
Cytochrome P450 Enzymes and Drug Metabolism in Humans

 ,
, 
Abstract
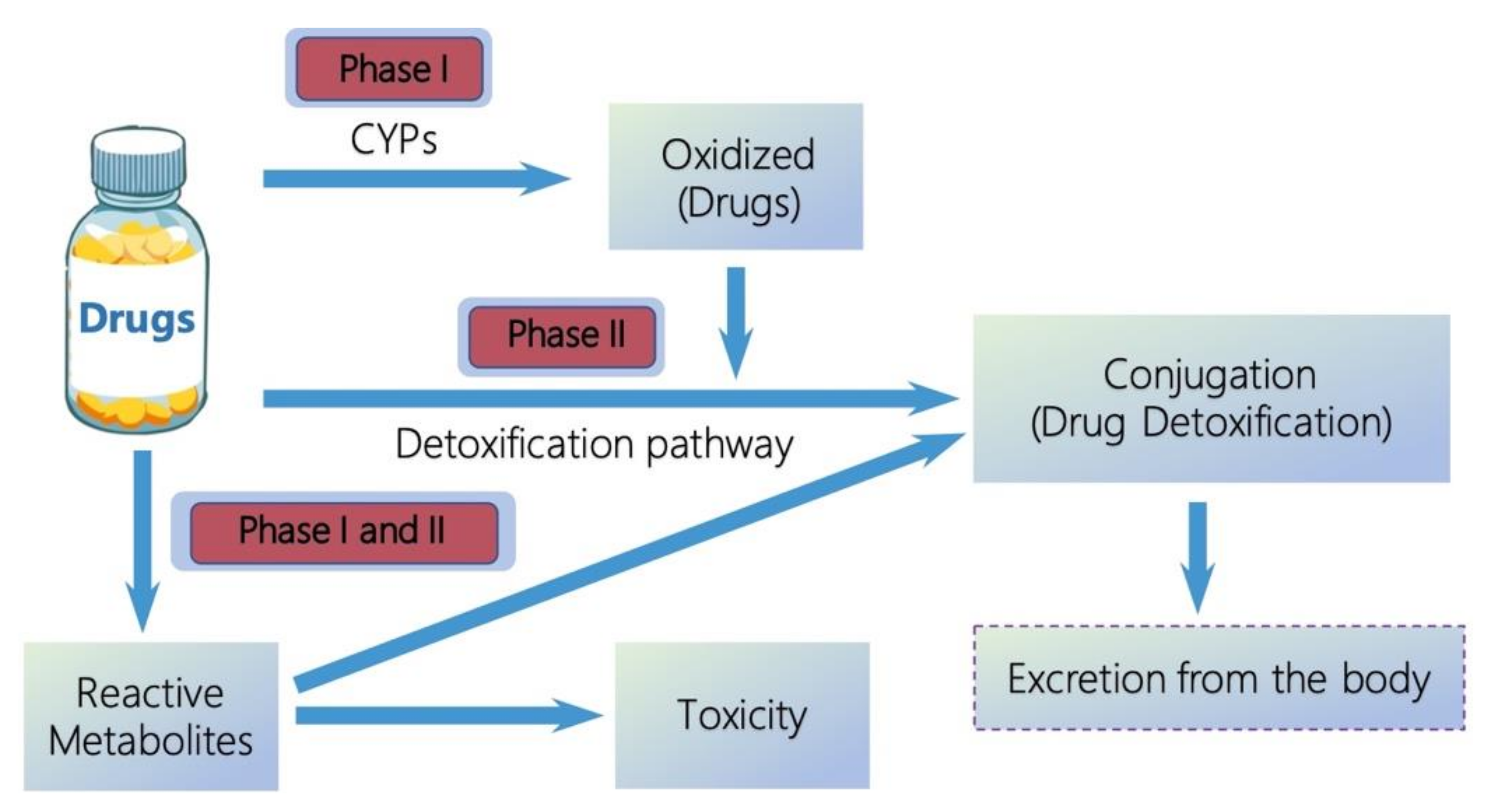
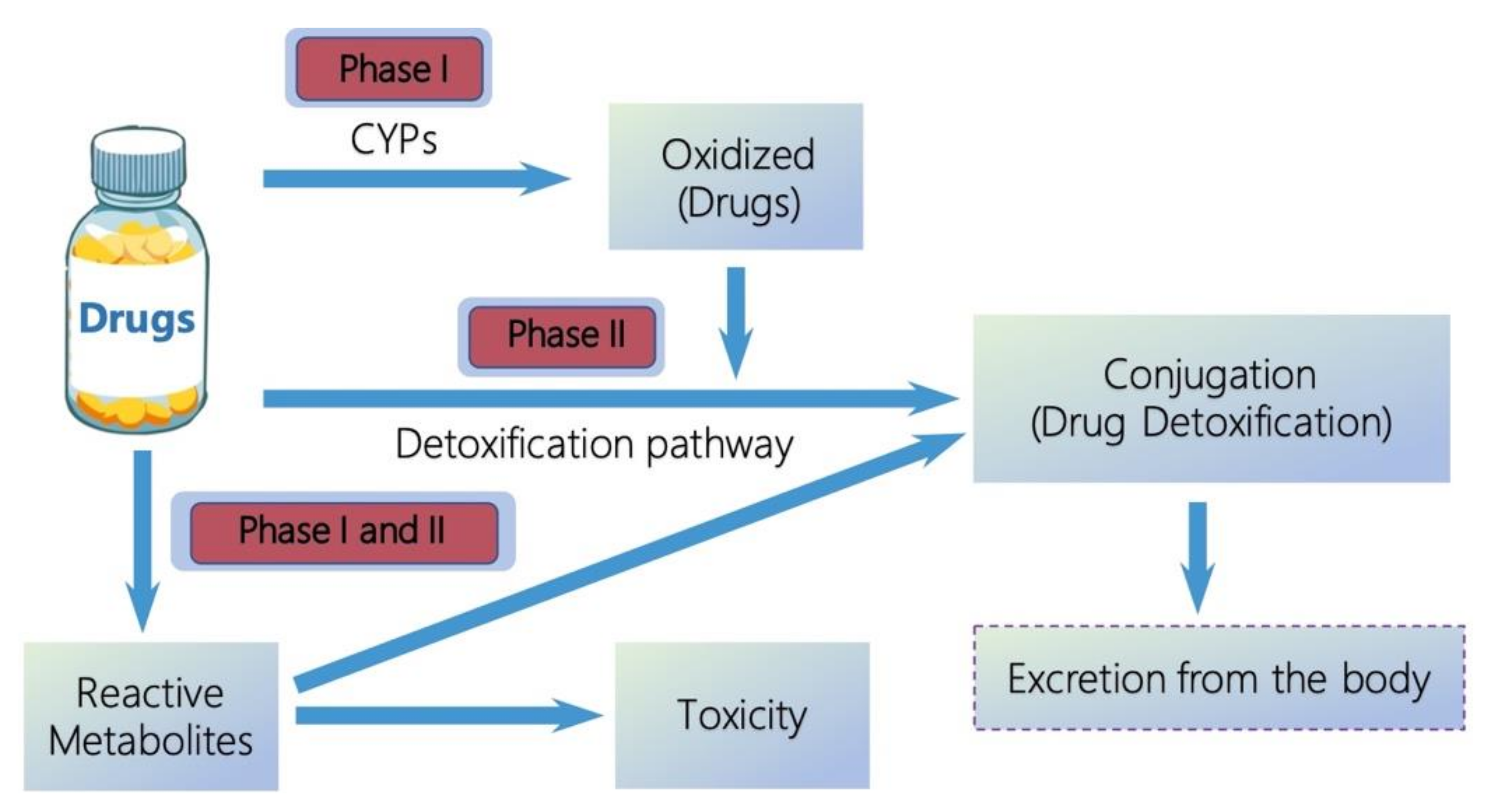
:1. Introduction
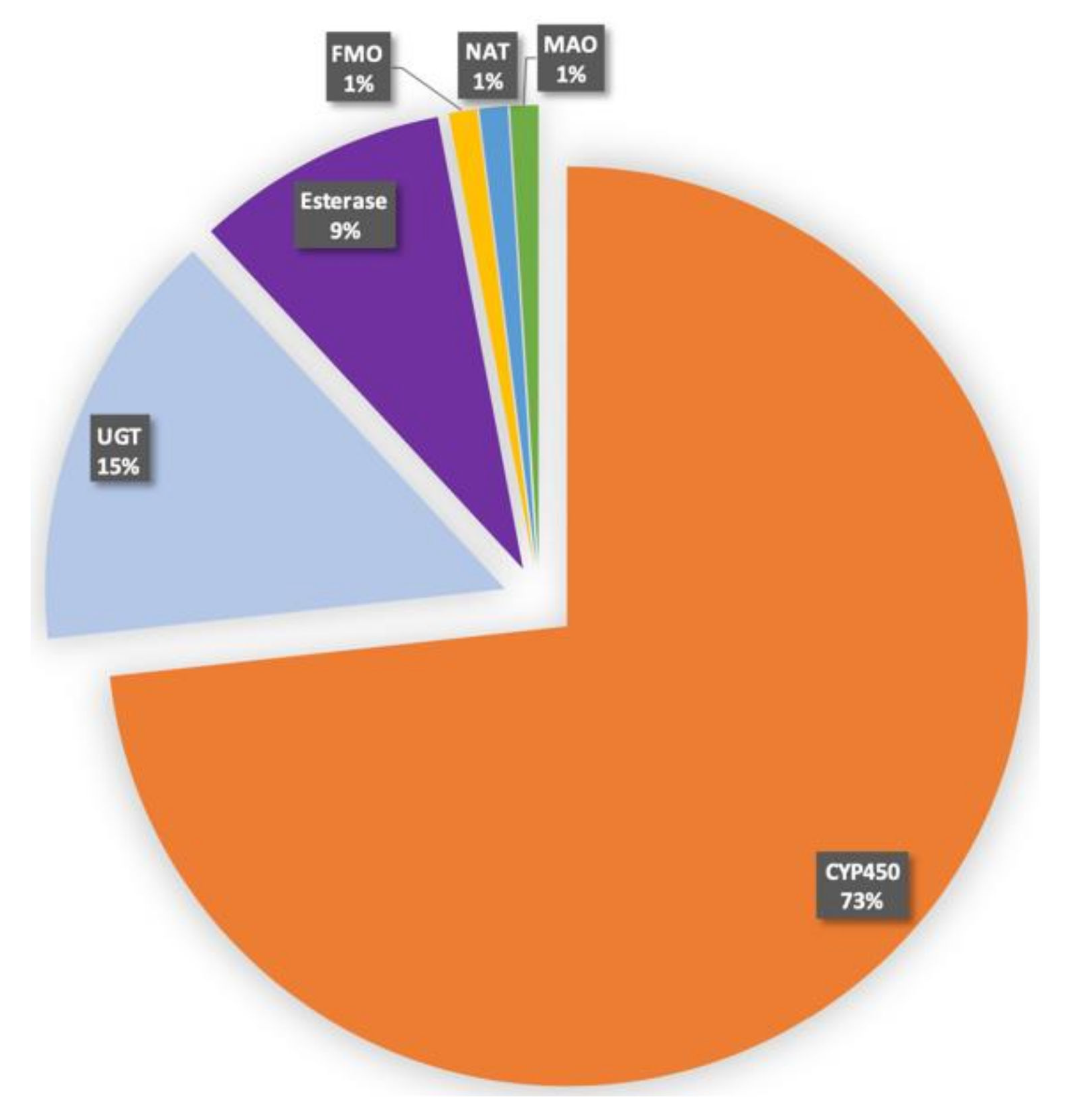
2. Human CYPs
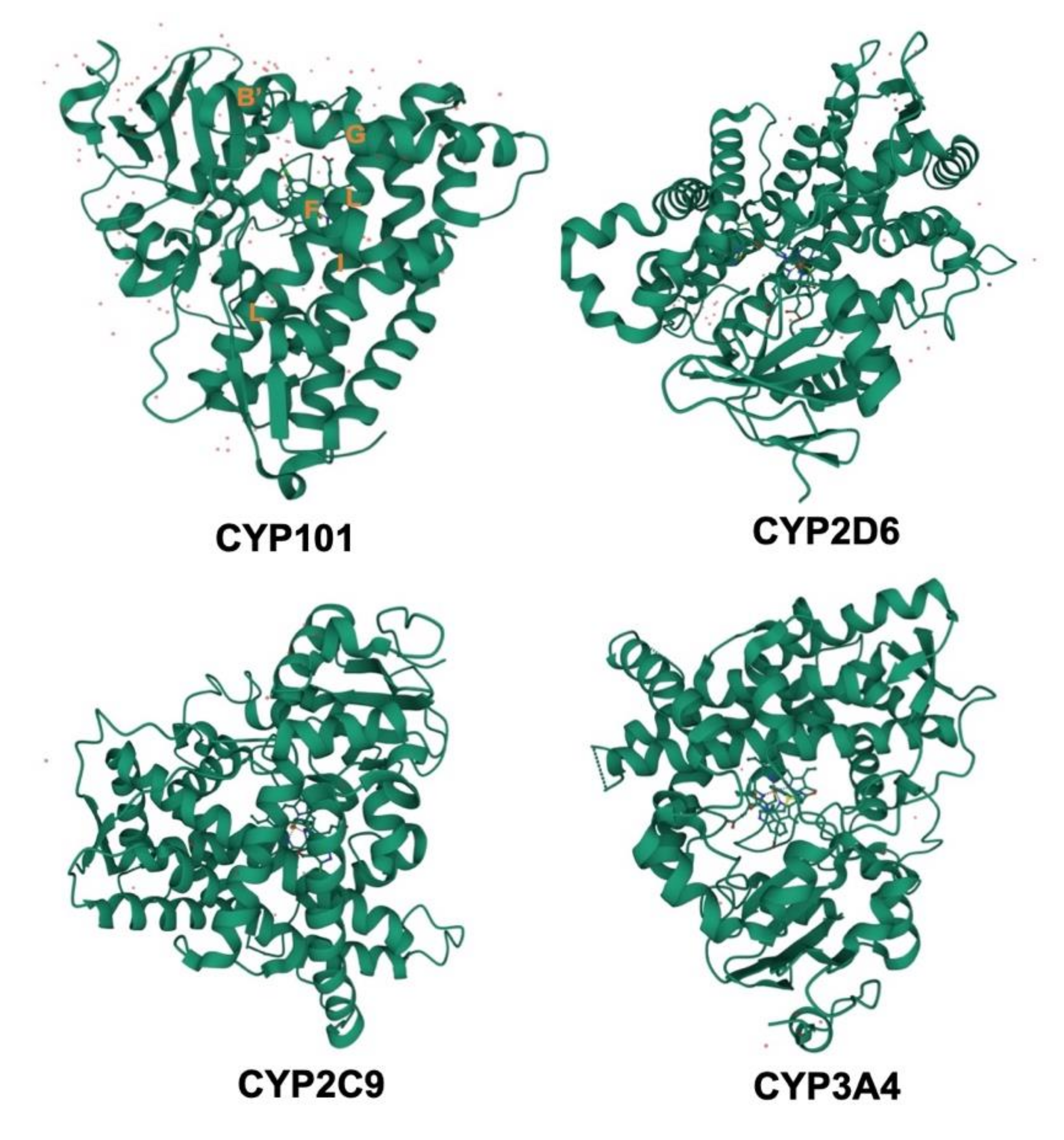
3. Structures of CYPs
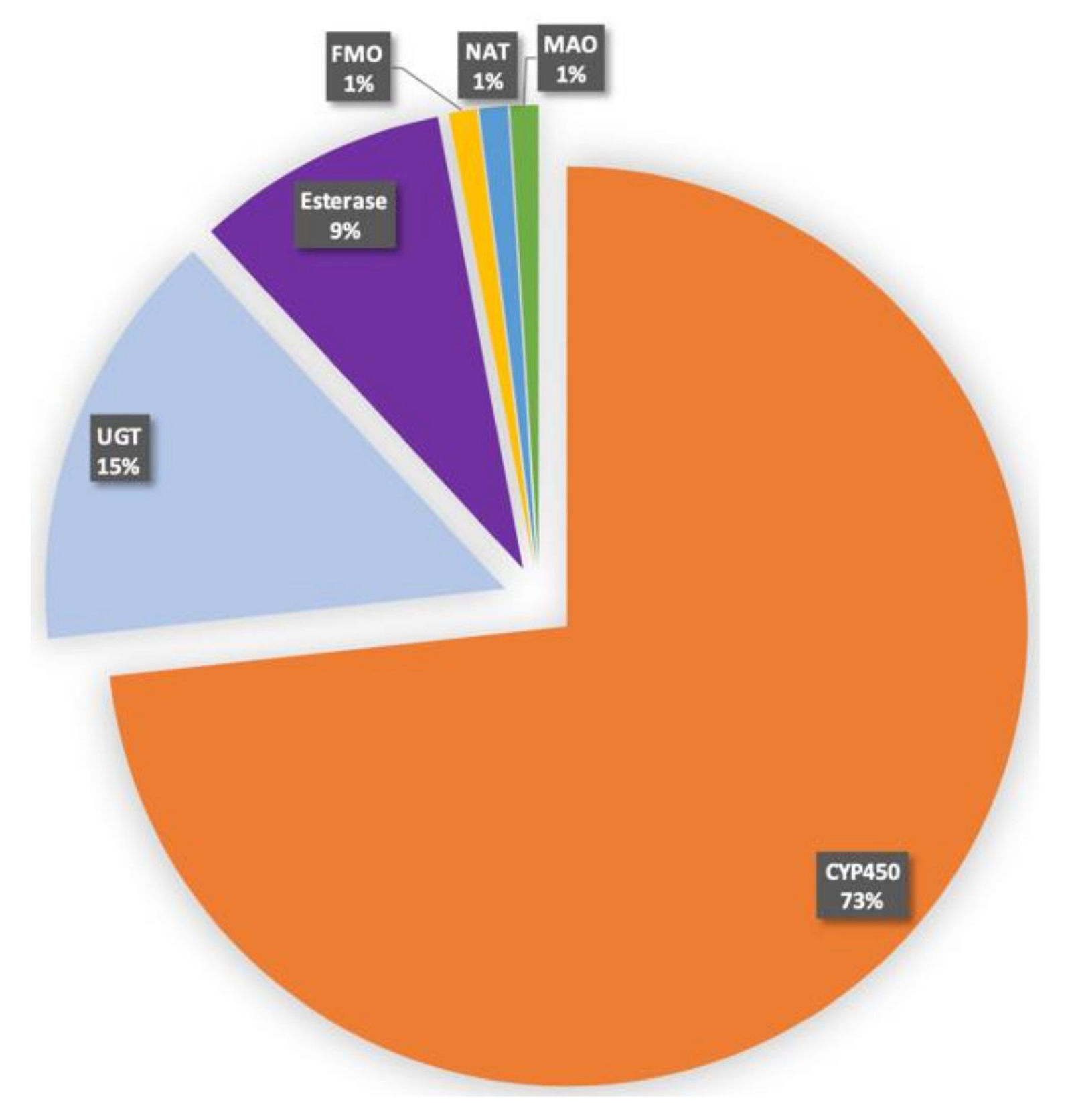
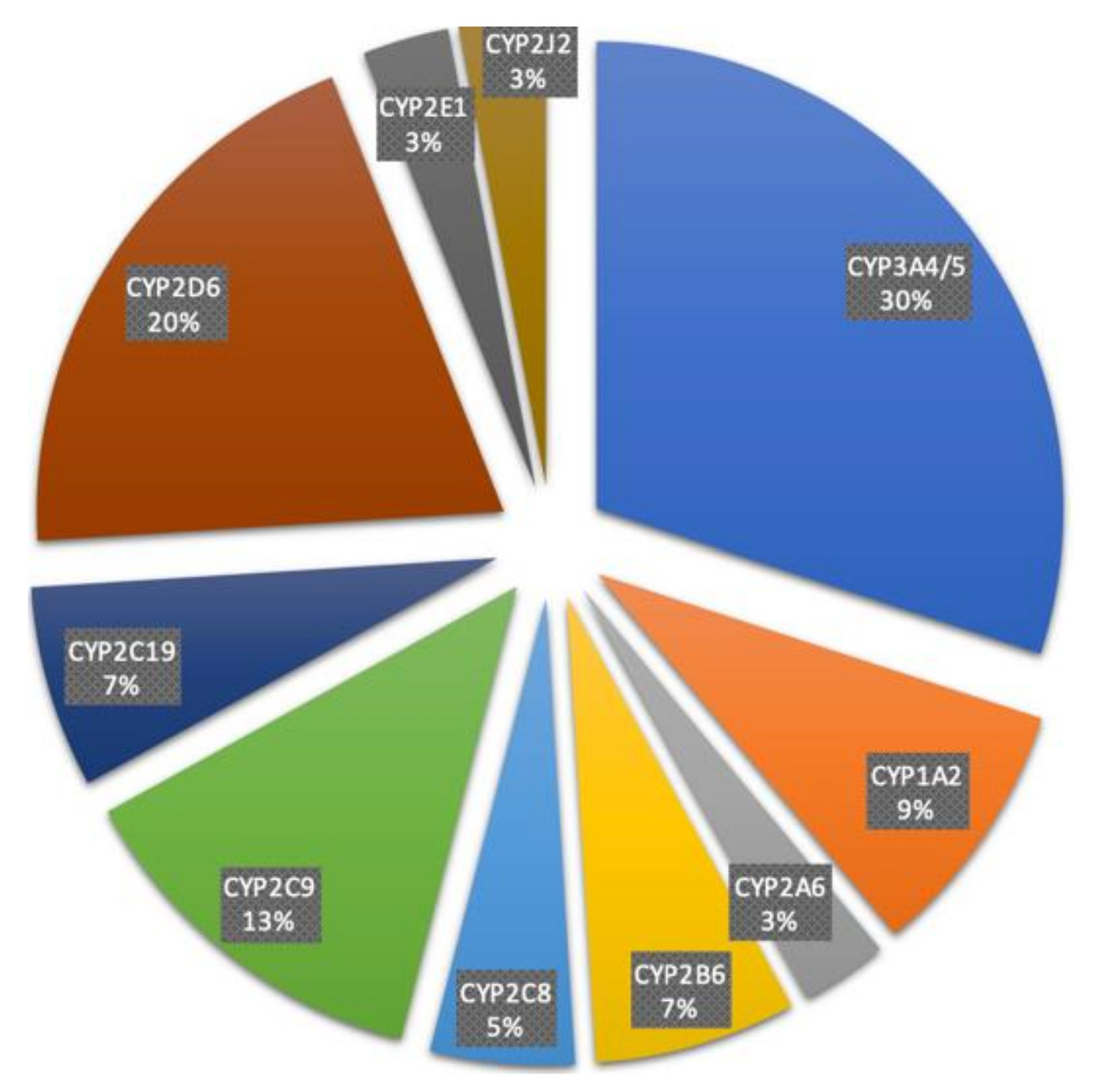
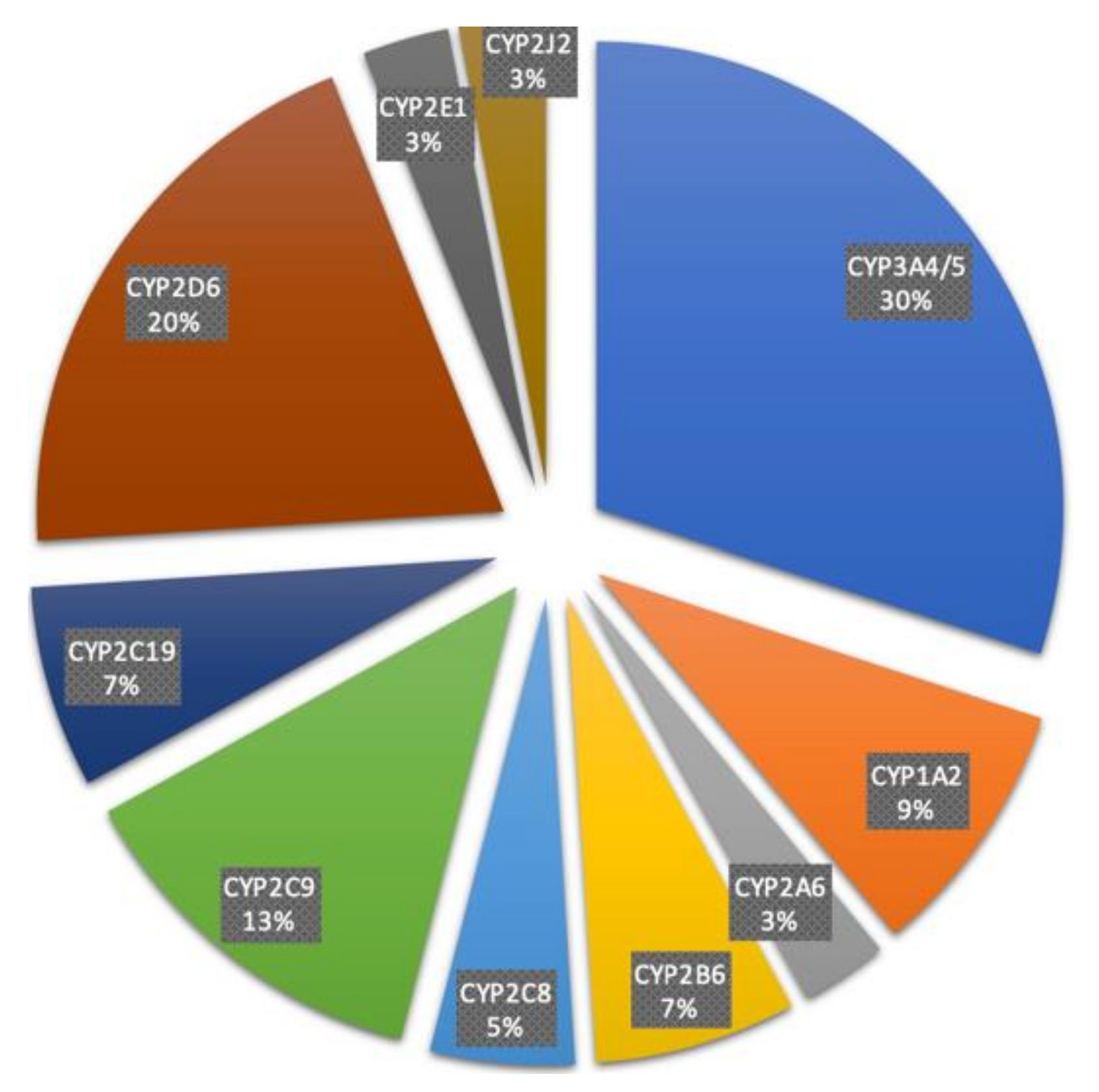
4. Characteristics of Major Drug Metabolizing CYPs
5. Individual Variation of CYP-Mediated Drug Metabolism
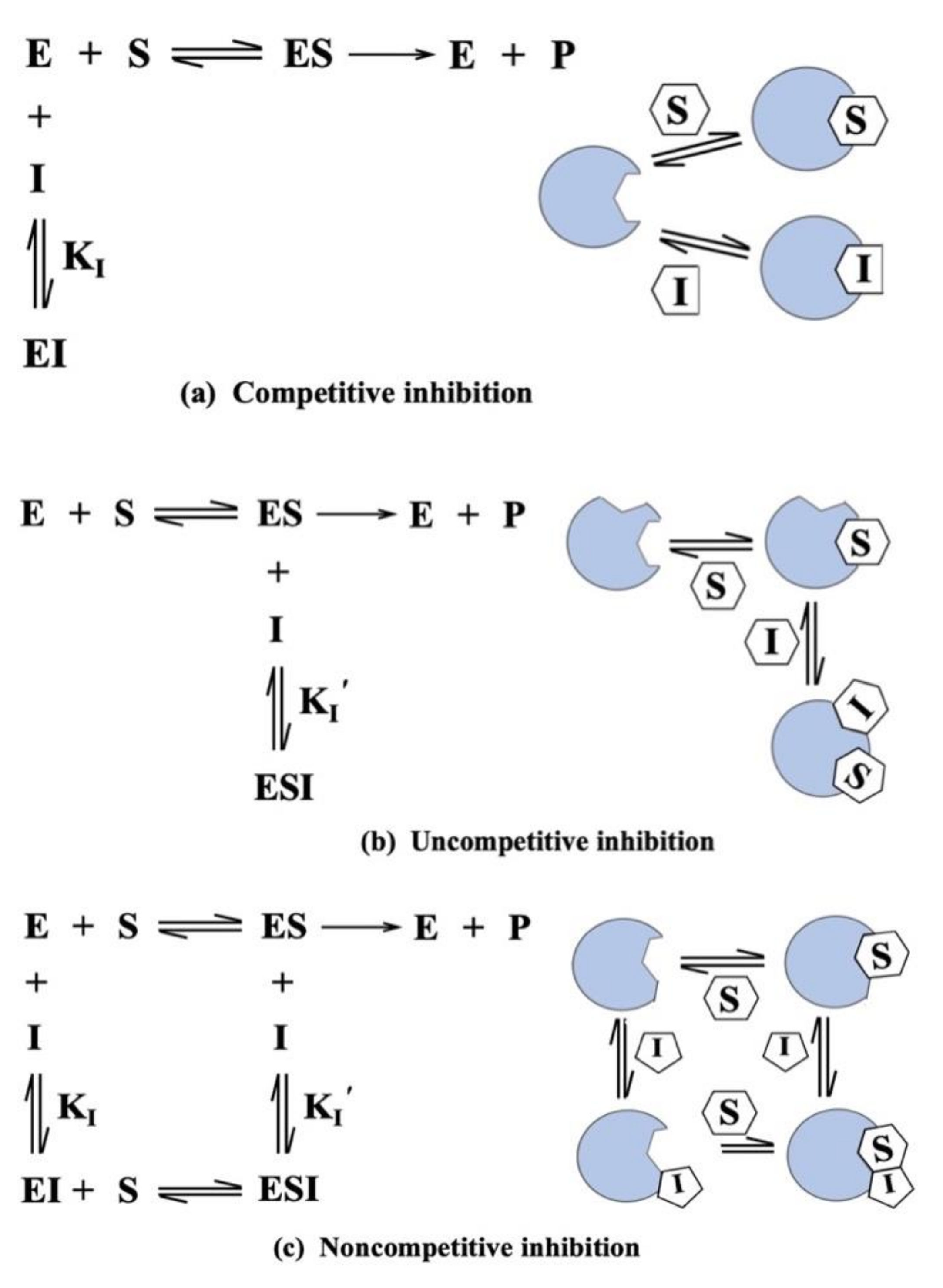
6. Clinical Implications and Therapeutic Benefits
7. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Patel, R.; Barker, J.; ElShaer, A. Pharmaceutical excipients and drug metabolism: A mini-review. Int. J. Mol. Sci. 2020, 21, 8224. [Google Scholar] [CrossRef]
- Tao, G.; Huang, J.; Moorthy, B.; Wang, C.; Hu, M.; Gao, S.; Ghose, R. Potential role of drug metabolizing enzymes in chemotherapy-induced gastrointestinal toxicity and hepatotoxicity. Expert Opin. Drug Metab. Toxicol. 2020, 16, 1109–1124. [Google Scholar] [CrossRef]
- Rendic, S.; Guengerich, F.P. Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem. Res. Toxicol. 2015, 28, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.M.; Fernandes, C.; Remião, F.; Tiritan, M.E. Enantioselectivity in drug pharmacokinetics and toxicity: Pharmacological relevance and analytical methods. Molecules 2021, 26, 3113. [Google Scholar] [CrossRef]
- Rendic, S.P. Metabolism and interactions of Ivermectin with human cytochrome P450 enzymes and drug transporters, possible adverse and toxic effects. Arch. Toxicol. 2021, 95, 1535–1546. [Google Scholar] [CrossRef]
- Jancova, P.; Anzenbacher, P.; Anzenbacherova, E. Phase II drug metabolizing enzymes. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2010, 154, 103–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, B.; Pedretti, A.; Vistoli, G. Reactions and enzymes in the metabolism of drugs and other xenobiotics. Drug Discov. Today 2012, 17, 549–560. [Google Scholar] [CrossRef] [PubMed]
