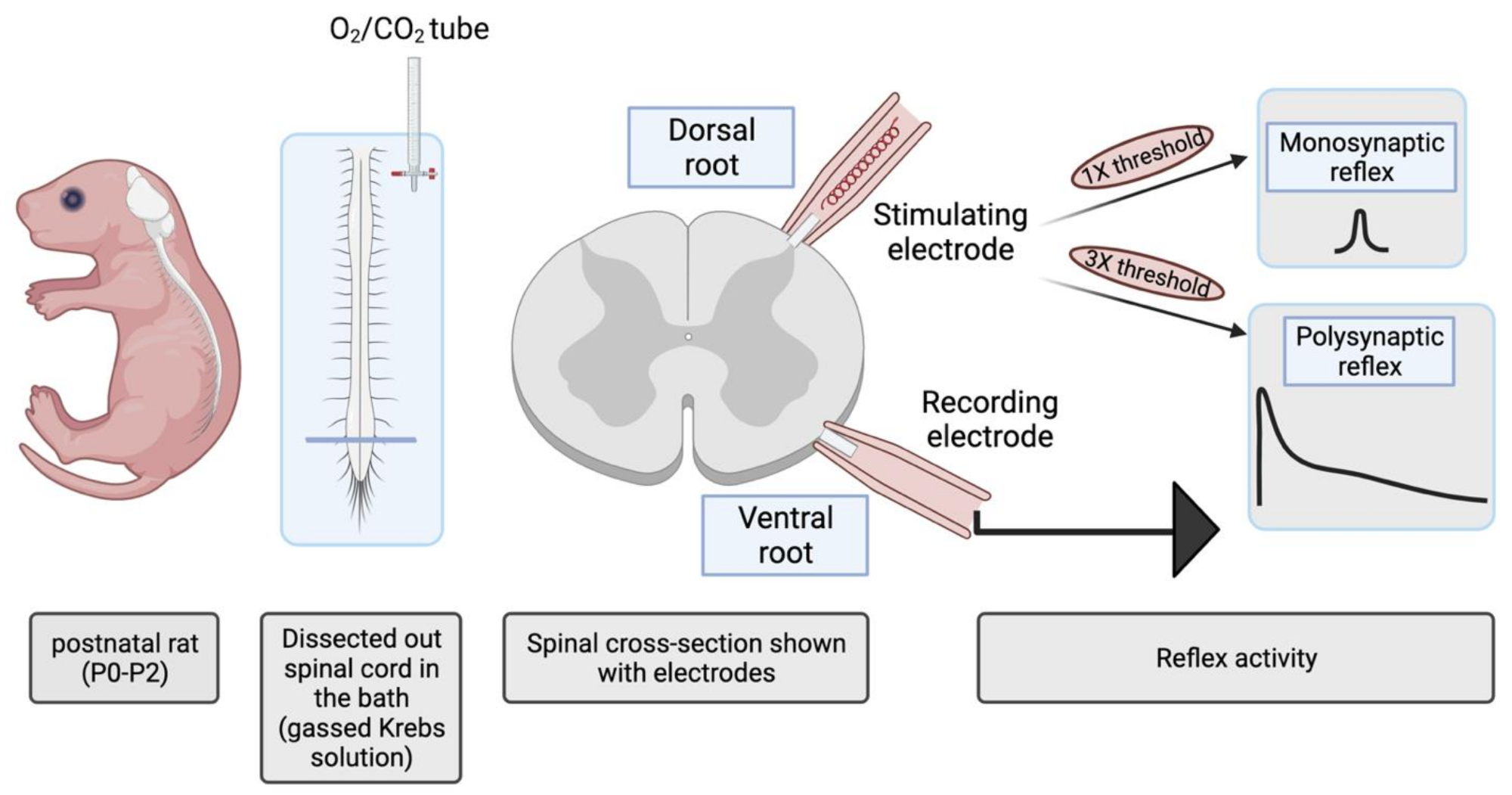
4.1. Wild-Type Rat Spinal Cord Preparation
4.2. Transgenic Mice Spinal Cord Preparation
4.3. Protocol for Drug Application and Lesioning the Spinal Cord
4.4. Electrophysiology
4.5. Immunohistochemistry
4.6. Drugs Used
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethical Statement
Abbreviations
| BSA | Bovine serum albumin |
| CO2 | Carbon dioxide |
| Cre | Cre recombinase enzyme |
| D-DRP | Dorsal–dorsal root potential |
| DR | Dorsal root |
| Elephys | Electrophysiology |
| FBS | Fetal bovine serum |
| iNOS | inducible nitric oxide synthase |
| KA | Kainate |
| L | Lumbar |
| n | total number of rats/mice |
| N | number of spinal slices |
| nAChRs | nicotinic acetylcholine receptors |
| Nic | Nicotine |
| NGS | Normal goat serum |
| NMDA | N-methyl-D-aspartate (NMDA) |
| O2 | Oxygen |
| PBS | Phosphate buffer solution |
| PFA | Paraformaldehyde |
| SCI | Spinal cord injury |
| VR | Ventral root |
| Wnt | Wingless related MMTV integration site |
| 5HT | 5-hydroxytryptamine |
References
- Corsini, S.; Tortora, M.; Rauti, R.; Nistri, A. Nicotine protects rat hypoglossal motoneurons from excitotoxic death via downregulation of connexin 36. Cell Death Dis. 2017, 8, e2881. [Google Scholar] [CrossRef]
- Kaur, J.; Rauti, R.; Nistri, A. Nicotine-mediated neuroprotection of rat spinal networks against excitotoxicity. Eur. J. Neurosci. 2018, 47, 1353–1374. [Google Scholar] [CrossRef]
- Bajrektarevic, D.; Corsini, S.; Nistri, A.; Tortora, M. Nicotine Neuroprotection of Brain Neurons: The Other Side of Nicotine Addiction. In Neuroscience of Nicotine; Preedy, V.R., Ed.; Academic Press: London, UK, 2019; pp. 79–86. [Google Scholar]
- Taccola, G.; Margaryan, G.; Mladinic, M.; Nistri, A. Kainate and metabolic perturbation mimicking spinal injury differentially contribute to early damage of locomotor networks in the in vitro neonatal rat spinal cord. Neuroscience 2008, 155, 538–555. [Google Scholar] [CrossRef] [PubMed]
- Taccola, G.; Mladinic, M.; Nistri, A. Dynamics of early locomotor network dysfunction following a focal lesion in an in vitro model of spinal injury. Eur. J. Neurosci. 2010, 31, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Richardson, E.J.; Ness, T.J.; Redden, D.T.; Stewart, C.C.; Richards, J.S. Effects of nicotine on spinal cord injury pain vary among subtypes of pain and smoking status: Results from a randomized, controlled experiment. J. Pain 2012, 13, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Shefner, J.M.; Berman, S.A.; Young, R.R. The effect of nicotine on recurrent inhibition in the spinal cord. Neurology 1993, 43, 2647–2651. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Erausquin, M.; Pons, S.; Faure, P.; Changeux, J.P. Nicotine differentially activates inhibitory and excitatory neurons in the dorsal spinal cord. Pain 2004, 109, 308–318. [Google Scholar] [CrossRef]
- Matsumoto, M.; Xie, W.; Inoue, M.; Ueda, H. Evidence for the tonic inhibition of spinal pain by nicotinic cholinergic transmission through primary afferents. Mol. Pain 2007, 3, 1744–8069. [Google Scholar] [CrossRef]
- Preedy, V.R. Neuroscience of Nicotine; Academic Press: London, UK, 2019; pp. 1–544. [Google Scholar] [CrossRef]
