
Etifoxine Restores Mitochondrial Oxidative Phosphorylation and Improves Cognitive Recovery Following Traumatic Brain Injury


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
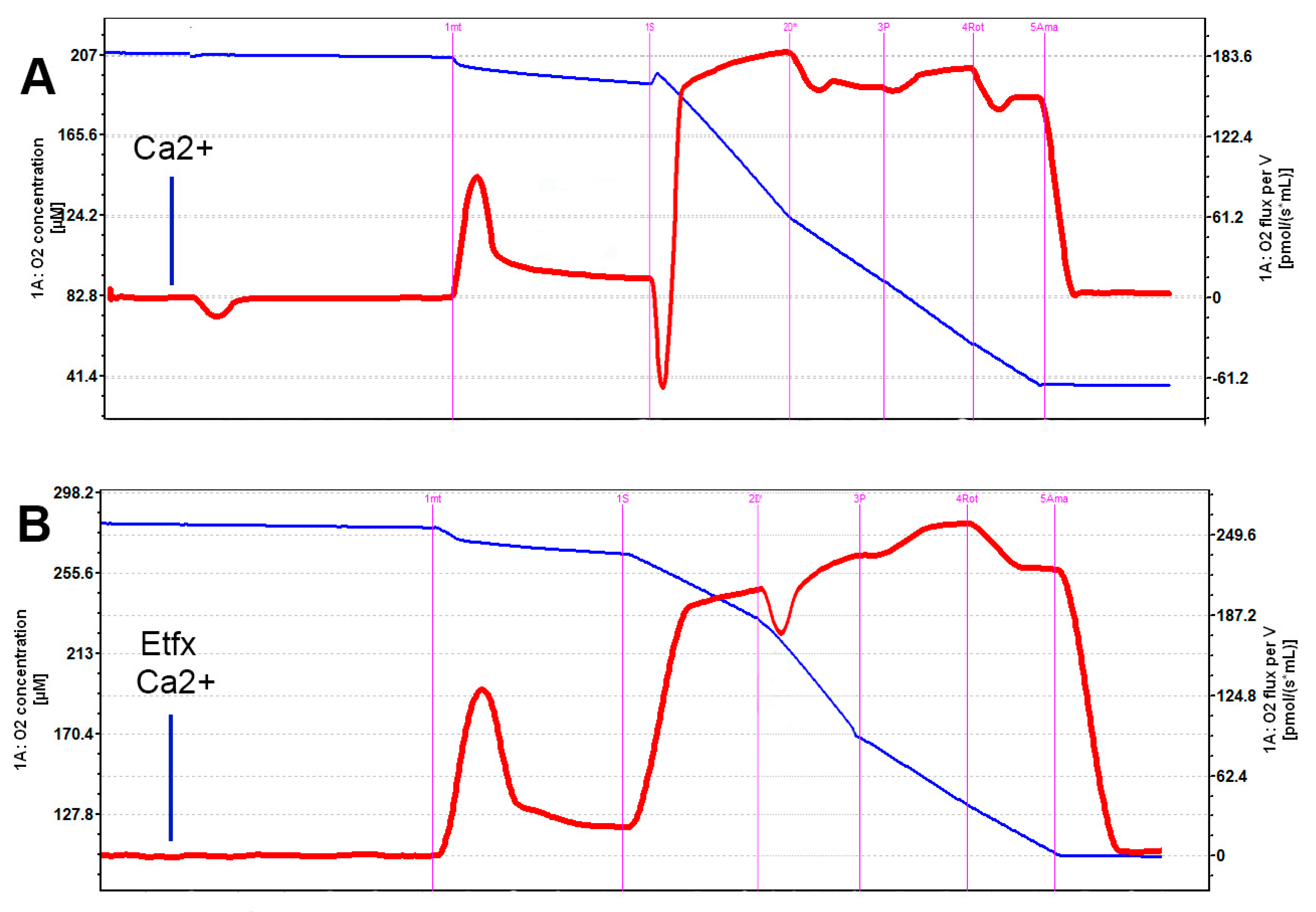
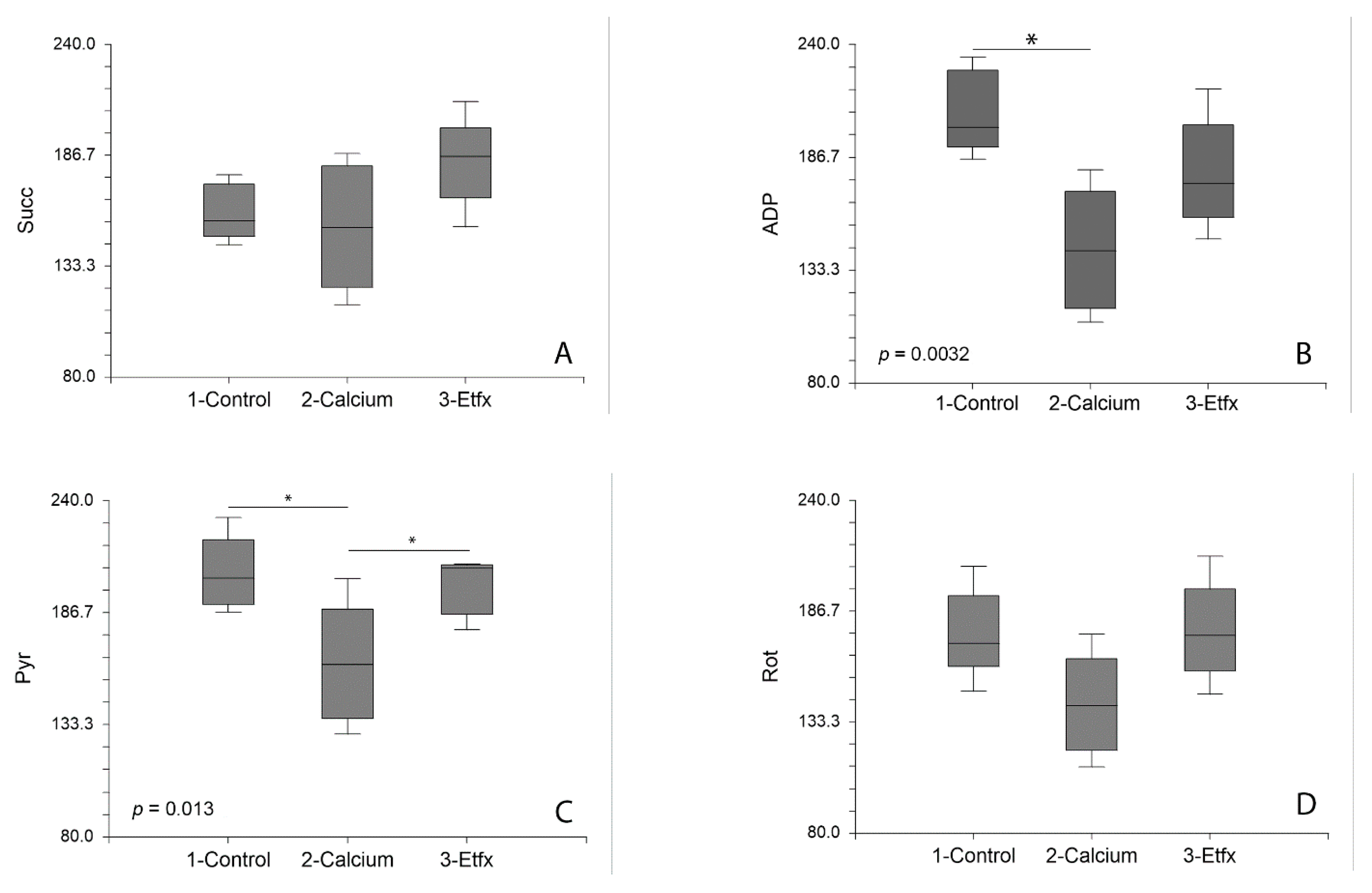
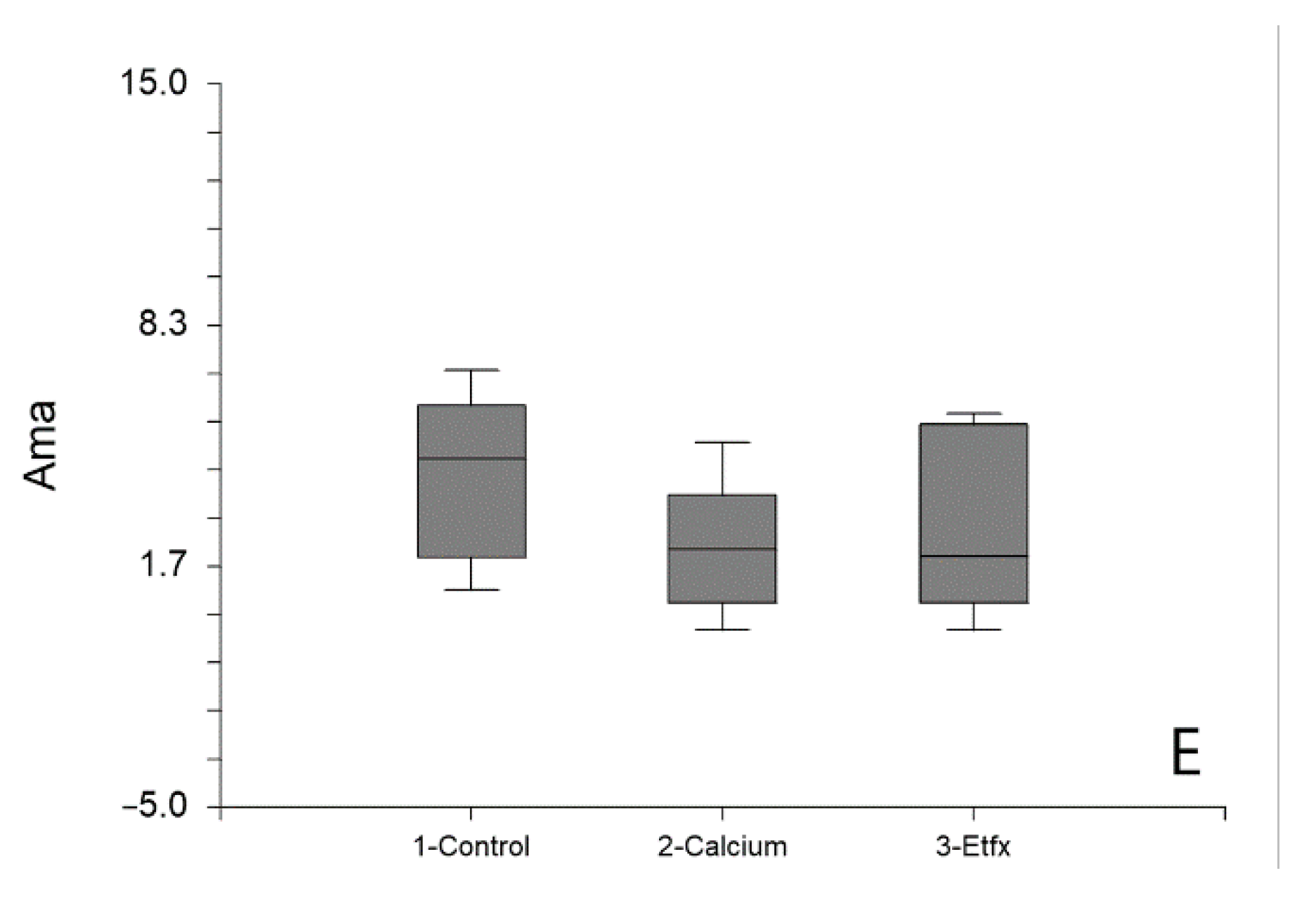
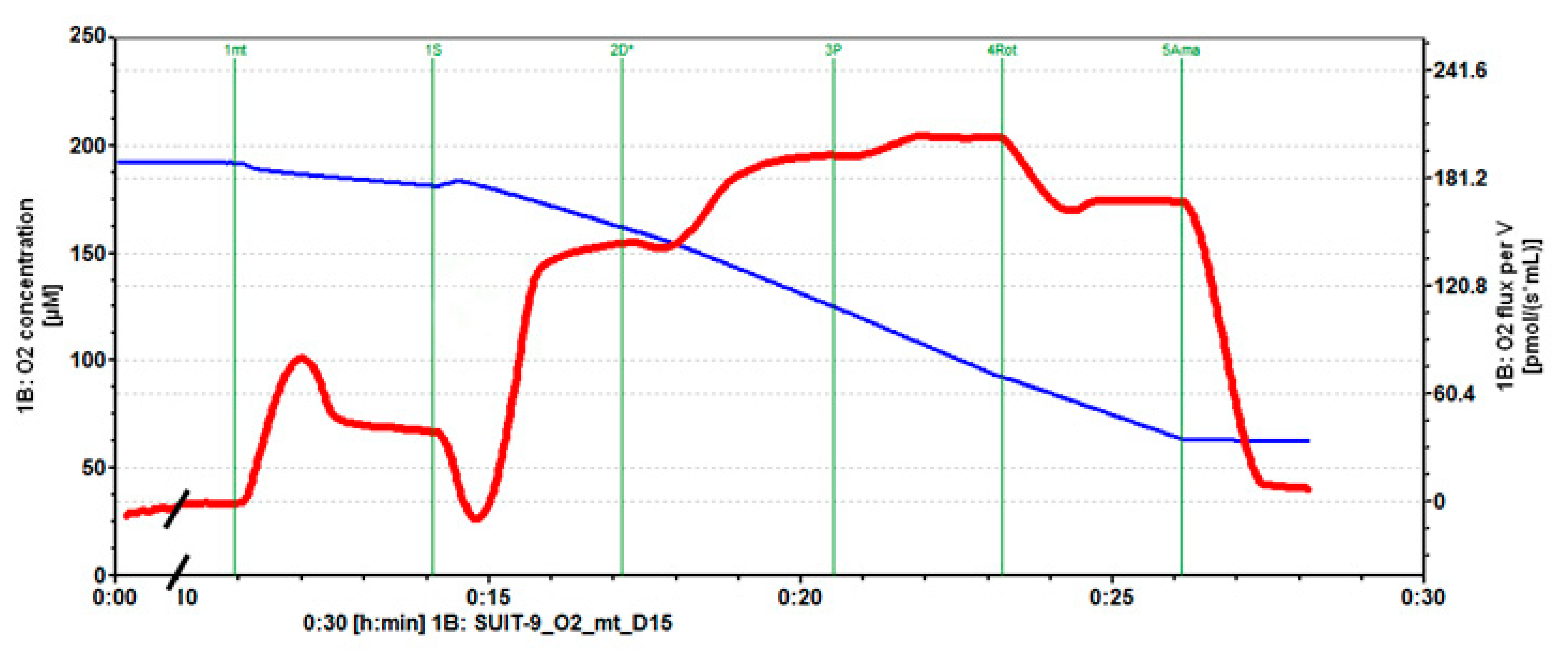
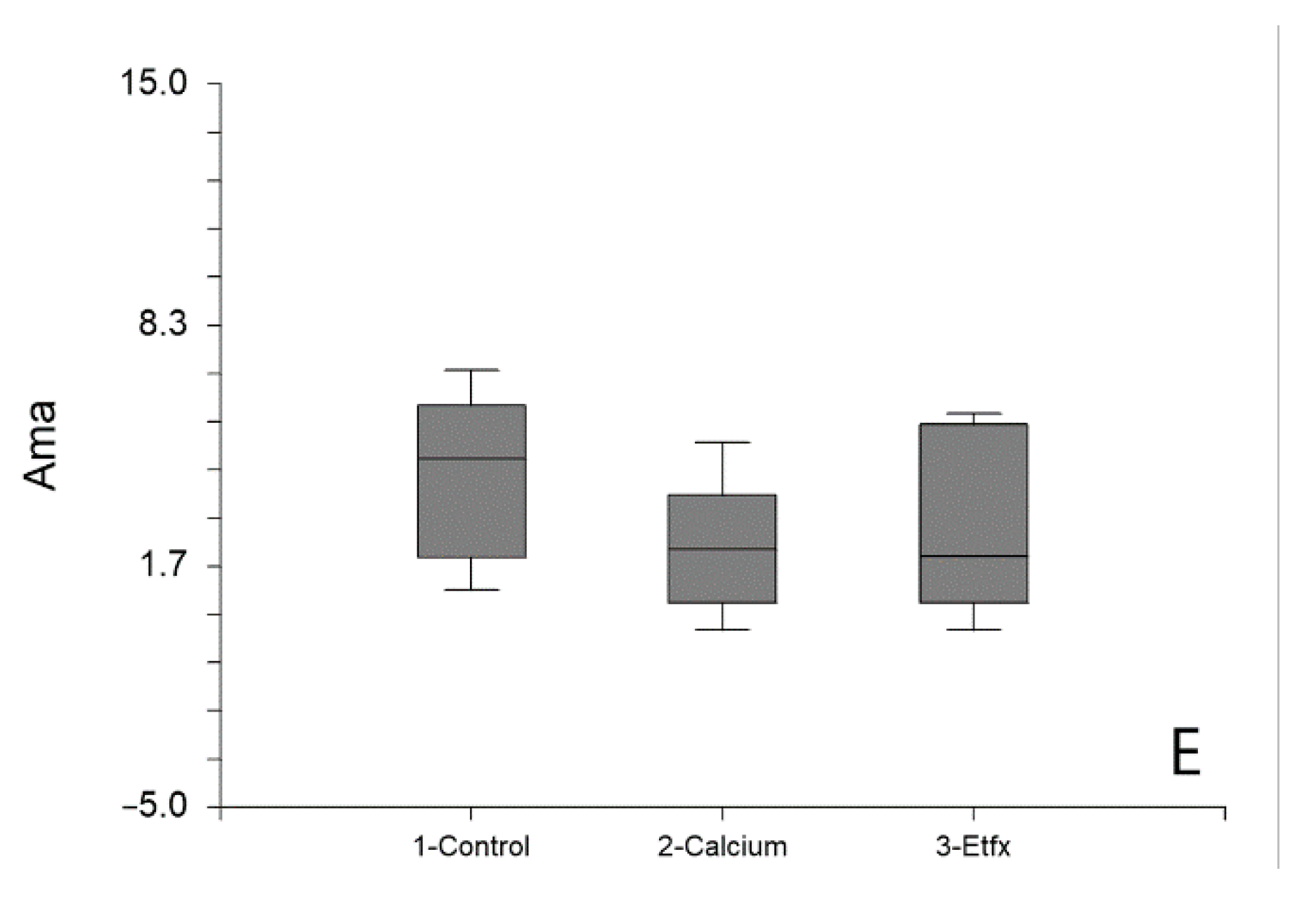
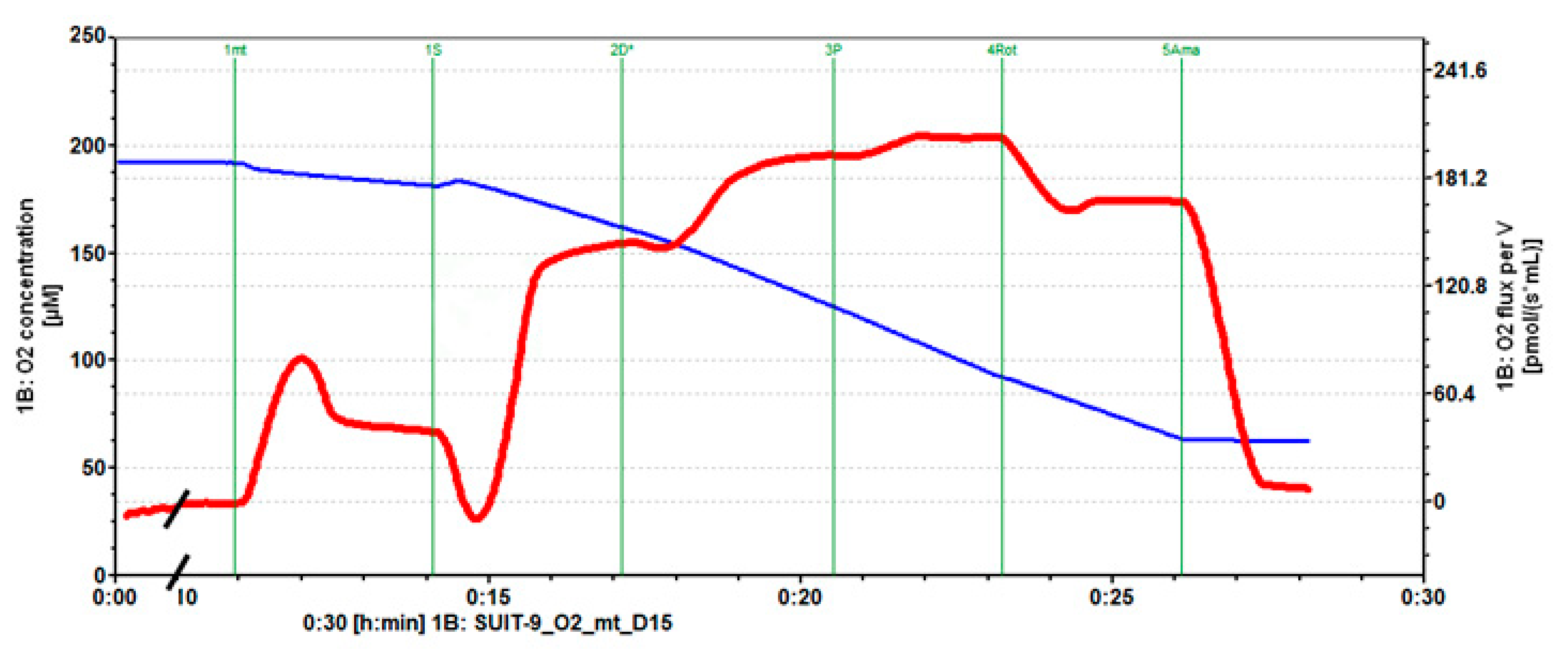
2.1. Mitochondrial Respiration
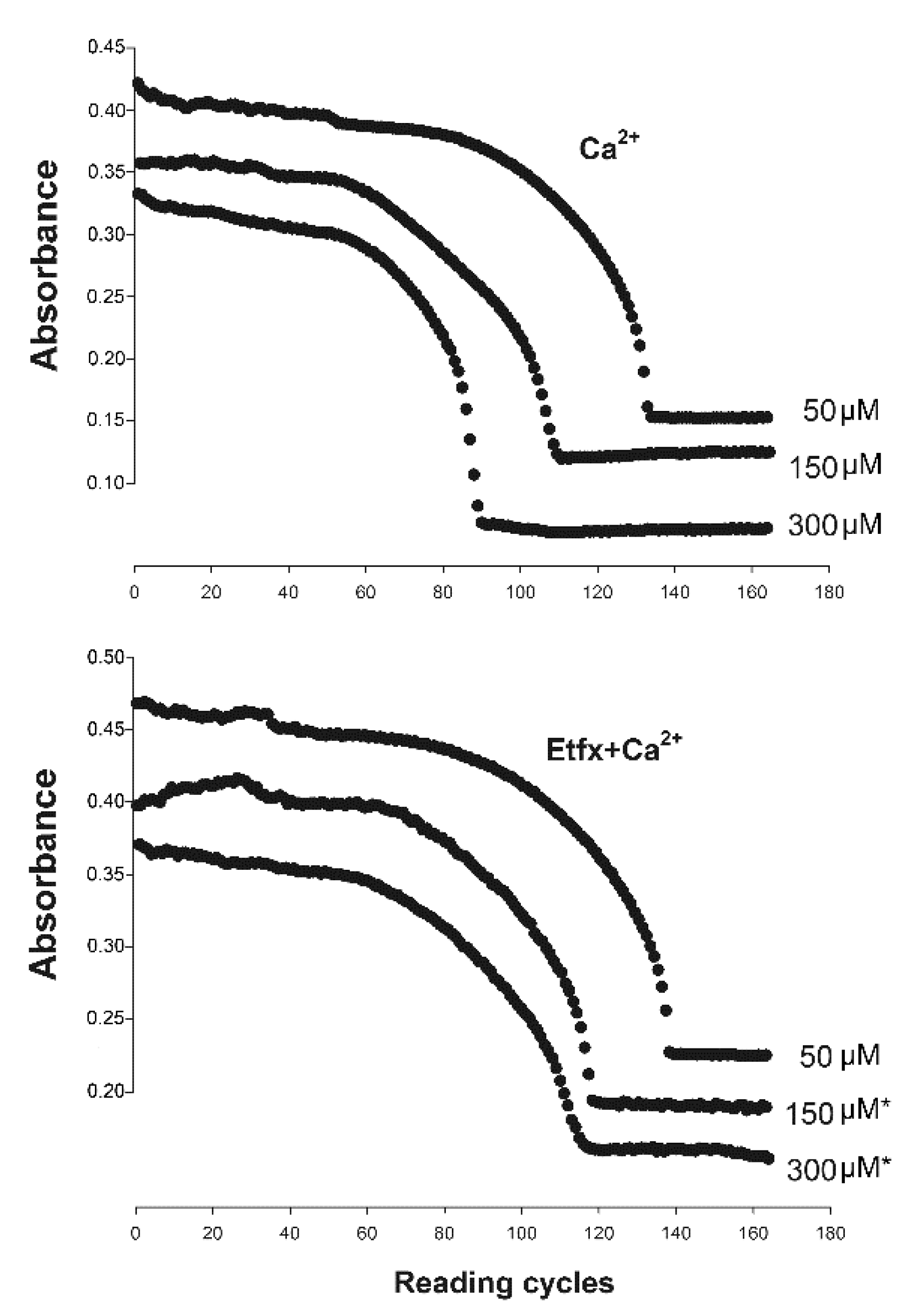
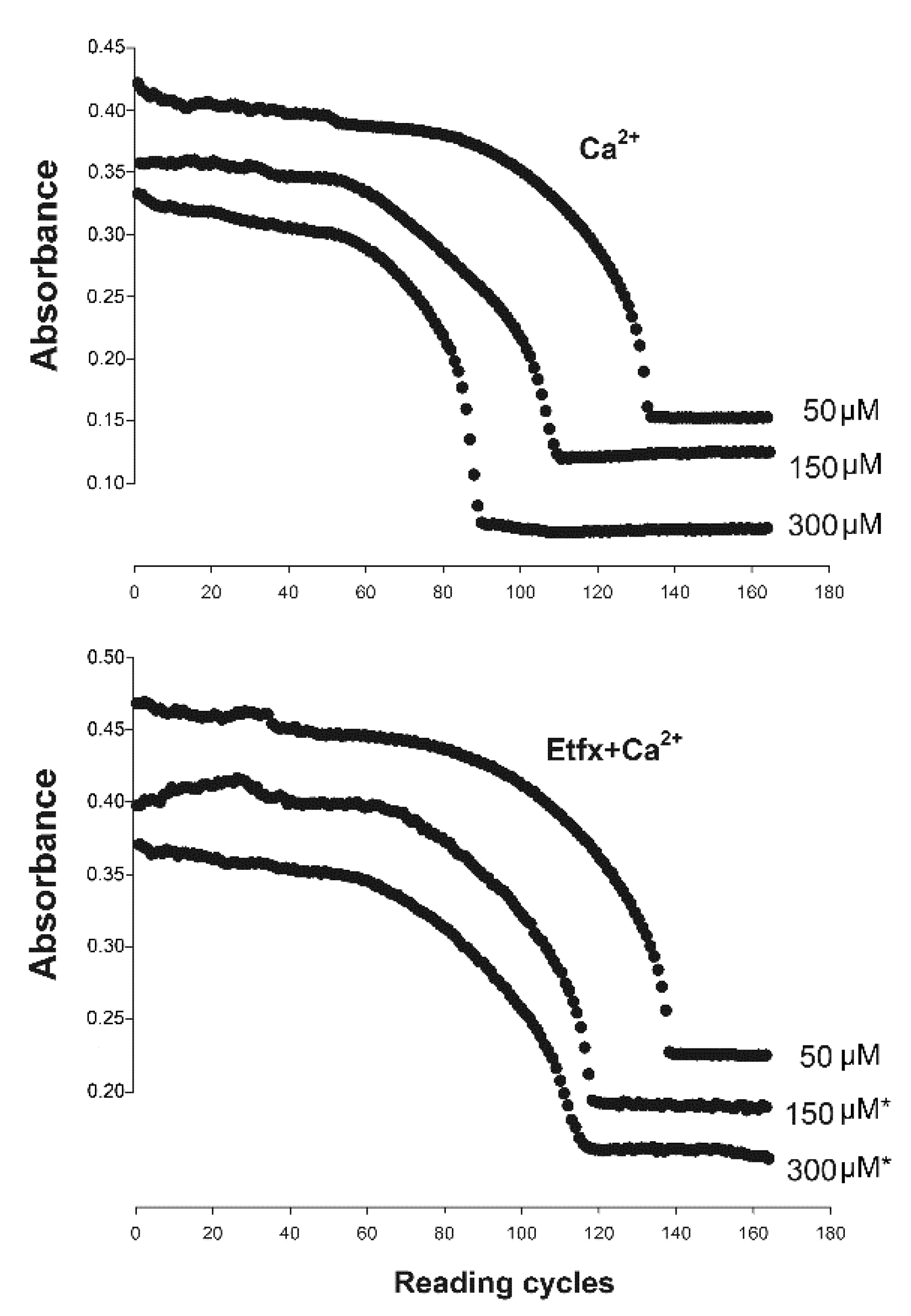
2.2. Mitochondrial Swelling
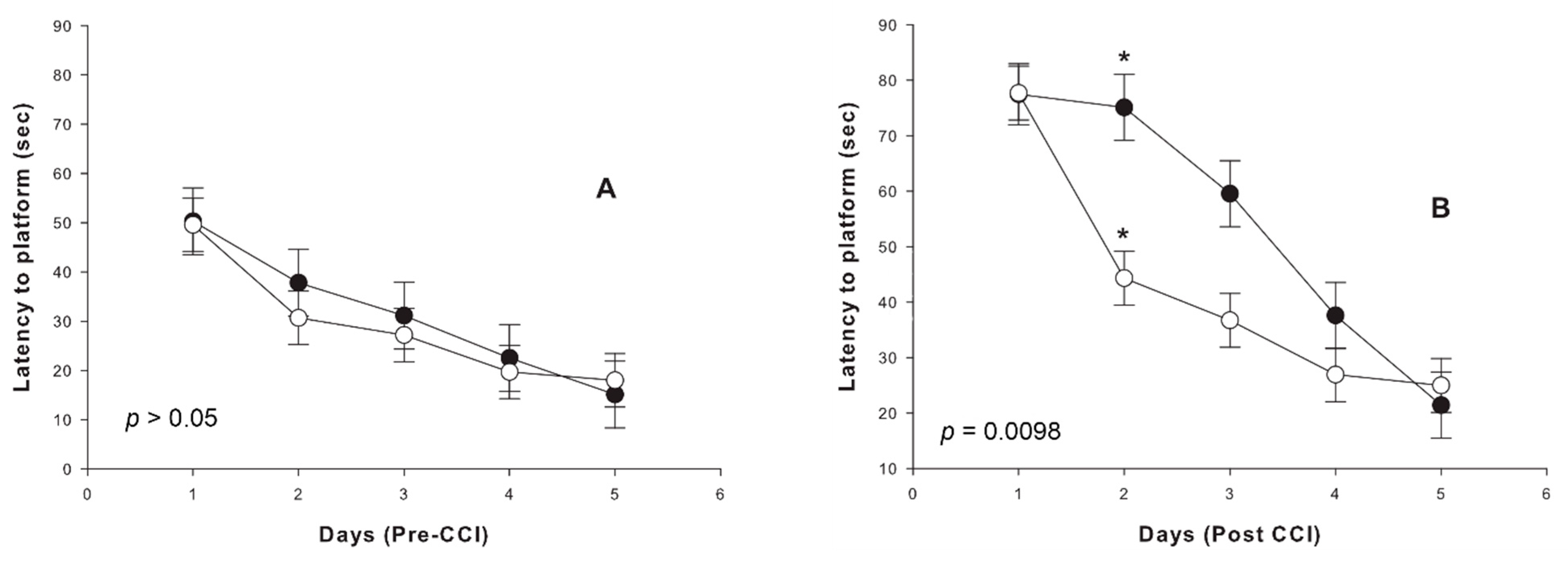
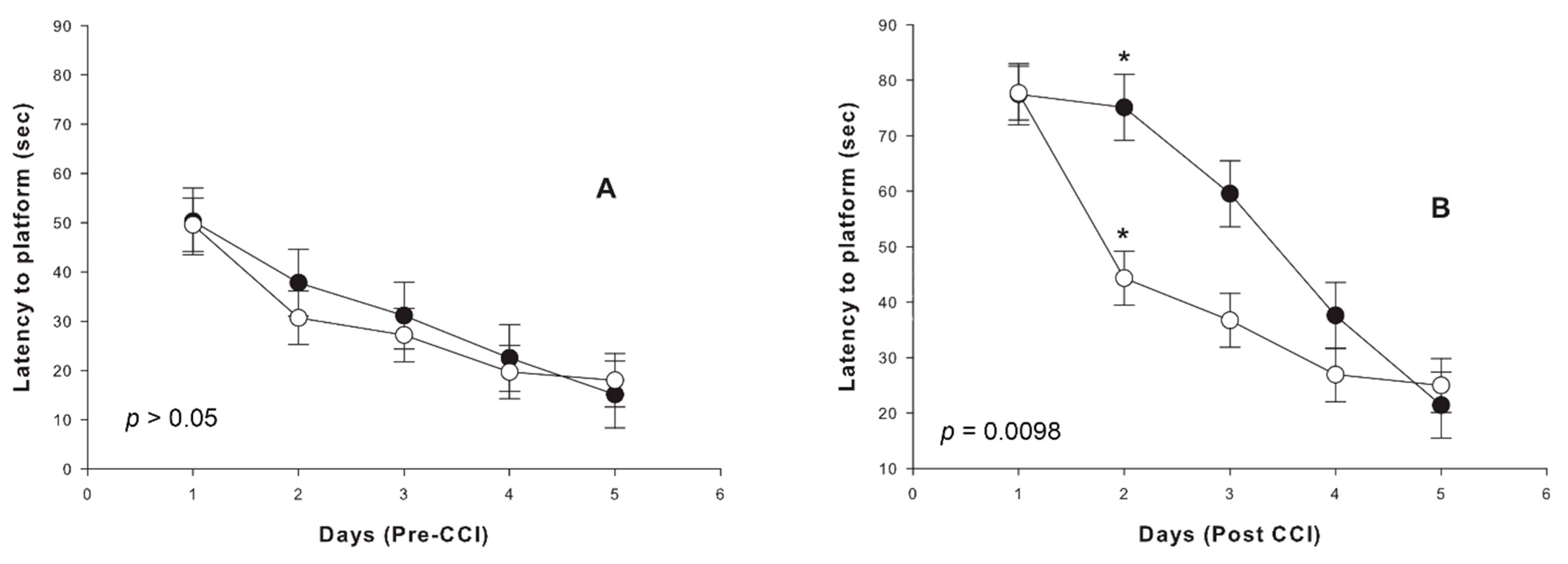
2.3. Cognitive Outcome
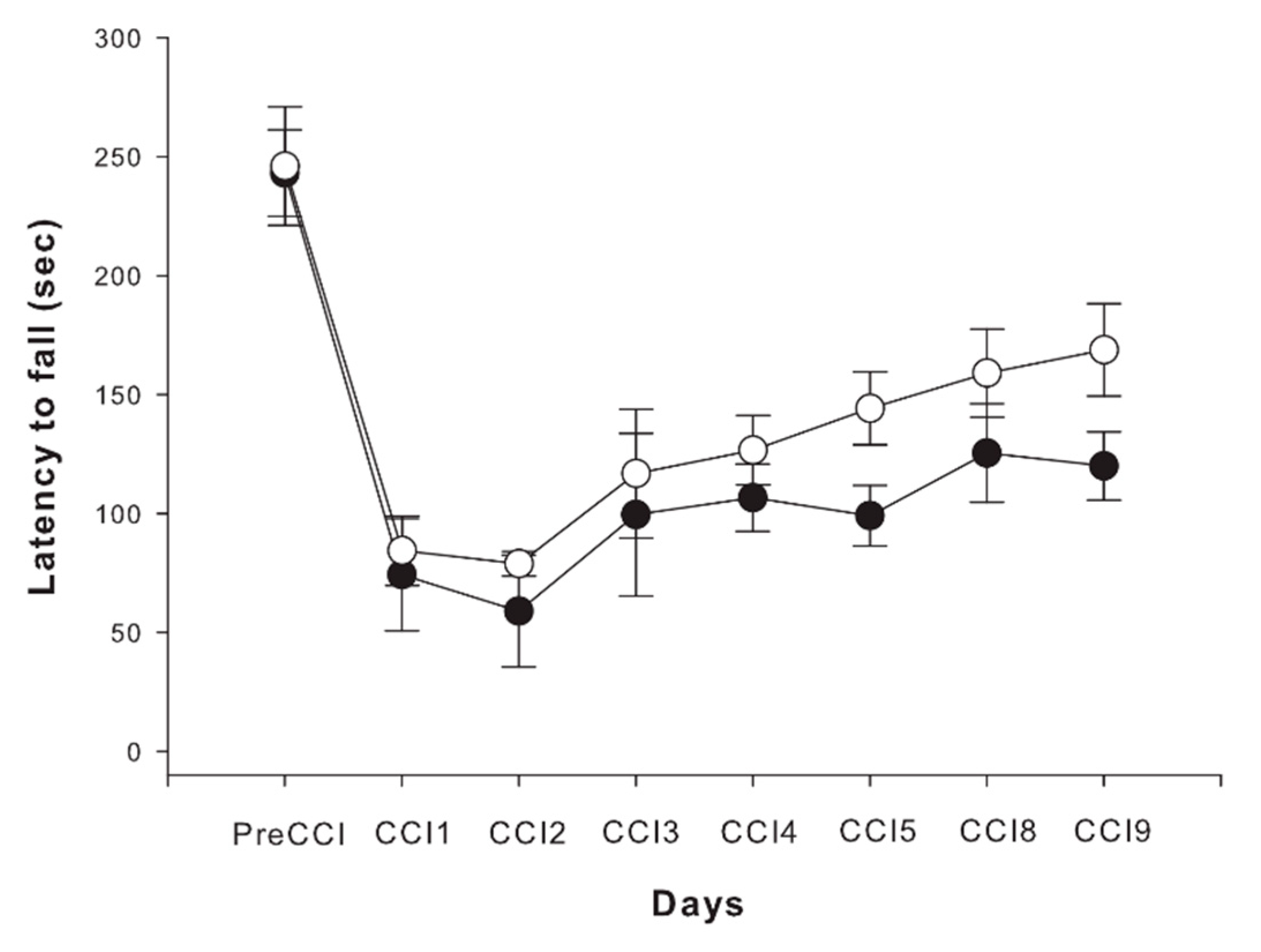
2.4. Motor Outcome
3. Discussion
4. Methods
4.1. Animals
4.2. Brain Injury Model
4.2.1. Model Description
4.2.2. Animal Grouping and Treatment
4.3. Isolation of Mitochondria from Rat Brain
4.4. Mitochondrial Respiration Measurements
4.5. Assessment of Ca2+-Induced Mitochondrial Swelling
4.6. Cognitive Outcome
4.7. Motor Outcome
4.8. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peeters, W.; van den Brande, R.; Polinder, S.; Brazinova, A.; Steyerberg, E.W.; Lingsma, H.F.; Maas, A.I.R. Epidemiology of traumatic brain injury in Europe. Acta Neurochir. 2015, 157, 1683–1696. [Google Scholar] [CrossRef] [Green Version]
- Roozenbeek, B.; Maas, A.I.R.; Menon, D.K. Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 2013, 9, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Reilly, P.; Graham, D.; Hume Adams, J.; Jennett, B. Patients with head injury who talk and die. Lancet 1975, 306, 375–377. [Google Scholar] [CrossRef]
- Doppenberg, E.M.R.; Choi, S.C.; Bullock, R. Clinical Trials in Traumatic Brain Injury: Lessons for the Future. J. Neurosurg. Anesthesiol. 2004, 16, 87–94. [Google Scholar] [CrossRef]