- De Groot, M.J. Designing better drugs: Predicting cytochrome P450 metabolism. Drug Discov. Today 2006, 11, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug Metabolism in the Liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Danielson, P.B. The cytochrome P450 superfamily: Biochemistry, evolution and drug metabolism in humans. Curr. Drug Metab. 2002, 3, 561–597. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M. Human drug metabolising cytochrome P450 enzymes: Properties and polymorphisms. Naunyn Schmiedeberg’s Arch. Pharmacol. 2004, 369, 89–104. [Google Scholar] [CrossRef]
- Zahoor, I.; Rui, B.; Khan, J.; Datta, I.; Giri, S. An emerging potential of metabolomics in multiple sclerosis: A comprehensive overview. Cell Mol. Life Sci. 2021, 78, 3181–3203. [Google Scholar] [CrossRef] [PubMed]
- Eichelbaum, M.; Ingelman-Sundberg, M.; Evans, W.E. Pharmacogenomics and individualized drug therapy. Annu. Rev. Med. 2006, 57, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Lu, A.Y. Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 535–567. [Google Scholar] [CrossRef] [PubMed]
- Arranz, M.J.; Salazar, J.; Hernández, M.H. Pharmacogenetics of antipsychotics: Clinical utility and implementation. Behav. Brain Res. 2021, 401, 113058. [Google Scholar] [CrossRef] [PubMed]
- Sathyanarayanan, G.; Haapala, M.; Sikanen, T. Digital microfluidics-enabled analysis of individual variation in liver cytochrome P450 activity. Anal. Chem. 2020, 92, 14693–14701. [Google Scholar] [CrossRef] [PubMed]
- Puszkiel, A.; Arellano, C.; Vachoux, C.; Evrard, A.; Le Morvan, V.; Boyer, J.C.; Robert, J.; Delmas, C.; Dalenc, F.; Debled, M.; et al. Model-based quantification of impact of genetic polymorphisms and co-medications on pharmacokinetics of tamoxifen and six metabolites in breast cancer. Clin. Pharmacol. Ther. 2021, 109, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, Y.F.; Corton, J.C.; Klaassen, C.D. Expression of cytochrome P450 isozyme transcripts and activities in human livers. Xenobiotica 2021, 51, 279–286. [Google Scholar] [CrossRef]
- Stipp, M.C.; Acco, A. Involvement of cytochrome P450 enzymes in inflammation and cancer: A review. Cancer Chemother. Pharmacol. 2021, 87, 295–309. [Google Scholar] [CrossRef]
