
LSSmScarlet, dCyRFP2s, dCyOFP2s and CRISPRed2s, Genetically Encoded Red Fluorescent Proteins with a Large Stokes Shift

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Developing Red Fluorescent Proteins with Large Stokes Shift Based on mScarlet, mCyRFP1, CyOFP1 and mCRISPRed Proteins in E. coli
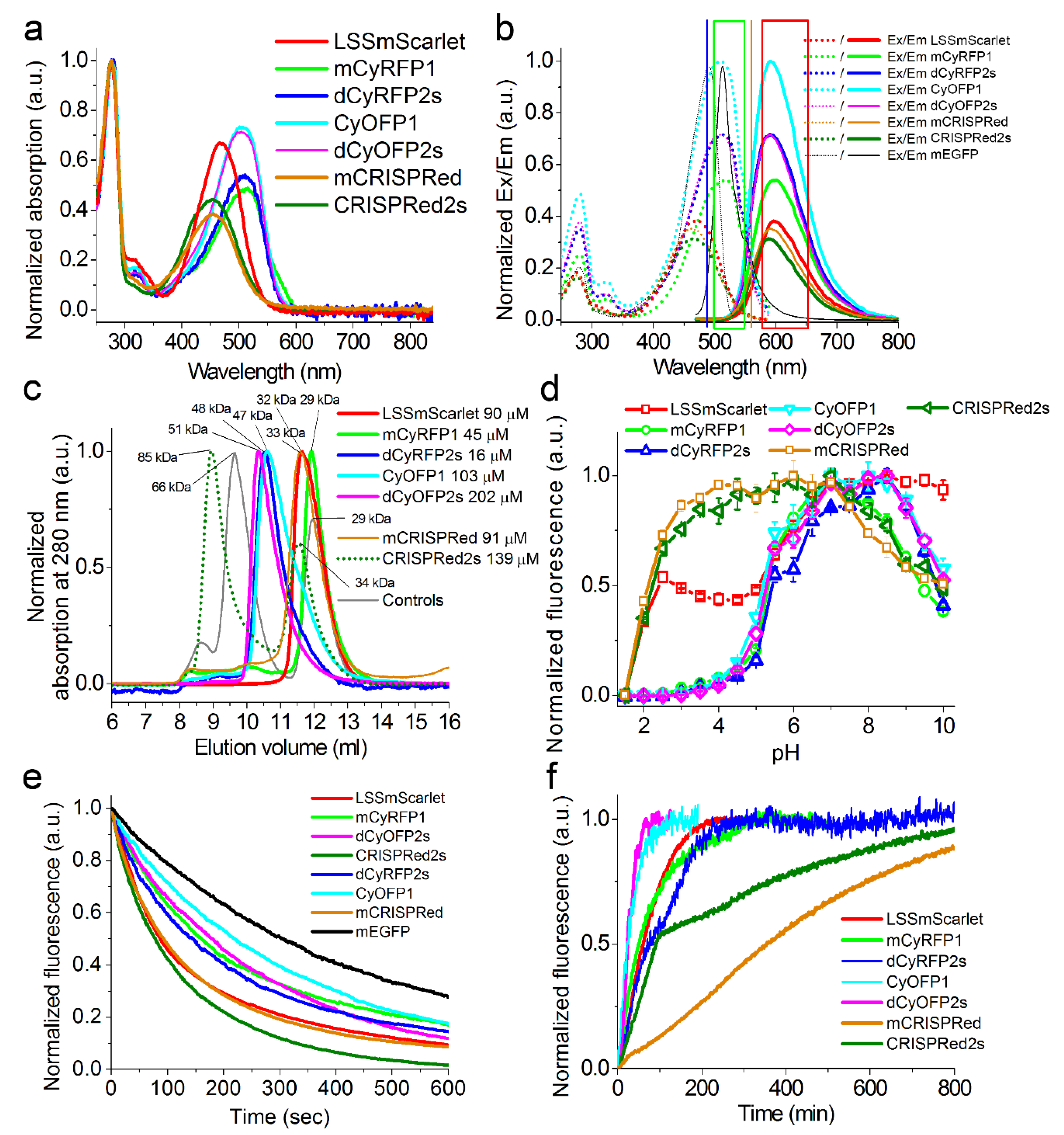
2.2. In Vitro Characterization of the Purified LSSmScarlet, dCyRFP2s, dCyOFP2s and CRISPRed2s LSSRFPs
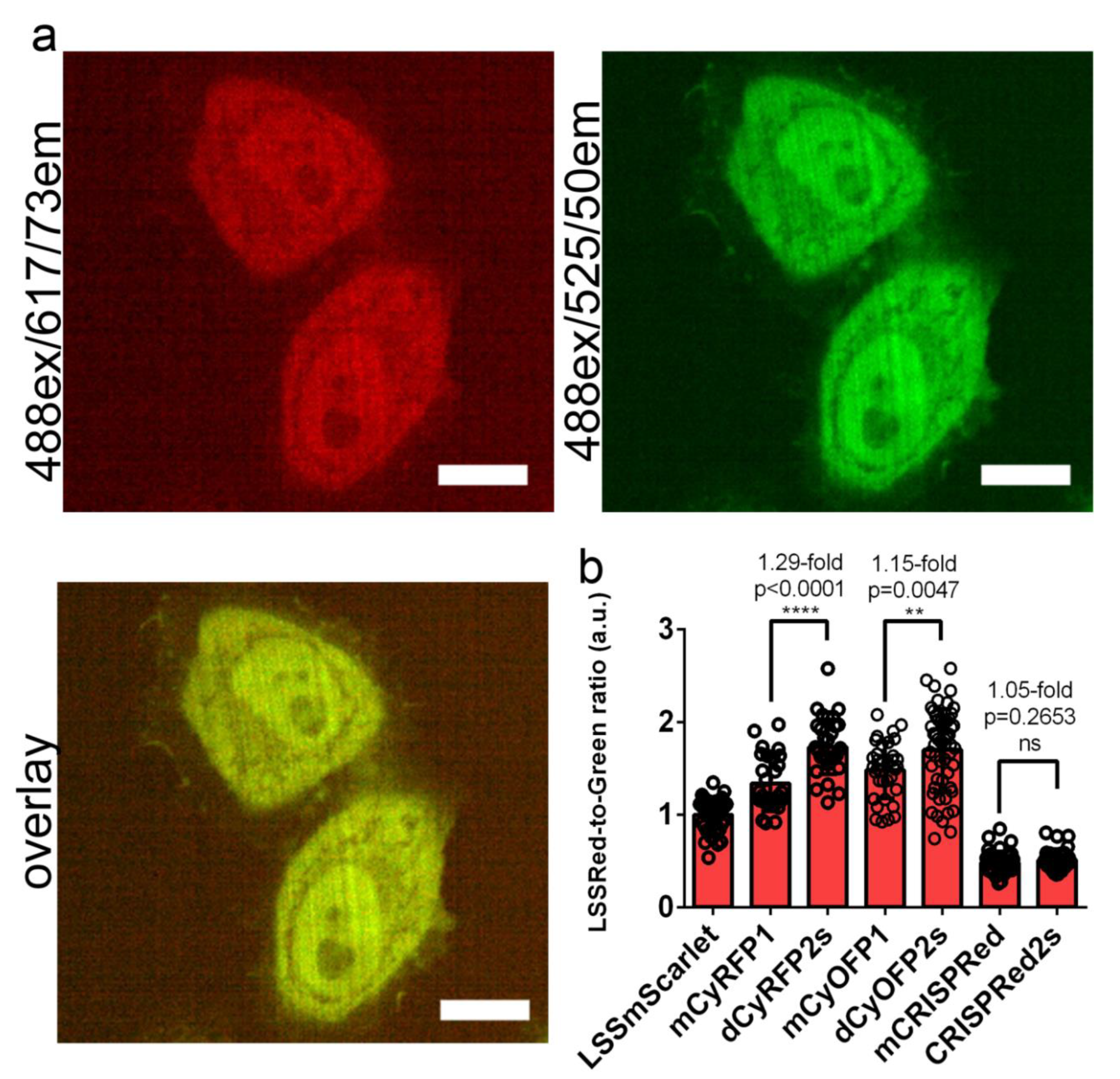
2.3. Brightness of the LSSmScarlet, dCyRFP2s, dCyOFP2s and CRISPRed2s LSSRFPs in Cultured Mammalian Cells
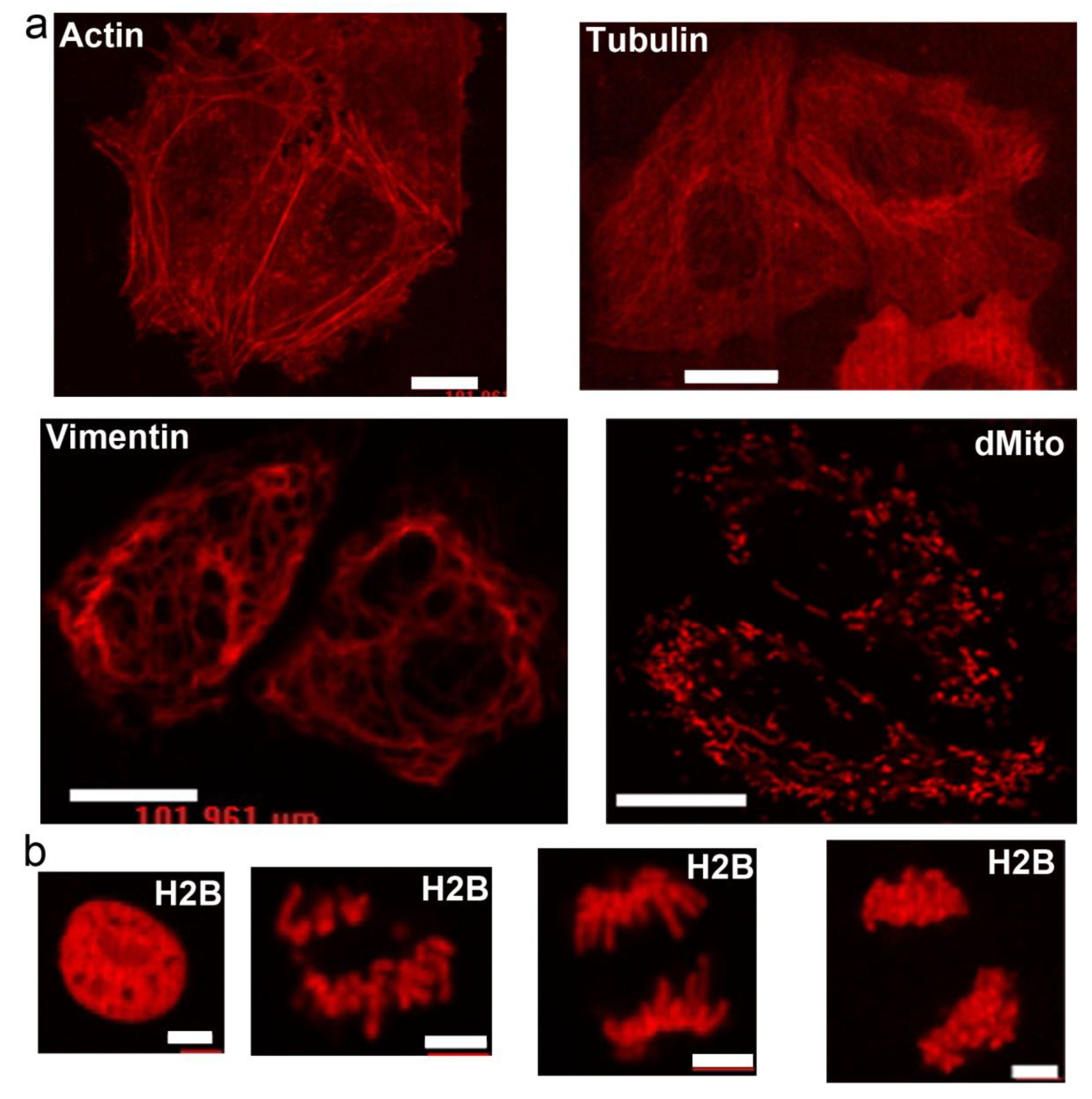
2.4. Behavior of LSSmScarlet in Mammalian Cells in Fusions with Structural Proteins
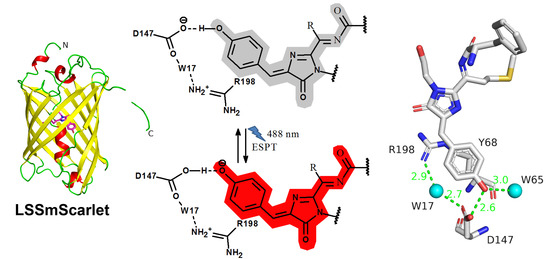
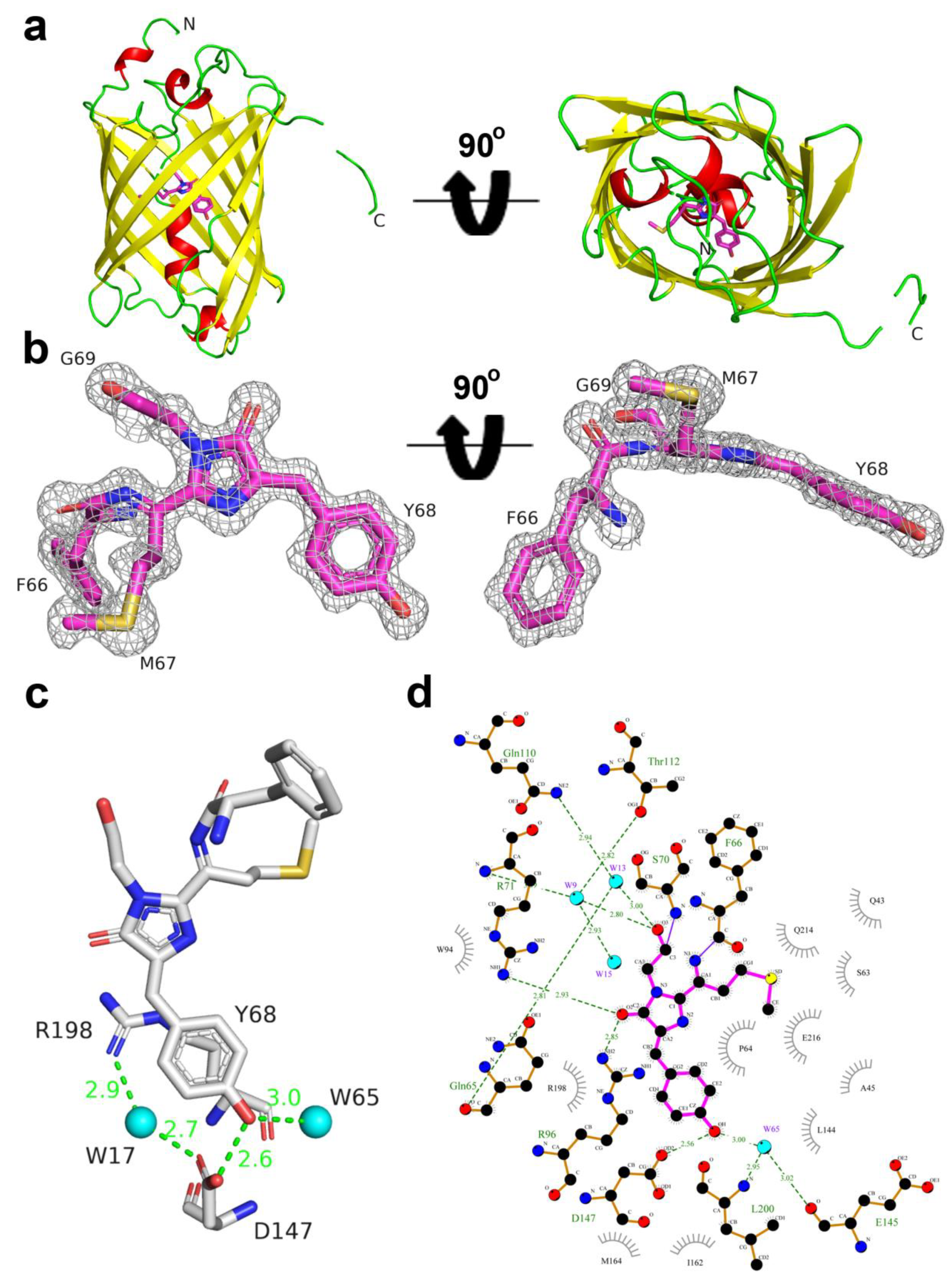
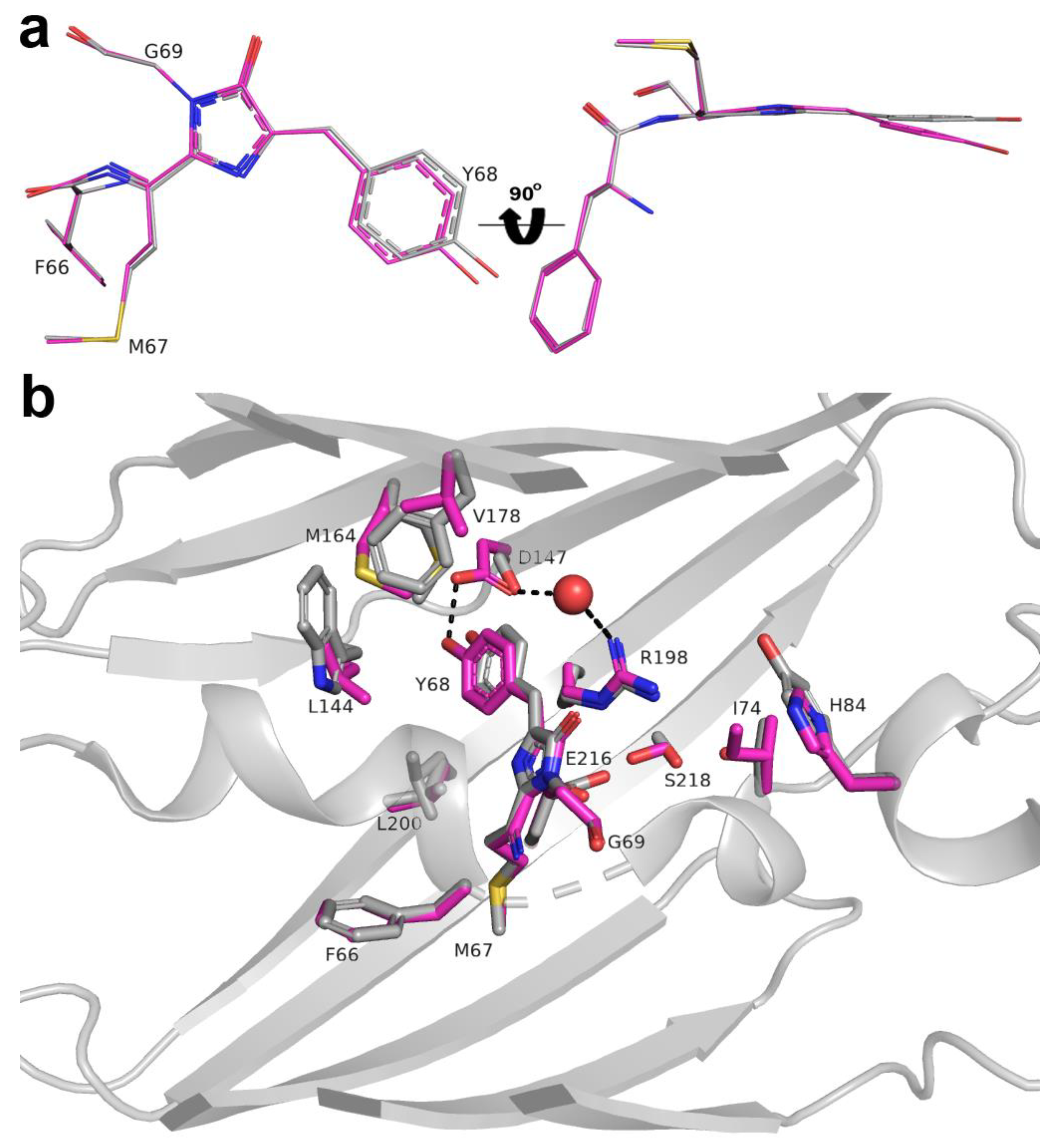
2.5. Structural Characterization of LSSmScarlet
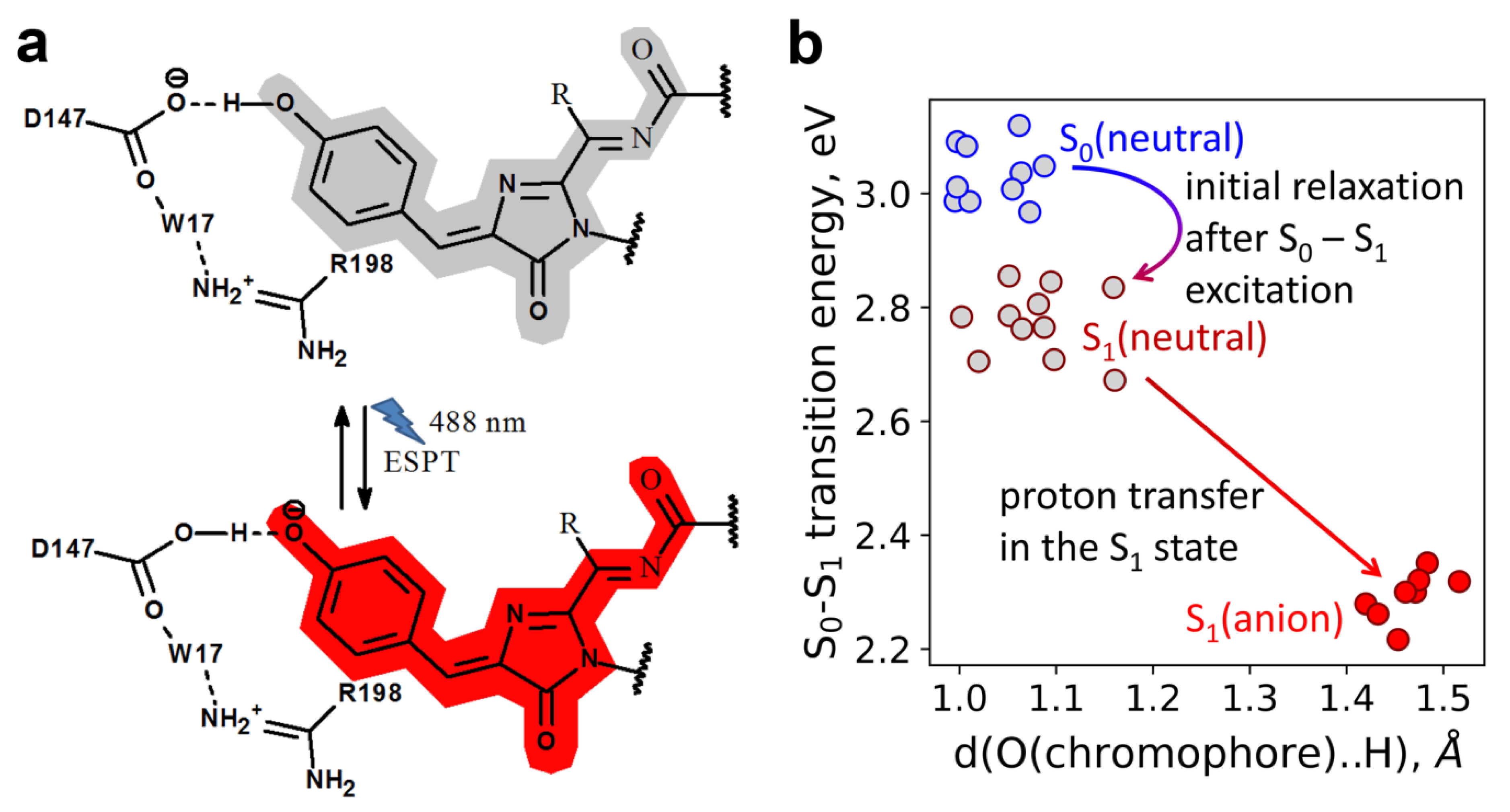
2.6. Molecular Dynamics Simulations of the ESPT Mechanism in LSSmScarlet
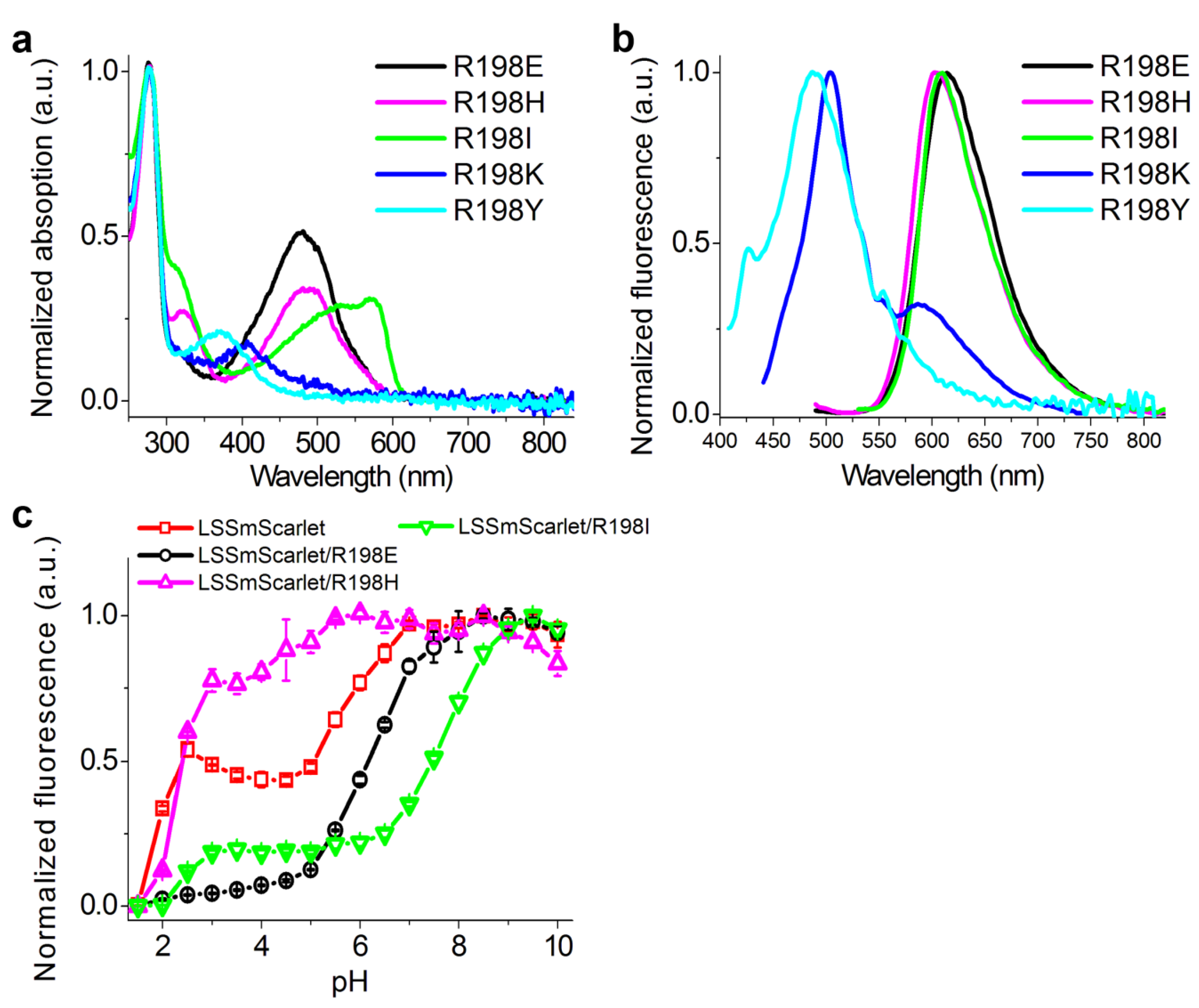
2.7. The Role of R198 Residue in ESPT of the LSSmScarlet Protein
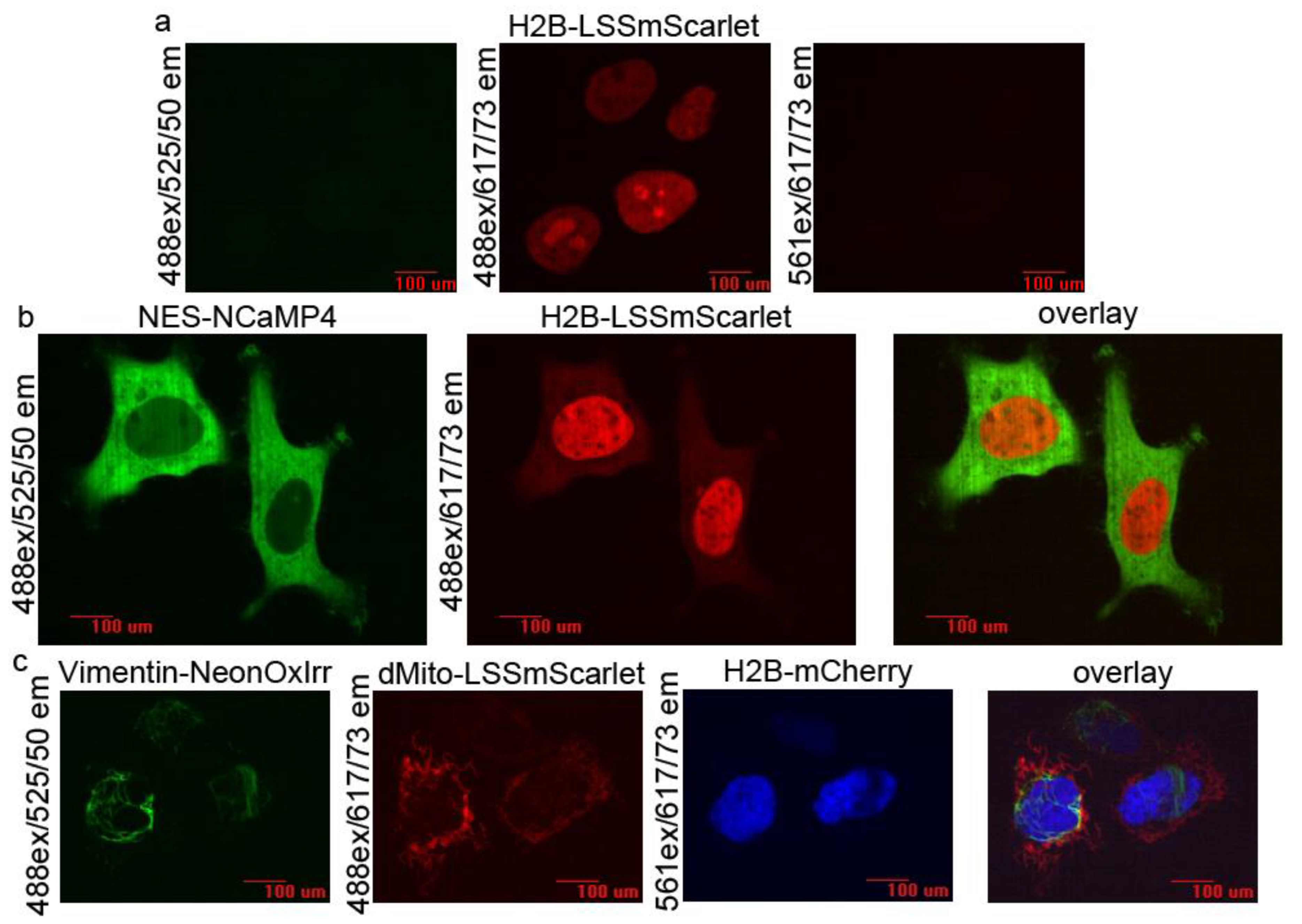
2.8. Two- and Three-Color Confocal Imaging with the LSSmScarlet Protein
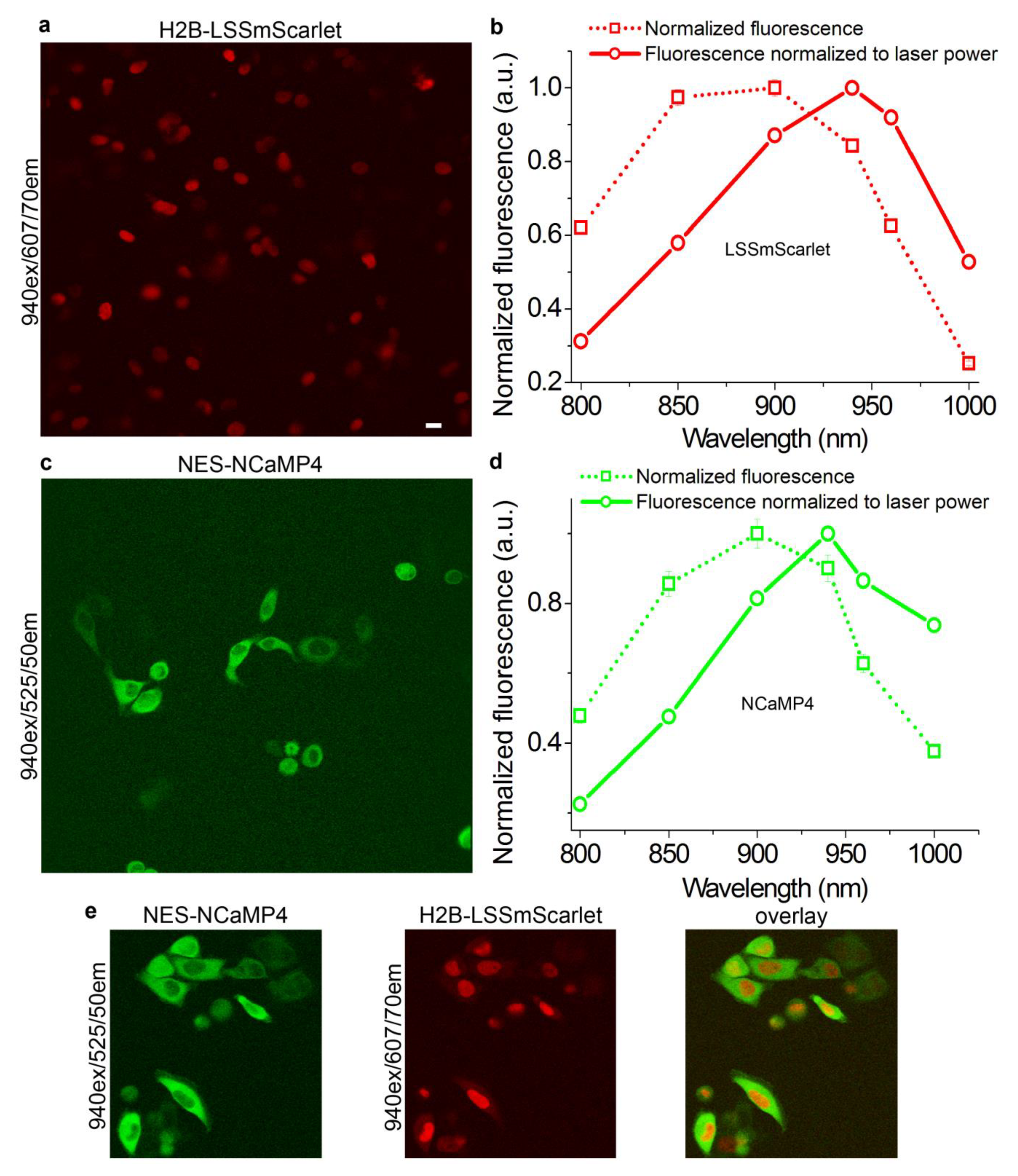
2.9. Two-Photon Imaging of the LSSmScarlet Protein
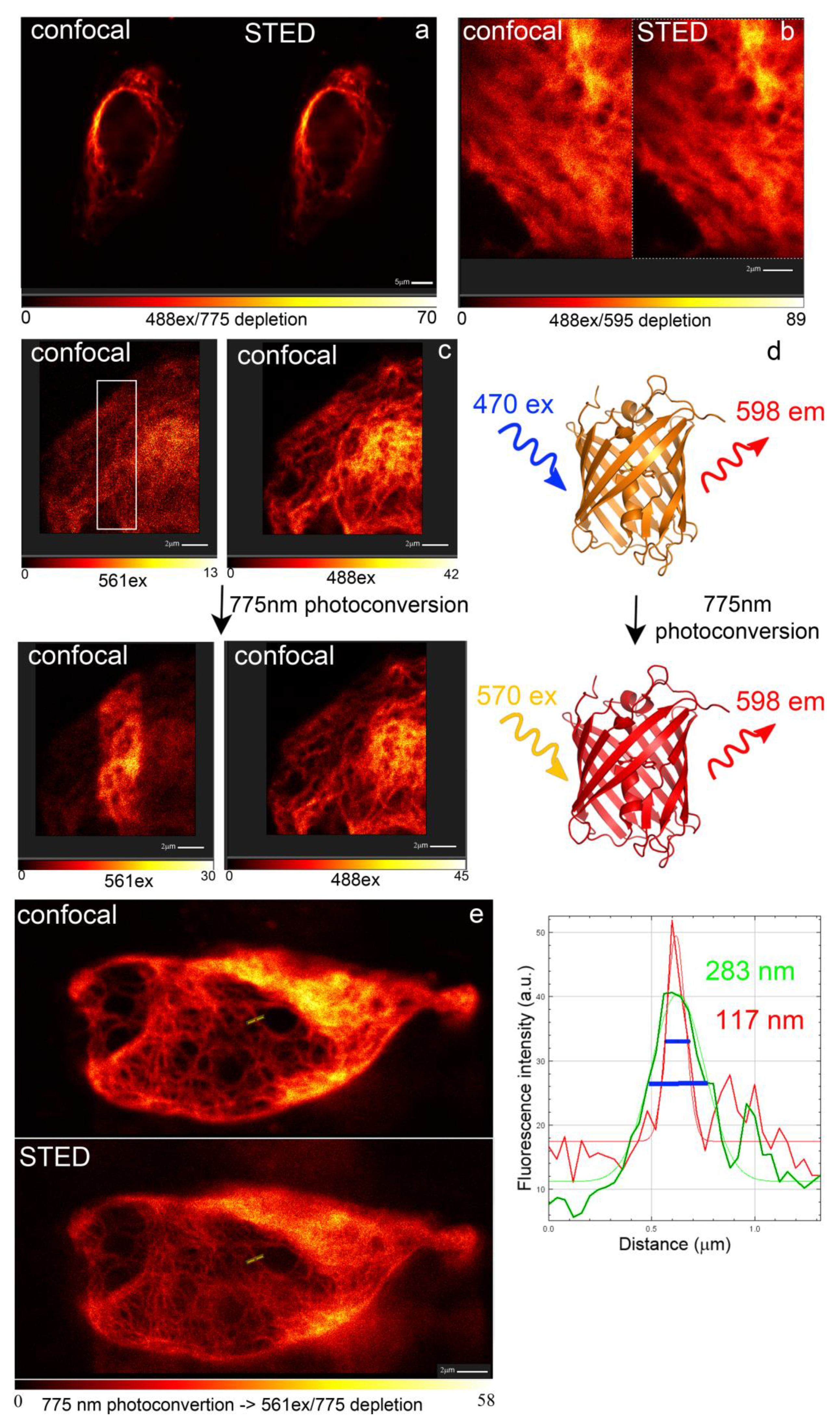
2.10. LSSRed-to-Red Photoconversion of the LSSmScarlet Protein in the Conditions of the Super-Resolution STED Imaging
3. Materials and Methods
3.1. Cloning of Bacterial Vectors, Mutagenesis and Library Screening
3.2. Proteins’ Purification and Characterization
3.3. Protein Crystallization
3.4. Data Collection, Processing, Structure Solution and Refinement
3.5. Structure Analysis and Validation