- Jobson, C.L.M.; Renard, J.; Szkudlarek, H.; Rosen, L.G.; Pereira, B.; Wright, D.J.; Rushlow, W.; Laviolette, S.R. Adolescent Nicotine Exposure Induces Dysregulation of Mesocorticolimbic Activity States and Depressive and Anxiety-like Prefrontal Cortical Molecular Phenotypes Persisting into Adulthood. Cereb. Cortex 2019, 29, 3140–3153. [Google Scholar] [CrossRef]
- Damaj, M.I.; Glassco, W.; Dukat, M.; Martin, B.R. Pharmacological characterization of nicotine-induced seizures in mice. J. Pharmacol. Exp. Ther. 1999, 291, 1284–1291. [Google Scholar]
- Iha, H.A.; Kunisawa, N.; Shimizu, S.; Tokudome, K.; Mukai, T.; Kinboshi, M.; Ikeda, A.; Ito, H.; Serikawa, T.; Ohno, Y. Nicotine Elicits Convulsive Seizures by Activating Amygdalar Neurons. Front. Pharmacol. 2017, 8, 57. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Godoy, J.A.; Vargas, J.Y.; Arrazola, M.S.; Rios, J.A.; Carvajal, F.J.; Serrano, F.G.; Farias, G.G. Nicotine prevents synaptic impairment induced by amyloid-beta oligomers through alpha7-nicotinic acetylcholine receptor activation. Neuromolecular Med. 2013, 15, 549–569. [Google Scholar] [CrossRef]
- Sakurai, R.; Cerny, L.M.; Torday, J.S.; Rehan, V.K. Mechanism for nicotine-induced up-regulation of Wnt signaling in human alveolar interstitial fibroblasts. Exp. Lung Res. 2011, 37, 144–154. [Google Scholar] [CrossRef]
- Gordon, M.D.; Nusse, R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006, 281, 22429–22433. [Google Scholar] [CrossRef]
- Shinozuka, T.; Takada, R.; Yoshida, S.; Yonemura, S.; Takada, S. Wnt produced by stretched roof-plate cells is required for the promotion of cell proliferation around the central canal of the spinal cord. Development 2019, 146, dev159343. [Google Scholar] [CrossRef]
- Megason, S.G.; McMahon, A.P. A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development 2002, 129, 2087–2098. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Muroyama, Y.; Fujihara, M.; Ikeya, M.; Kondoh, H.; Takada, S. Wnt signaling plays an essential role in neuronal specification of the dorsal spinal cord. Genes Dev. 2002, 16, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.; Lu, C.C.; Kerman, R.; Steward, O.; Xu, X.M.; Zou, Y. Repulsive Wnt signaling inhibits axon regeneration after CNS injury. J. Neurosci. 2008, 28, 8376–8382. [Google Scholar] [CrossRef]
- Yang, Q.O.; Yang, W.J.; Li, J.; Liu, F.T.; Yuan, H.; Ou Yang, Y.P. Ryk receptors on unmyelinated nerve fibers mediate excitatory synaptic transmission and CCL2 release during neuropathic pain induced by peripheral nerve injury. Mol. Pain 2017, 13, 1744806917709372. [Google Scholar] [CrossRef]
- González, P.; Fernández-Martos, C.M.; Arenas, E.; Rodríguez, F.J. The Ryk receptor is expressed in glial and fibronectin-expressing cells after spinal cord injury. J. Neurotrauma 2013, 30, 806–817. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Ji, Q.; Jin, L.; Tian, M.; Zhang, L.D.; Liu, X.Y. Secreted frizzled-related protein 1 regulates the progression of neuropathic pain in mice following spinal nerve ligation. J. Cell. Physiol. 2018, 233, 5815–5822. [Google Scholar] [CrossRef]