- Burlaka, V.S.; Burlaka, A.A. Cytochrome P450 content in primary tumors and liver metastases of patients with metastatic colorectal cancer. Exp. Oncol. 2020, 42, 1–3. [Google Scholar]
- Krkoška, M.; Svobodová, J.; Kabátková, M.; Zapletal, O.; Hyršlová Vaculová, A.; Nekvindová, J.; Vondráček, J. Deregulation of signaling pathways controlling cell survival and proliferation in cancer cells alters induction of cytochrome P450 family 1 enzymes. Toxicology 2021, 461, 152897. [Google Scholar] [CrossRef]
- Van Schaik, R.H. CYP450 pharmacogenetics for personalizing cancer therapy. Drug Resist. Update 2008, 11, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Barros-Oliveira, M.D.C.; Costa-Silva, D.R.; Dos Santos, A.R.; Pereira, R.O.; Soares-Júnior, J.M.; Silva, B.B.D. Influence of CYP19A1 gene expression levels in women with breast cancer: A systematic review of the literature. Clinics 2021, 76, e2846. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Johnson, V.; Barrera, J.; Porras, M.; Hinojosa, D.; Hernández, I.; McGarrah, P.; Potter, D.A. Targeting cytochrome P450-dependent cancer cell mitochondria: Cancer associated CYPs and where to find them. Cancer Metastasis Rev. 2018, 37, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Verma, H.; Singh Bahia, M.; Choudhary, S.; Kumar Singh, P.; Silakari, O. Drug metabolizing enzymes-associated chemo resistance and strategies to overcome it. Drug Metab. Rev. 2019, 51, 196–223. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, M.; Li, D.; Li, C.; Luo, C.; Wang, Z.; Zhang, W.; Yang, Z.; Feng, Y.; Wang, S.; et al. Cytochrome P450 enzyme-mediated auto-enhanced photodynamic cancer therapy of co-nanoassembly between clopidogrel and photosensitizer. Theranostics 2020, 10, 5550–5564. [Google Scholar] [CrossRef]
- Paolini, M.; Poul, L.; Darmon, A.; Germain, M.; Pottier, A.; Levy, L.; Vibert, E. A new opportunity for nanomedicines: Micellar cytochrome P450 inhibitors to improve drug efficacy in a cancer therapy model. Nanomedicine 2017, 13, 1715–1723. [Google Scholar] [CrossRef]
- Wang, Y.; He, X.; Li, C.; Ma, Y.; Xue, W.; Hu, B.; Wang, J.; Zhang, T.; Zhang, F. Carvedilol serves as a novel CYP1B1 inhibitor, a systematic drug repurposing approach through structure-based virtual screening and experimental verification. Eur. J. Med. Chem. 2020, 193, 112235. [Google Scholar] [CrossRef]