- Maas, A.I.R.; Steyerberg, E.W.; Murray, G.D.; Bullock, R.; Baethmann, A.; Marshall, L.F.; Teasdale, G.M. Why Have Recent Trials of Neuroprotective Agents in Head Injury Failed to Show Convincing Efficacy? A Pragmatic Analysis and Theoretical Considerations. Neurosurgery 1999, 44, 1286–1298. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.K.; Michel, M.E.; Ansell, B.; Baethmann, A.; Biegon, A.; Bracken, M.B.; Bullock, M.R.; Choi, S.C.; Clifton, G.L.; Contant, C.F.; et al. Clinical Trials in Head Injury. J. Neurotrauma 2002, 19, 503–557. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Becker, D.P.; Ward, J.D.; Sullivan, H.G.; Adams, W.E.; Rosner, M.J. Significance of intracranial hypertension in severe head injury. J. Neurosurg. 1977, 47, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Saul, T.G.; Ducker, T.B. Effect of intracranial pressure monitoring and aggressive treatment on mortality in severe head injury. J. Neurosurg. 1982, 56, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Bouma, G.J.; Muizelaar, J.P.; Bandoh, K.; Marmarou, A. Blood pressure and intracranial pressure-volume dynamics in severe head injury: Relationship with cerebral blood flow. J. Neurosurg. 1992, 77, 15–19. [Google Scholar] [CrossRef]
- Jaggi, J.L.; Obrist, W.D.; Gennarelli, T.A.; Langfitt, T.W. Relationship of early cerebral blood flow and metabolism to outcome in acute head injury. J. Neurosurg. 1990, 72, 176–182. [Google Scholar] [CrossRef]
- Obrist, W.D.; Langfitt, T.W.; Jaggi, J.L.; Cruz, J.; Gennarelli, T.A. Cerebral blood flow and metabolism in comatose patients with acute head injury. J. Neurosurg. 1984, 61, 241–253. [Google Scholar] [CrossRef]
- Chieregato, A.; Tanfani, A.; Compagnone, C.; Turrini, C.; Sarpieri, F.; Ravaldini, M.; Targa, L.; Fainardi, E. Global cerebral blood flow and CPP after severe head injury: A xenon-CT study. Intensive Care Med. 2007, 33, 856–862. [Google Scholar] [CrossRef]
- Cottenceau, V.; Masson, F.; Mahamid, E.; Petit, L.; Shik, V.; Sztark, F.; Zaaroor, M.; Soustiel, J.F. Comparison of Effects of Equiosmolar Doses of Mannitol and Hypertonic Saline on Cerebral Blood Flow and Metabolism in Traumatic Brain Injury. J. Neurotrauma 2011, 28, 2003–2012. [Google Scholar] [CrossRef]