3.6. Mammalian Plasmids Construction
3.7. Mammalian Live- and Fixed-Cell Imaging
3.8. Statistics
3.9. Molecular Modeling
3.10. Fixation of the Cells with 4% PFA
3.11. STED Imaging
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| RFP | Red fluorescent protein |
| LSSRFP | Red fluorescent protein with large Stokes shift |
| FP | Fluorescent protein |
| PBS | Phosphate Buffered Saline |
| QY | Quantum yield |
| SD | Standard deviation |
References
- Piatkevich, K.D.; Malashkevich, V.N.; Almo, S.C.; Verkhusha, V.V. Engineering ESPT pathways based on structural analysis of LSSmKate red fluorescent proteins with large Stokes shift. J. Am. Chem. Soc. 2010, 132, 10762–10770. [Google Scholar] [CrossRef] [Green Version]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Oh, Y.; Sens, A.; Ataie, N.; Dana, H.; Macklin, J.J.; Laviv, T.; Welf, E.S.; Dean, K.M.; Zhang, F.; et al. A bright cyan-excitable orange fluorescent protein facilitates dual-emission microscopy and enhances bioluminescence imaging in vivo. Nat. Biotechnol. 2016, 34, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Subach, F.V.; Verkhusha, V.V. Chromophore transformations in red fluorescent proteins. Chem. Rev. 2012, 112, 4308–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subach, F.V.; Piatkevich, K.D.; Verkhusha, V.V. Directed molecular evolution to design advanced red fluorescent proteins. Nat. Methods 2011, 8, 1019–1026. [Google Scholar] [CrossRef]
- Subach, O.M.; Subach, F.V. GAF-CaMP3-sfGFP, An Enhanced Version of the Near-Infrared Genetically Encoded Positive Phytochrome-Based Calcium Indicator for the Visualization of Neuronal Activity. Int. J. Mol. Sci. 2020, 21, 6883. [Google Scholar] [CrossRef]