- Mitsui, Y.; Chang, I.; Fukuhara, S.; Hiraki, M.; Arichi, N.; Yasumoto, H.; Hirata, H.; Yamamura, S.; Shahryari, V.; Deng, G.; et al. CYP1B1 promotes tumorigenesis via altered expression of CDC20 and DAPK1 genes in renal cell carcinoma. BMC Cancer 2015, 15, 942. [Google Scholar] [CrossRef]
- Mu, W.; Hu, C.; Zhang, H.; Qu, Z.; Cen, J.; Qiu, Z.; Li, C.; Ren, H.; Li, Y.; He, X.; et al. miR-27b synergizes with anticancer drugs via p53 activation and CYP1B1 suppression. Cell Res. 2015, 25, 477–495. [Google Scholar] [CrossRef] [Green Version]
- Dutour, R.; Poirier, D. Inhibitors of cytochrome P450 (CYP) 1B1. Eur. J. Med. Chem. 2017, 135, 296–306. [Google Scholar] [CrossRef]
- Karkhanis, A.; Hong, Y.; Chan, E.C.Y. Inhibition and inactivation of human CYP2J2: Implications in cardiac pathophysiology and opportunities in cancer therapy. Biochem. Pharmacol. 2017, 135, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlgren, M.; Ingelman-Sundberg, M. Tumour-specific expression of CYP2W1: Its potential as a drug target in cancer therapy. Expert Opin. Ther. Targets 2007, 11, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Hill, H.; Rder, A.; Williams, R. The chemical nature and reactivity of cytochrome P-450. In Biochemistry; Springer: Berlin/Heidelberg, Germany, 1970. [Google Scholar]
- Nelson, D.R.; Koymans, L.; Kamataki, T.; Stegeman, J.J.; Feyereisen, R.; Waxman, D.J.; Waterman, M.R.; Gotoh, O.; Coon, M.J.; Estabrook, R.W.; et al. P450 superfamily: Update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 1996, 6, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Park, S.Y.; Shiro, Y.; Adachi, S. X-ray structure of nitric oxide reductase (cytochrome P450nor) at atomic resolution. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Chang, Z.; Pan, Z.; Fu, Z.Q.; Wang, X. Modes of heme binding and substrate access for cytochrome P450 CYP74A revealed by crystal structures of allene oxide synthase. Proc. Natl. Acad. Sci. USA 2008, 105, 13883–13888. [Google Scholar] [CrossRef] [Green Version]