- Soustiel, J.F.; Sviri, G.E.; Mahamid, E.; Shik, V.; Abeshaus, S.; Zaaroor, M. Cerebral Blood Flow and Metabolism Following Decompressive Craniectomy for Control of Increased Intracranial Pressure. Neurosurgery 2010, 67, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Soustiel, J.F.; Sviri, G.E. Monitoring of cerebral metabolism: Non-ischemic impairment of oxidative metabolism following severe traumatic brain injury. Neurol. Res. 2007, 29, 654–660. [Google Scholar] [CrossRef]
- Vespa, P.; Bergsneider, M.; Hattori, N.; Wu, H.-M.; Huang, S.-C.; Martin, N.A.; Glenn, T.C.; McArthur, D.L.; Hovda, D.A. Metabolic Crisis without Brain Ischemia is Common after Traumatic Brain Injury: A Combined Microdialysis and Positron Emission Tomography Study. J. Cereb. Blood Flow Metab. 2005, 25, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donkin, J.J.; Vink, R. Mechanisms of cerebral edema in traumatic brain injury: Therapeutic developments. Curr. Opin. Neurol. 2010, 23, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Simard, J.M.; Staley, K.J.; Nahed, B.V.; Jones, P.S.; Sun, D. Molecular mechanisms of ischemic cerebral edema: Role of electroneutral ion transport. Physiology 2009, 24, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Szabó, C. Mechanisms of cell necrosis. Crit. Care Med. 2005, 33, S530–S534. [Google Scholar] [CrossRef]
- Kinnally, K.W.; Zorov, D.B.; Antonenko, Y.N.; Snyder, S.H.; McEnery, M.W.; Tedeschi, H. Mitochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions of ligands. Proc. Natl. Acad. Sci. USA 1993, 90, 1374–1378. [Google Scholar] [CrossRef] [Green Version]
- McEnery, M.W.; Snowman, A.M.; Trifiletti, R.R.; Snyder, S.H. Isolation of the mitochondrial benzodiazepine receptor: Association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. USA 1992, 89, 3170–3174. [Google Scholar] [CrossRef] [Green Version]