- Gonzalez, P.; Rodríguez, F.J. Analysis of the expression of the Wnt family of proteins and its modulatory role on cytokine expression in non activated and activated astroglial cells. Neurosci. Res. 2017, 114, 16–29. [Google Scholar] [CrossRef]
- Fernández-Martos, C.M.; González-Fernández, C.; González, P.; Maqueda, A.; Arenas, E.; Rodríguez, F.J. Differential expression of Wnts after spinal cord contusion injury in adult rats. PLoS ONE 2011, 6, e27000. [Google Scholar] [CrossRef]
- Wei, L.; Chen, C.; Ding, L.; Mo, M.; Zou, J.; Lu, Z.; Li, H.; Wu, H.; Dai, Y.; Xu, P.; et al. Wnt1 Promotes EAAT2 Expression and Mediates the Protective Effects of Astrocytes on Dopaminergic Cells in Parkinson’s Disease. Neural Plast. 2019, 1247276. [Google Scholar] [CrossRef]
- Wang, X.; Shi, S.H.; Yao, H.J.; Jing, Q.K.; Mo, Y.P.; Lv, W.; Song, L.Y.; Yuan, X.C.; Li, Z.G.; Qin, L.N. Electroacupuncture at Dazhui (GV14) and Mingmen (GV4) protects against spinal cord injury: The role of the Wnt/β-catenin signaling pathway. Neural Regen. Res. 2016, 11, 2004–2011. [Google Scholar]
- Lewis, A.E.; Vasudevan, H.N.; O’Neill, A.K.; Soriano, P.; Bush, J.O. The widely used Wnt1-Cre transgene causes developmental phenotypes by ectopic activation of Wnt signaling. Dev. Biol. 2013, 379, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wu, F.; Yuan, H.; Wang, A.; Kang, G.J.; Truong, T.; Chen, L.; McCallion, A.S.; Gong, X.; Li, S. Sox10+ Cells Contribute to Vascular Development in Multiple Organs-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1727–1731. [Google Scholar] [CrossRef]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef] [PubMed]
- Chikazawa, N.; Tanaka, H.; Tasaka, T.; Nakamura, M.; Tanaka, M.; Onishi, H.; Katano, M. Inhibition of Wnt Signaling Pathway Decreases Chemotherapy-resistant Side-population Colon Cancer Cells. Anticancer Res. 2010, 30, 2041–2048. [Google Scholar] [PubMed]
- Ki, D.H.; Oppel, F.; Durbin, A.D.; Look, A.T. Mechanisms underlying synergy between DNA topoisomerase I-targeted drugs and mTOR kinase inhibitors in NF1-associated malignant peripheral nerve sheath tumors. Oncogene 2019, 38, 6585–6598. [Google Scholar] [CrossRef]
- Grillner, S. Biological pattern generation: The cellular and computational logic of networks in motion. Neuron 2006, 52, 751–766. [Google Scholar] [CrossRef]
- Kiehn, O. Locomotor circuits in the mammalian spinal cord. Annu. Rev. Neurosci. 2006, 29, 279–306. [Google Scholar] [CrossRef]
- Blutstein, T.; Castello, M.A.; Viechweg, S.S.; Hadjimarkou, M.M.; McQuail, J.A.; Holder, M.; Thompson, L.P.; Mong, J.A. Differential Responses of Hippocampal Neurons and Astrocytes to Nicotine and Hypoxia in the Fetal Guinea Pig. Neurotox. Res. 2013, 24, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A. Cholinergic systems in brain development and disruption by neurotoxicants: Nicotine, environmental tobacco smoke, organophosphates. Toxicol. Appl. Pharmacol. 2004, 198, 132–151. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Beato, M.; Nistri, A. Alternating rhythmic activity induced by dorsal root stimulation in the neonatal rat spinal cord in vitro. J. Physiol. 2001, 530, 105–112. [Google Scholar] [CrossRef]