- Brash, A.R. Mechanistic aspects of CYP74 allene oxide synthases and related cytochrome P450 enzymes. Phytochemistry 2009, 70, 1522–1531. [Google Scholar] [CrossRef] [Green Version]
- Cryle, M.J.; Schlichting, I. Structural insights from a P450 Carrier Protein complex reveal how specificity is achieved in the P450(BioI) ACP complex. Proc. Natl. Acad. Sci. USA 2008, 105, 15696–15701. [Google Scholar] [CrossRef] [Green Version]
- Nagano, S.; Li, H.; Shimizu, H.; Nishida, C.; Ogura, H.; Ortiz de Montellano, P.R.; Poulos, T.L. Crystal structures of epothilone D-bound, epothilone B-bound, and substrate-free forms of cytochrome P450epoK. J. Biol. Chem. 2003, 278, 44886–44893. [Google Scholar] [CrossRef] [Green Version]
- Shimada, T.; Yamazaki, H.; Mimura, M.; Wakamiya, N.; Ueng, Y.F.; Guengerich, F.P.; Inui, Y. Characterization of microsomal cytochrome P450 enzymes involved in the oxidation of xenobiotic chemicals in human fetal liver and adult lungs. Drug Metab. Dispos. 1996, 24, 515–522. [Google Scholar]
- Conney, A.H. Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons: G. H. A. Clowes Memorial Lecture. Cancer Res. 1982, 42, 4875–4917. [Google Scholar]
- Lang, N.P.; Butler, M.A.; Massengill, J.; Lawson, M.; Stotts, R.C.; Hauer-Jensen, M.; Kadlubar, F.F. Rapid metabolic phenotypes for acetyltransferase and cytochrome P4501A2 and putative exposure to food-borne heterocyclic amines increase the risk for colorectal cancer or polyps. Cancer Epidemiol. Biomark. Prev. 1994, 3, 675–682. [Google Scholar]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Ohtsuki, S.; Kamiie, J.; Suzuki, T.; Abe, T.; Terasaki, T. Simultaneous absolute quantification of 11 cytochrome P450 isoforms in human liver microsomes by liquid chromatography tandem mass spectrometry with in silico target peptide selection. J. Pharm. Sci. 2011, 100, 341–352. [Google Scholar] [CrossRef]
- Gonzalez, F.J. Cytochrome P450 humanised mice. Hum. Genom. 2004, 1, 300–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, M.; Kapelyukh, Y.; Tahara, H.; Seibler, J.; Rode, A.; Krueger, S.; Lee, D.N.; Wolf, C.R.; Scheer, N. Quantitative prediction of human pregnane X receptor and cytochrome P450 3A4 mediated drug-drug interaction in a novel multiple humanized mouse line. Mol. Pharmacol. 