- Subach, O.M.; Barykina, N.V.; Anokhin, K.V.; Piatkevich, K.D.; Subach, F.V. Near-Infrared Genetically Encoded Positive Calcium Indicator Based on GAF-FP Bacterial Phytochrome. Int. J. Mol. Sci. 2019, 20, 3488. [Google Scholar] [CrossRef] [Green Version]
- Pedelacq, J.D.; Cabantous, S.; Tran, T.; Terwilliger, T.C.; Waldo, G.S. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006, 24, 79–88. [Google Scholar] [CrossRef]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef]
- Grabe, M.; Oster, G. Regulation of organelle acidity. J. Gen. Physiol. 2001, 117, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Bindels, D.S.; Haarbosch, L.; van Weeren, L.; Postma, M.; Wiese, K.E.; Mastop, M.; Aumonier, S.; Gotthard, G.; Royant, A.; Hink, M.A.; et al. mScarlet: A bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 2017, 14, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Bajar, B.T.; Wang, E.S.; Lam, A.J.; Kim, B.B.; Jacobs, C.L.; Howe, E.S.; Davidson, M.W.; Lin, M.Z.; Chu, J. Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Sci. Rep. 2016, 6, 20889. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.; Nabel, E.M.; Murdock, M.H.; Lao-Peregrin, C.; Tsoulfas, P.; Blackmore, M.G.; Lee, F.S.; Liston, C.; Morishita, H.; Petsko, G.A. mGreenLantern: A bright monomeric fluorescent protein with rapid expression and cell filling properties for neuronal imaging. Proc. Natl. Acad. Sci. USA 2020, 117, 30710–30721. [Google Scholar] [CrossRef]
- Balleza, E.; Kim, J.M.; Cluzel, P. Systematic characterization of maturation time of fluorescent proteins in living cells. Nat. Methods 2018, 15, 47–51. [Google Scholar] [CrossRef]
- Matlashov, M.E.; Shcherbakova, D.M.; Alvelid, J.; Baloban, M.; Pennacchietti, F.; Shemetov, A.A.; Testa, I.; Verkhusha, V.V. A set of monomeric near-infrared fluorescent proteins for multicolor imaging across scales. Nat. Commun. 2020, 11, 239. [Google Scholar] [CrossRef] [Green Version]