- Cazalets, J.R.; Sqalli-Houssaini, Y.; Clarac, F. Activation of the central pattern generators for locomotion by serotonin and excitatory amino acids in neonatal rat. J. Physiol. 1992, 455, 187–204. [Google Scholar] [CrossRef]
- Gillies, J.D.; Lance, J.W.; Neilson, P.D.; Tassinari, C.A. Presynaptic inhibition of the monosynaptic reflex by vibration. J. Physiol. 1969, 205, 329–339. [Google Scholar] [CrossRef]
- Kiehn, O. Decoding the organization of spinal circuits that control locomotion. Nat. Rev. Neurosci. 2016, 17, 224–238. [Google Scholar] [CrossRef]
- Wollman, L.B.; Levine, R.B.; Fregosi, R.F. Developmental plasticity of GABAergic neurotransmission to brainstem motoneurons. J. Physiol. 2018, 596, 5993–6008. [Google Scholar] [CrossRef]
- Wollman, L.B.; Levine, R.B.; Fregosi, R.F. Developmental nicotine exposure alters glycinergic neurotransmission to hypoglossal motoneurons in neonatal rats. J. Neurophysiol. 2018, 120, 1135–1142. [Google Scholar] [CrossRef]
- Wollman, L.B.; Clarke, J.; DeLucia, C.M.; Levine, R.B.; Fregosi, R.F. Developmental Nicotine Exposure Alters Synaptic Input to Hypoglossal Motoneurons and Is Associated with Altered Function of Upper Airway Muscles. eNeuro 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Ai, F.; Zhang, Y.; Peng, B. Restoration of miR-98 relieves the inhibitory effect of nicotine on the differentiation of P19 cells into cardiomyocytes. Biotechnol. Lett. 2016, 38, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Zou, Y.; Zhao, Z.; Li, B.; Ran, P. Nicotine-induced epithelial-mesenchymal transition via Wnt/b-catenin signaling in human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L199–L209. [Google Scholar] [CrossRef] [PubMed]
- Lemjabbar-Alaoui, H.; Dasari, V.; Sidhu, S.S.; Mengistab, A.; Finkbeiner, W.; Gallup, M.; Basbaum, C. Wnt and Hedgehog are critical mediators of cigarette smoke-induced lung cancer. PLoS ONE 2006, 1, e93. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ahmed, J.; Wang, G.; Hassan, I.; Strulovici-Barel, Y.; Hackett, N.R.; Crystal, R.G. Down-Regulation of the Canonical Wnt b-Catenin Pathway in the Airway Epithelium of Healthy Smokers and Smokers with COPD. PLoS ONE 2011, 6, e14793. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, G.L.; Nistri, A. S100β as an early biomarker of excitotoxic damage in spinal cord organotypic cultures. J. Neurochem. 2014, 130, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, Q.; Xi, J.; Xiao, B.; Li, Y.; Ma, C. Neuroprotective effect of fasudil on inflammation through PI3K/Akt and Wnt/β-catenin dependent pathways in a mice model of Parkinson’s disease. Int. J. Clin. Exp. Pathol. 2015, 8, 2354–2364. [Google Scholar]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Cossetti, C.; D’Adamo, P.; Zardini, E.; Andreoni, L.; Ihekwaba, A.E.; et al. Reactive astrocytes and Wnt/β-catenin signaling link nigrostriatal injury to repair in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Neurobiol. Dis. 2011, 41, 508–527. [Google Scholar] [CrossRef]
- Simonetti, M.; Kuner, R. Spinal Wnt5a Plays a Key Role in Spinal Dendritic Spine Remodeling in Neuropathic and Inflammatory Pain Models and in the Proalgesic Effects of Peripheral Wnt3a. J. Neurosci. 2020, 40, 6664–6677. [Google Scholar] [CrossRef] [PubMed]