2011, 80, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingelman-Sundberg, M.; Sim, S.C.; Gomez, A.; Rodriguez-Antona, C. Influence of cytochrome P450 polymorphisms on drug therapies: Pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol. Ther. 2007, 116, 496–526. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: The past, present and future. Trends Pharmacol. Sci. 2004, 25, 193–200. [Google Scholar] [CrossRef]
- Sadee, W.; Wang, D.; Papp, A.C.; Pinsonneault, J.K.; Smith, R.M.; Moyer, R.A.; Johnson, A.D. Pharmacogenomics of the RNA world: Structural RNA polymorphisms in drug therapy. Clin. Pharmacol. Ther. 2011, 89, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Ingelman-Sundberg, M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): Clinical consequences, evolutionary aspects and functional diversity. Pharm. J. 2005, 5, 6–13. [Google Scholar] [CrossRef]
- Hassani Idrissi, H.; Hmimech, W.; Khorb, N.E.; Akoudad, H.; Habbal, R.; Nadifi, S. A synergic effect between CYP2C19*2, CYP2C19*3 loss-of-function and CYP2C19*17 gain-of-function alleles is associated with Clopidogrel resistance among Moroccan Acute Coronary Syndromes patients. BMC Res. Notes 2018, 11, 46. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.A.; Pereira, N. Pharmacogenomic impact of CYP2C19 variation on clopidogrel therapy in precision cardiovascular medicine. J. Pers. Med. 2018, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Johansson, I.; Ingelman-Sundberg, M. CNVs of human genes and their implication in pharmacogenetics. Cytogenet. Genome Res. 2008, 123, 195–204. [Google Scholar] [CrossRef]
- Riaz, S.; Muhammad Din, S.; Usman Tareen, M.; Tariq, F.; Latif, Y.; Siddiqi, S.; Sultan, A.; Mansoor, A. genetic polymorphism of CYP2C19 in Pakistani population. Iran. J. Pharm. Res. 2019, 18, 1097–1102. [Google Scholar] [PubMed]