- Erdogan, M.; Fabritius, A.; Basquin, J.; Griesbeck, O. Targeted In Situ Protein Diversification and Intra-organelle Validation in Mammalian Cells. Cell Chem. Biol. 2020, 27, 610–621.e5. [Google Scholar] [CrossRef]
- Shu, X.; Shaner, N.C.; Yarbrough, C.A.; Tsien, R.Y.; Remington, S.J. Novel chromophores and buried charges control color in mFruits. Biochemistry 2006, 45, 9639–9647. [Google Scholar] [CrossRef] [PubMed]
- Subach, O.M.; Sotskov, V.P.; Plusnin, V.V.; Gruzdeva, A.M.; Barykina, N.V.; Ivashkina, O.I.; Anokhin, K.V.; Nikolaeva, A.Y.; Korzhenevskiy, D.A.; Vlaskina, A.V.; et al. Novel Genetically Encoded Bright Positive Calcium Indicator NCaMP7 Based on the mNeonGreen Fluorescent Protein. Int. J. Mol. Sci. 2020, 21, 1644. [Google Scholar] [CrossRef] [Green Version]
- Subach, O.M.; Kunitsyna, T.A.; Mineyeva, O.A.; Lazutkin, A.A.; Bezryadnov, D.V.; Barykina, N.V.; Piatkevich, K.D.; Ermakova, Y.G.; Bilan, D.S.; Belousov, V.V.; et al. Slowly Reducible Genetically Encoded Green Fluorescent Indicator for In Vivo and Ex Vivo Visualization of Hydrogen Peroxide. Int. J. Mol. Sci. 2019, 20, 3138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmstrom, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piatkevich, K.D.; English, B.P.; Malashkevich, V.N.; Xiao, H.; Almo, S.C.; Singer, R.H.; Verkhusha, V.V. Photoswitchable red fluorescent protein with a large Stokes shift. Chem. Biol. 2014, 21, 1402–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, S.N.; Hunt, H.D.; Horton, R.M.; Pullen, J.K.; Pease, L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989, 77, 51–59. [Google Scholar] [CrossRef]