- Azarashvili, T.; Grachev, D.; Krestinina, O.; Evtodienko, Y.; Yurkov, I.; Papadopoulos, V.; Reiser, G. The peripheral-type benzodiazepine receptor is involved in control of Ca2+-induced permeability transition pore opening in rat brain mitochondria. Cell Calcium 2007, 42, 27–39. [Google Scholar] [CrossRef]
- Soustiel, J.F.; Zaaroor, M.; Vlodavsky, E.; Veenman, L.; Weizman, A.; Gavish, M. Neuroprotective effect of Ro5-4864 following brain injury. Exp. Neurol. 2008, 214, 201–208. [Google Scholar] [CrossRef]
- Soustiel, J.F.; Vlodavsky, E.; Milman, F.; Gavish, M.; Zaaroor, M. Improvement of cerebral metabolism mediated by Ro5-4864 is associated with relief of intracranial pressure and mitochondrial protective effect in experimental brain injury. Pharm. Res. 2011, 28, 2945–2953. [Google Scholar] [CrossRef]
- Nakamoto, Y.; Watabe, S.; Shiotani, T.; Yoshii, M. Peripheral-type benzodiazepine receptors in association with epileptic seizures in EL mice. Brain Res. 1996, 717, 91–98. [Google Scholar] [CrossRef]
- Shiotani, T.; Nakamoto, Y.; Watabe, S.; Yoshii, M.; Nabeshima, T. Anticonvulsant actions of nefiracetam on epileptic EL mice and their relation to peripheral-type benzodiazepine receptors. Brain Res. 2000, 859, 255–261. [Google Scholar] [CrossRef]
- Stein, D.J. Etifoxine versus alprazolam for the treatment of adjustment disorder with anxiety: A randomized controlled trial. Adv. Ther. 2015, 32, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verleye, M.; Akwa, Y.; Liere, P.; Ladurelle, N.; Pianos, A.; Eychenne, B.; Schumacher, M.; Gillardin, J.-M. The anxiolytic etifoxine activates the peripheral benzodiazepine receptor and increases the neurosteroid levels in rat brain. Pharmacol. Biochem. Behav. 2005, 82, 712–720. [Google Scholar] [CrossRef]
- Girard, P.; Pansart, Y.; Gillardin, J.M. Preventive and curative effects of etifoxine in a rat model of brain oedema. Clin. Exp. Pharm. Physiol. 2009, 36, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Li, H.D.; Li, M.; Shi, E.; Jin, W.N.; Wood, K.; Gonzales, R.; Liu, Q. A translocator protein 18 kDa agonist protects against cerebral ischemia/reperfusion injury. J. Neuroinflammation 2017, 14, 151. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Ren, H.; Sheth, K.N.; Shi, F.D.; Liu, Q. A TSPO ligand attenuates brain injury after intracerebral hemorrhage. FASEB J. 2017, 31, 3278–3287. [Google Scholar] [CrossRef] [Green Version]
- Shehadeh, M.; Palzur, E.; Apel, L.; Soustiel, J.F. Reduction of Traumatic Brain Damage by Tspo Ligand Etifoxine. Int. J. Mol. Sci. 2019, 20, 2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochanek, P.M.; Bramlett, H.M.; Shear, D.A.; Dixon, C.E.; Mondello, S.; Dietrich, W.D.; Hayes, R.L.; Wang, K.K.; Poloyac, S.M.; Empey, P.E.; et al. Synthesis of Findings, Current Investigations, and Future Directions: Operation Brain Trauma Therapy. J. Neurotrauma 2016, 33, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.J.; Rosenfeld, J.V.; Murray, L.; Arabi, Y.M.; Davies, A.R.; D’Urso, P.; Kossmann, T.; Ponsford, J.; Seppelt, I.; Reilly, P.; et al. Decompressive craniectomy in diffuse traumatic brain injury. N. Engl. J. Med. 2011, 364, 1493–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, P.J.; Kolias, A.G.; Timofeev, I.S.; Corteen, E.A.; Czosnyka, M.; Timothy, J.; Anderson, I.; Bulters, D.O.; Belli, A.; Eynon, C.A.; et al. Trial of Decompressive Craniectomy for Traumatic Intracranial Hypertension. N. Engl. J. Med. 2016, 375, 1119–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Springer, J.E.; Prajapati, P.; Sullivan, P.G. Targeting the mitochondrial permeability transition pore in traumatic central nervous system injury. Neural Regen. Res. 2018, 13, 1338–1341. [Google Scholar] [CrossRef]
- Veech, R.L.; Valeri, C.R.; VanItallie, T.B. The mitochondrial permeability transition pore provides a key to the diagnosis and treatment of traumatic brain injury. IUBMB Life 2012, 64, 203–207. [Google Scholar] [CrossRef]
- Okonkwo, D.O.; Buki, A.; Siman, R.; Povlishock, J.T. Cyclosporin A limits calcium-induced axonal damage following traumatic brain injury. Neuroreport 1999, 10, 353–358. [Google Scholar] [CrossRef]