- Foster, A.; Mobley, E.; Wang, Z. Complicated pain management in a CYP450 2D6 poor metabolizer. Pain Pract. 2007, 7, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Chen, S. Epigenetic regulation of cytochrome P450 enzymes and clinical implication. Curr. Drug Metab. 2015, 16, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M.; Gomez, A. The past, present and future of pharmacoepigenomics. Pharmacogenomics 2010, 11, 625–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokizane, T.; Shiina, H.; Igawa, M.; Enokida, H.; Urakami, S.; Kawakami, T.; Ogishima, T.; Okino, S.T.; Li, L.C.; Tanaka, Y.; et al. Cytochrome P450 1B1 is overexpressed and regulated by hypomethylation in prostate cancer. Clin. Cancer Res. 2005, 11, 5793–5801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.J.; Choi, Y.J.; Kim, J.W.; Chun, H.S.; Im, I.; Yoon, S.; Han, Y.M.; Song, C.W.; Kim, H. Differences in the epigenetic regulation of cytochrome P450 genes between human embryonic stem cell-derived hepatocytes and primary hepatocytes. PLoS ONE 2015, 10, e0132992. [Google Scholar]
- Tsuchiya, N.; Inoue, T.; Narita, S.; Kumazawa, T.; Saito, M.; Obara, T.; Tsuruta, H.; Horikawa, Y.; Yuasa, T.; Satoh, S.; et al. Drug related genetic polymorphisms affecting adverse reactions to methotrexate, vinblastine, doxorubicin and cisplatin in patients with urothelial cancer. J. Urol. 2008, 180, 2389–2395. [Google Scholar] [CrossRef]
- Singh, D.; Kashyap, A.; Pandey, R.V.; Saini, K.S. Novel advances in cytochrome P450 research. Drug Discov. Today 2011, 16, 793–799. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Nakajima, M.; Takagi, S.; Taniya, T.; Yokoi, T. MicroRNA regulates the expression of human cytochrome P450 1B1. Cancer Res. 2006, 66, 9090–9098. [Google Scholar] [CrossRef] [Green Version]
- Takamizawa, J.; Konishi, H.; Yanagisawa, K.; Tomida, S.; Osada, H.; Endoh, H.; Harano, T.; Yatabe, Y.; Nagino, M.; Nimura, Y.; et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004, 64, 3753–3756. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Mabarak, C.; Camacho-Carranza, R.; Espinosa-Aguirre, J.J. Cytochrome P450 in the central nervous system as a therapeutic target in neurodegenerative diseases. Drug Metab. Rev. 2018, 50, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Miksys, S.; Tyndale, R.F. Cytochrome P450-mediated drug metabolism in the brain. J. Psychiatry Neurosci. 2013, 38, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Mann, A.; Miksys, S.L.; Gaedigk, A.; Kish, S.J.; Mash, D.C.; Tyndale, R.F. The neuroprotective enzyme CYP2D6 increases in the brain with age and is lower in Parkinson’s disease patients. Neurobiol. Aging 2012, 33, 2160–2171. [Google Scholar] [CrossRef]