- Subach, F.V.; Patterson, G.H.; Manley, S.; Gillette, J.M.; Lippincott-Schwartz, J.; Verkhusha, V.V. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat. Methods 2009, 6, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Svetogorov, R.; Dorovatovskii, P.; Lazarenko, V. Belok/XSA Diffraction Beamline for Studying Crystalline Samples at Kurchatov Synchrotron Radiation Source. Cryst. Res. Technol. 2020, 55, 1900184. [Google Scholar] [CrossRef]
- Winter, G.; Waterman, D.G.; Parkhurst, J.M.; Brewster, A.S.; Gildea, R.J.; Gerstel, M.; Fuentes-Montero, L.; Vollmar, M.; Michels-Clark, T.; Young, I.D.; et al. DIALS: Implementation and evaluation of a new integration package. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62 (Pt 1), 72–82. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 1994, 50 (Pt 5), 760–763. [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60 (Pt 12 Pt 1), 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60 (Pt 12 Pt 1), 2256–2268. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Vriend, G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Denning, E.J.; Priyakumar, U.D.; Nilsson, L.; Mackerell, A.D., Jr. Impact of 2′-hydroxyl sampling on the conformational properties of RNA: Update of the CHARMM all-atom additive force field for RNA. J. Comput. Chem. 2011, 32, 1929–1943. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Seritan, S.; Bannwarth, C.; Fales, B.S.; Hohenstein, E.G.; Isborn, C.M.; Kokkila-Schumacher, S.I.L.; Li, X.; Liu, F.; Luehr, N.; Snyder, J.W.; et al. TeraChem: A graphical processing unit-accelerated electronic structure package for large-scale ab initio molecular dynamics. WIREs Comput. Mol. Sci. 2020, 11, e1494. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.; Handy, N. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Zeng, Q.; Zhuang, W.; Liang, W. Characterizing the Structures, Spectra, and Energy Landscapes Involved in the Excited-State Proton Transfer Process of Red Fluorescent Protein LSSmKate1. J. Phys. Chem. B 2016, 120, 9833–9842. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Melo, M.C.R.; Bernardi, R.C.; Rudack, T.; Scheurer, M.; Riplinger, C.; Phillips, J.C.; Maia, J.D.C.; Rocha, G.B.; Ribeiro, J.V.; Stone, J.E.; et al. NAMD goes quantum: An integrative suite for hybrid simulations. Nat. Methods 2018, 15, 351–354. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | Abs, Ex/Em (nm) | QY (%) b | ε (mM−1·cm−1) c | Brightness vs. EGFP (%) | pKa | Monomeric State | Photobleaching Half-Time (s) f | Maturation Half-Time (min) | |
|---|---|---|---|---|---|---|---|---|---|
| ex. at Max d | ex. at 488 nm e | ||||||||
| LSSmScarlet | 466, 470/598 | 43 ± 2 | 30.2 ± 0.6 | 39 | 34 | 1.91 ± 0.01; 5.78 ± 0.06 | Monomer | 87 ± 16 | 61 |
| mCyRFP1 | 514, 520/600 | 61.2 ± 0.4 | 30.6 ± 0.5 | 55 | 49 | 5.33 ± 0.03; 9.41 ± 0.12 | Monomer | 161 ± 26 | 51 |
| dCyRFP2s | 508, 516/592 | 58 ± 1 | 42 ± 2 | 73 | 65 | 5.44 ± 0.04; 9.817 ± 0.004 | Dimer | 144 ± 20 | 75 |
| CyOFP1 | 503, 510/594 | 76 a | 45.1 ± 0.2 | 102 | 97 | 5.22 ± 0.07; >10 | Dimer | 223 ± 37 | 25 |
| dCyOFP2s | 503, 510/592 | 69 ± 7 | 36 ± 4 | 73 | 73 | 5.29 ± 0.03; >10 | Dimer | 176 ± 30 | 24 |
| mCRISPRed | 452, 462/592 | 42 ± 2 | 29.1 ± 0.9 | 36 | 27 | 2.14 ± 0.02 9.8 ± 0.3 | Monomer | 93 ± 7 | 354 |
| CRISPRed2s | 452, 464/590 | 38.2 ± 0.2 | 28.7 ± 0.9 | 32 | 24 | 2.23 ± 0.03 10.0 ± 0.1 | Monomer-Tetramer | 79 ± 13 | 90 |
| Proteins | Normalized Brightness vs. EGFP in HeLa Cells a (p Value b) | Normalized Brightness vs. EGFP for Pure protein c | Difference in Brightness between Pure Protein and HeLa Cells, Fold |