- Okonkwo, D.O.; Povlishock, J.T. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J. Cereb. Blood Flow Metab. 1999, 19, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.G.; Thompson, M.; Scheff, S.W. Continuous infusion of cyclosporin A postinjury significantly ameliorates cortical damage following traumatic brain injury. Exp. Neurol. 2000, 161, 631–637. [Google Scholar] [CrossRef]
- Sullivan, P.G.; Thompson, M.B.; Scheff, S.W. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp. Neurol. 1999, 160, 226–234. [Google Scholar] [CrossRef]
- Vlodavsky, E.; Palzur, E.; Shehadeh, M.; Soustiel, J.F. Post-traumatic cytotoxic edema is directly related to mitochondrial function. J. Cereb. Blood Flow Metab. 2017, 37, 166–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, C.E.; Bramlett, H.M.; Dietrich, W.D.; Shear, D.A.; Yan, H.Q.; Deng-Bryant, Y.; Mondello, S.; Wang, K.K.; Hayes, R.L.; Empey, P.E.; et al. Cyclosporine Treatment in Traumatic Brain Injury: Operation Brain Trauma Therapy. J. Neurotrauma 2016, 33, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.; Shohami, E.; Alexandrovich, A.; Yatsiv, I.; Kloog, Y.; Biegon, A. Increase in peripheral benzodiazepine receptors and loss of glutamate NMDA receptors in a mouse model of closed head injury: A quantitative autoradiographic study. Neuroimage 2003, 20, 1971–1981. [Google Scholar] [CrossRef] [PubMed]
- Soustiel, J.F.; Palzur, E.; Vlodavsky, E.; Veenman, L.; Gavish, M. The effect of oxygenation level on cerebral post-traumatic apoptotsis is modulated by the 18-kDa translocator protein (also known as peripheral-type benzodiazepine receptor) in a rat model of cortical contusion. Neuropathol. Appl. Neurobiol. 2008, 34, 412–423. [Google Scholar] [CrossRef]