- Alrefaei, A.F.; Münsterberg, A.E.; Wheeler, G.N. FZD10 regulates cell proliferation and mediates Wnt1 induced neurogenesis in the developing spinal cord. PLoS ONE 2020, 15, e0219721. [Google Scholar] [CrossRef]
- Cifra, A.; Mazzone, G.L.; Nani, F.; Nistri, A.; Mladinic, M. Postnatal developmental profile of neurons and glia in motor nuclei of the brainstem and spinal cord, and its comparison with organotypic slice cultures. Dev. Neurobiol. 2012, 72, 1140–1160. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.K.; Conroy, W.G. Nicotinic alpha 7 receptors: Synaptic options and downstream signaling in neurons. J. Neurobiol. 2002, 53, 512–523. [Google Scholar] [CrossRef]
- Hsu, Y.N.; Edwards, S.C.; Wecker, L. Nicotine enhances the cyclic AMP-dependent protein kinase-mediated phosphorylation of alpha4 subunits of neuronal nicotinic receptors. J. Neurochem. 1997, 69, 2427–2431. [Google Scholar] [CrossRef]
- Takeda, D.; Nakatsuka, T.; Gu, J.G.; Yoshida, M. The activation of nicotinic acetylcholine receptors enhances the inhibitory synaptic transmission in the deep dorsal horn neurons of the adult rat spinal cord. Mol. Pain 2007, 3, 1744–8069. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, G.L.; Margaryan, G.; Kuzhandaivel, A.; Nasrabady, S.E.; Mladinic, M.; Nistri, A. Kainate-induced delayed onset of excitotoxicity with functional loss unrelated to the extent of neuronal damage in the in vitro spinal cord. Neuroscience 2010, 168, 451–462. [Google Scholar] [CrossRef]
- Mazzone, G.L.; Veeraraghavan, P.; Gonzalez-Inchauspe, C.; Nistri, A.; Uchitel, O.D. ASIC channel inhibition enhances excitotoxic neuronal death in an in vitro model of spinal cord injury. Neuroscience 2017, 343, 398–410. [Google Scholar] [CrossRef]
- Kuzhandaivel, A.; Nistri, A.; Mazzone, G.L.; Mladinic, M. Molecular Mechanisms Underlying Cell Death in Spinal Networks in Relation to Locomotor Activity after Acute Injury in vitro. Front. Cell. Neurosci. 2011, 5, 9. [Google Scholar] [CrossRef]
- Margaryan, G.; Mladinic, M.; Mattioli, C.; Nistri, A. Extracellular magnesium enhances the damage to locomotor net-works produced by metabolic perturbation mimicking spinal injury in the neonatal rat spinal cord in vitro. Neuroscience 2009, 163, 669–682. [Google Scholar] [CrossRef]
- Petrović, A.; Kaur, J.; Tomljanović, I.; Nistri, A.; Mladinic, M. Pharmacological induction of Heat Shock Protein 70 by celastrol protects motoneurons from excitotoxicity in rat spinal cord in vitro. Eur. J. Neurosci. 2019, 49, 215–231. [Google Scholar] [CrossRef]
- Sierra, R.; Bustillo, S.G.; Kameneva, P.; Fiore, E.J.; Mazzone, G.L.; Borda, M.; Blanco, M.V.; Usuardi, C.; Furlan, A.; Ernfors, P.; et al. Contribution of neural crest and GLAST+ Wnt1+ bone marrow pericytes with liver fibrogenesis and/or regeneration. Liver Int. 2020, 40, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Baranauskas, G.; Nistri, A. Effects of RP 67580 on substance P-elicited responses and postsynaptic potentials of motoneurones of the rat isolated spinal cord. Peptides 1995, 16, 357–359. [Google Scholar]