|---|---|---|---|
| LSSmScarlet | 1.00 ± 0.04 (NA) | 1.00 | 0.93 |
| mCyRFP1 | 1.34 ± 0.08 (<0.0001) | 1.44 | 1.08 |
| dCyRFP2s | 1.73 ± 0.08 (<0.0001) | 1.91 | 1.11 |
| CyOFP1 | 1.48 ± 0.08 (<0.0001) | 2.85 | 1.93 |
| dCyOFP2s | 1.70 ± 0.11 (<0.0001) | 2.15 | 1.26 |
| mCRISPRed | 0.48 ± 0.03 (<0.0001) | 1.00 | 2.08 |
| CRISPRed2s | 0.51 ± 0.03 (<0.0001) | 0.71 | 1.38 |
| Data Collection | |
| Diffraction Source | “Belok-RSA” Beamline, NRC “Kurchatov Institute” |
| Wavelength (Å) | 0.75 |
| Temperature (K) | 100 |
| Detector | CCD |
| Crystal-to-detector distance (mm) | 120.00 |
| Rotation range per image (°) | 1.0 |
| Total rotation range (°) | 306 |
| Space group | C2 |
| a, b, c (Å) | 84.52; 45.36; 59.04 |
| α, β, γ (°) | 90.0; 102.34; 90.0 |
| Average mosaicity (°) | 1.4 |
| Unique reflections | 42,242 (2137) |
| Resolution range (Å) | 34.4–1.40 (1.42–1.40) |
| Completeness (%) | 98.0 (100.0) |
| Average redundancy | 6.0 (6.2) |
| 〈I/σ(I)〉 | 7.2 (1.7) |
| Rmeas (%) | 15.0 (67.2) |
| CC1/2 | 99.1 (81.3) |
| Refinement | |
| Rfact (%) | 16.4 |
| Rfree (%) | 19.0 |
| Bonds (Å) | 0.02 |
| Angles (°) | 2.16 |
| Ramachandran plot | |
| Most favored (%) | 98.6 |
| Allowed (%) | 1.4 |
| No. atoms | |
| Protein | 1850 |
| Water | 272 |
| Chromophore | 23 |
| Sodium ion | 2 |
| Other ligands | 5 |
| B-factors (Å2) | |
| Protein | 10.77 |
| Water | 21.34 |
| Chromophore | 7.06 |
| Calcium ion | 10.02 |
| Other ligands | 12.32 |
| Proteins | Abs/Em (nm) | pKa |
|---|---|---|
| LSSmScarlet | 466/598 | 1.91 ± 0.01; 5.78 ± 0.06 |
| LSSmScarlet/R198E | 480/614 | 6.181 ± 0.006 |
| LSSmScarlet/R198H | 487/604 | 2.403 ± 0.008 |
| LSSmScarlet/R198I | 571 (472)/610 | 7.76 ± 0.03 |