- Veiga, S.; Azcoitia, I.; Garcia-Segura, L.M. Ro5-4864, a peripheral benzodiazepine receptor ligand, reduces reactive gliosis and protects hippocampal hilar neurons from kainic acid excitotoxicity. J. Neurosci. Res. 2005, 80, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Daugherty, D.J.; Selvaraj, V.; Chechneva, O.V.; Liu, X.B.; Pleasure, D.E.; Deng, W. A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Mol. Med. 2013, 5, 891–903. [Google Scholar] [CrossRef]
- Aouad, M.; Charlet, A.; Rodeau, J.L.; Poisbeau, P. Reduction and prevention of vincristine-induced neuropathic pain symptoms by the non-benzodiazepine anxiolytic etifoxine are mediated by 3alpha-reduced neurosteroids. Pain 2009, 147, 54–59. [Google Scholar] [CrossRef]
- Girard, C.; Liu, S.; Cadepond, F.; Adams, D.; Lacroix, C.; Verleye, M.; Gillardin, J.M.; Baulieu, E.E.; Schumacher, M.; Schweizer-Groyer, G. Etifoxine improves peripheral nerve regeneration and functional recovery. Proc. Natl. Acad. Sci. USA 2008, 105, 20505–20510. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, P.T.; Thomas, A.P.; Armston, A.E.; Halestrap, A.P. Measurement of the intramitochondrial volume in hepatocytes without cell disruption and its elevation by hormones and valinomycin. Biochem. J. 1983, 214, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sileikyte, J.; Blachly-Dyson, E.; Sewell, R.; Carpi, A.; Menabo, R.; Di Lisa, F.; Ricchelli, F.; Bernardi, P.; Forte, M. Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (Translocator Protein of 18 kDa (TSPO)). J. Biol. Chem. 2014, 289, 13769–13781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleary, J.; Johnson, K.M.; Opipari, A.W., Jr.; Glick, G.D. Inhibition of the mitochondrial F1F0-ATPase by ligands of the peripheral benzodiazepine receptor. Bioorg. Med. Chem. Lett. 2007, 17, 1667–1670. [Google Scholar] [CrossRef]
- Johnson, K.M.; Chen, X.; Boitano, A.; Swenson, L.; Opipari, A.W., Jr.; Glick, G.D. Identification and validation of the mitochondrial F1F0-ATPase as the molecular target of the immunomodulatory benzodiazepine Bz-423. Chem. Biol. 2005, 12, 