- Ostroumov, K.; Grandolfo, M.; Nistri, A. The effects induced by the sulphonylurea glibenclamide on the neonatal rat spinal cord indicate a novel mechanism to control neuronal excitability and inhibitory neurotransmission. Br. J. Pharmacol. 2007, 150, 47–57. [Google Scholar] [CrossRef]
- Kaur, J.; Flores, G.J.; Nistri, A. Neuroprotective effect of propofol against excitotoxic injury to locomotor networks of the rat spinal cord in vitro. Eur. J. Neurosci. 2016, 44, 2418–2430. [Google Scholar] [CrossRef]
- Sámano, C.; Kaur, J.; Nistri, A. A study of methylprednisolone neuroprotection against acute injury to the rat spinal cord in vitro. Neuroscience 2016, 315, 136–149. [Google Scholar] [CrossRef] [PubMed]
- El Waly, B.; Escarrat, V.; Perez-Sanchez, J.; Kaur, J.; Pelletier, F.; Collazos-Castro, J.E.; Debarbieux, F. Intravital Assessment of Cells Responses to Conducting Polymer-Coated Carbon Microfibres for Bridging Spinal Cord Injury. Cells 2021, 10, 73. [Google Scholar] [CrossRef]
- Kaur, J.; Conti, E. Dataset on inflammation induced after lumbar puncture. Data Brief 2021, 34, 106729. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, G.L.; Mladinic, M.; Nistri, A. Excitotoxic cell death induces delayed proliferation of endogenous neuroprogenitor cells in organotypic slice cultures of the rat spinal cord. Cell Death Dis. 2013, 4, e902. [Google Scholar] [CrossRef][Green Version]
- Nasrabady, S.E.; Kuzhandaivel, A.; Nistri, A. Studies of locomotor network neuroprotection by the selective poly (ADP-ribose) polymerase-1 inhibitor PJ-34 against excitotoxic injury to the rat spinal cord in vitro. Eur. J. Neurosci. 2011, 33, 2216–2227. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, A.; Bianchetti, E.; Nistri, A. The volatile anesthetic methoxyflurane protects motoneurons against excitotoxicity in an in vitro model of rat spinal cord injury. Neuroscience 2015, 285, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Mojumdar, A. A Mechanistic Overview of Spinal Cord Injury, Oxidative DNA Damage Repair and Neuroprotective Therapies. Int. J. Neurosci. 2021. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, J.; Mazzone, G.L.; Aquino, J.B.; Nistri, A. Nicotine Neurotoxicity Involves Low Wnt1 Signaling in Spinal Locomotor Networks of the Postnatal Rodent Spinal Cord. Int. J. Mol. Sci. 2021, 22, 9572. https://doi.org/10.3390/ijms22179572
Kaur J, Mazzone GL, Aquino JB, Nistri A. Nicotine Neurotoxicity Involves Low Wnt1 Signaling in Spinal Locomotor Networks of the Postnatal Rodent Spinal Cord. International Journal of Molecular Sciences. 2021; 22(17):9572. https://doi.org/10.3390/ijms22179572
Chicago/Turabian StyleKaur, Jaspreet, Graciela L. Mazzone, Jorge B. Aquino, and Andrea Nistri. 2021. "Nicotine Neurotoxicity Involves Low Wnt1 Signaling in Spinal Locomotor Networks of the Postnatal Rodent Spinal Cord" International Journal of Molecular Sciences 22, no. 17: 9572. https://doi.org/10.3390/ijms22179572
APA StyleKaur, J., Mazzone, G. L., Aquino, J. B., & Nistri, A. (2021). Nicotine Neurotoxicity Involves Low Wnt1 Signaling in Spinal Locomotor Networks of the Postnatal Rodent Spinal Cord. International Journal of Molecular Sciences, 22(17), 9572. https://doi.org/10.3390/ijms22179572