| LSSmScarlet/R198K | 405/503 | NA |
| LSSmScarlet/R198Y | 372/488 | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subach, O.M.; Vlaskina, A.V.; Agapova, Y.K.; Dorovatovskii, P.V.; Nikolaeva, A.Y.; Ivashkina, O.I.; Popov, V.O.; Piatkevich, K.D.; Khrenova, M.G.; Smirnova, T.A.; et al. LSSmScarlet, dCyRFP2s, dCyOFP2s and CRISPRed2s, Genetically Encoded Red Fluorescent Proteins with a Large Stokes Shift. Int. J. Mol. Sci. 2021, 22, 12887. https://doi.org/10.3390/ijms222312887
Subach OM, Vlaskina AV, Agapova YK, Dorovatovskii PV, Nikolaeva AY, Ivashkina OI, Popov VO, Piatkevich KD, Khrenova MG, Smirnova TA, et al. LSSmScarlet, dCyRFP2s, dCyOFP2s and CRISPRed2s, Genetically Encoded Red Fluorescent Proteins with a Large Stokes Shift. International Journal of Molecular Sciences. 2021; 22(23):12887. https://doi.org/10.3390/ijms222312887
Chicago/Turabian StyleSubach, Oksana M., Anna V. Vlaskina, Yuliya K. Agapova, Pavel V. Dorovatovskii, Alena Y. Nikolaeva, Olga I. Ivashkina, Vladimir O. Popov, Kiryl D. Piatkevich, Maria G. Khrenova, Tatiana A. Smirnova, and et al. 2021. "LSSmScarlet, dCyRFP2s, dCyOFP2s and CRISPRed2s, Genetically Encoded Red Fluorescent Proteins with a Large Stokes Shift" International Journal of Molecular Sciences 22, no. 23: 12887. https://doi.org/10.3390/ijms222312887
APA StyleSubach, O. M., Vlaskina, A. V., Agapova, Y. K., Dorovatovskii, P. V., Nikolaeva, A. Y., Ivashkina, O. I., Popov, V. O., Piatkevich, K. D., Khrenova, M. G., Smirnova, T. A., Boyko, K. M., & Subach, F. V. (2021). LSSmScarlet, dCyRFP2s, dCyOFP2s and CRISPRed2s, Genetically Encoded Red Fluorescent Proteins with a Large Stokes Shift. International Journal of Molecular Sciences, 22(23), 12887. https://doi.org/10.3390/ijms222312887