485–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochanek, P.M.; Bramlett, H.M.; Dixon, C.E.; Shear, D.A.; Dietrich, W.D.; Schmid, K.E.; Mondello, S.; Wang, K.K.; Hayes, R.L.; Povlishock, J.T.; et al. Approach to Modeling, Therapy Evaluation, Drug Selection, and Biomarker Assessments for a Multicenter Pre-Clinical Drug Screening Consortium for Acute Therapies in Severe Traumatic Brain Injury: Operation Brain Trauma Therapy. J. Neurotrauma 2016, 33, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Sumbalova, Z.; Kucharska, J.; Kristek, F. Losartan improved respiratory function and coenzyme Q content in brain mitochondria of young spontaneously hypertensive rats. Cell. Mol. Neurobiol. 2010, 30, 751–758. [Google Scholar] [CrossRef]
- Gnaiger, E. Bioenergetics at low oxygen: Dependence of respiration and phosphorylation on oxygen and adenosine diphosphate supply. Respir. Physiol. 2001, 128, 277–297. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palzur, E.; Edelman, D.; Sakas, R.; Soustiel, J.F. Etifoxine Restores Mitochondrial Oxidative Phosphorylation and Improves Cognitive Recovery Following Traumatic Brain Injury. Int. J. Mol. Sci. 2021, 22, 12881. https://doi.org/10.3390/ijms222312881
Palzur E, Edelman D, Sakas R, Soustiel JF. Etifoxine Restores Mitochondrial Oxidative Phosphorylation and Improves Cognitive Recovery Following Traumatic Brain Injury. International Journal of Molecular Sciences. 2021; 22(23):12881. https://doi.org/10.3390/ijms222312881
Chicago/Turabian StylePalzur, Eilam, Doron Edelman, Reem Sakas, and Jean Francois Soustiel. 2021. "Etifoxine Restores Mitochondrial Oxidative Phosphorylation and Improves Cognitive Recovery Following Traumatic Brain Injury" International Journal of Molecular Sciences 22, no. 23: 12881. https://doi.org/10.3390/ijms222312881
APA StylePalzur, E., Edelman, D., Sakas, R., & Soustiel, J. F. (2021). Etifoxine Restores Mitochondrial Oxidative Phosphorylation and Improves Cognitive Recovery Following Traumatic Brain Injury. International Journal of Molecular Sciences, 22(23), 12881. https://doi.org/10.3390/ijms222312881