- Parkinson, A.; Mudra, D.R.; Johnson, C.; Dwyer, A.; Carroll, K.M. The effects of gender, age, ethnicity, and liver cirrhosis on cytochrome P450 enzyme activity in human liver microsomes and inducibility in cultured human hepatocytes. Toxicol. Appl. Pharmacol. 2004, 199, 193–209. [Google Scholar] [CrossRef] [PubMed]
- McMillan, D.M.; Tyndale, R.F. CYP-mediated drug metabolism in the brain impacts drug response. Pharmacol. Ther. 2018, 184, 189–200. [Google Scholar] [CrossRef]
- Banerjee, B.D.; Kumar, R.; Thamineni, K.L.; Shah, H.; Thakur, G.K.; Sharma, T. Effect of environmental exposure and pharmacogenomics on drug metabolism. Curr. Drug Metab. 2019, 20, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Sneha, S.; Baker, S.C.; Green, A.; Storr, S.; Aiyappa, R.; Martin, S.; Pors, K. Intratumoural cytochrome P450 expression in breast cancer: Impact on standard of care treatment and new efforts to develop tumour-selective therapies. Biomedicines 2021, 9, 290. [Google Scholar] [CrossRef]
- McFadyen, M.C.; McLeod, H.L.; Jackson, F.C.; Melvin, W.T.; Doehmer, J.; Murray, G.I. Cytochrome P450 CYP1B1 protein expression: A novel mechanism of anticancer drug resistance. Biochem. Pharmacol. 2001, 62, 207–212. [Google Scholar] [CrossRef]
- Raunio, H.; Juvonen, R.; Pasanen, M.; Pelkonen, O.; Pääkkö, P.; Soini, Y. Cytochrome P4502A6 (CYP2A6) expression in human hepatocellular carcinoma. Hepatology 1998, 27, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Yamakawa, K.; Saoo, K.; Hosokawa, K.; Yokohira, M.; Kuno, T.; Iwai, J.; Shirai, T.; Obika, K.; Kamataki, T.; et al. CYP2A6 overexpression in human lung cancers correlates with a high malignant status. Oncol. Rep. 2007, 18, 53–57. [Google Scholar] [CrossRef]
- Guo, Y.; Hu, B.; Xie, Y.; Billiar, T.R.; Sperry, J.L.; Huang, M.; Xie, W. Regulation of drug-metabolizing enzymes by local and systemic liver injuries. Expert Opin. Drug Metab. Toxicol. 2016, 12, 245–251. [Google Scholar] [CrossRef]
- Morgan, E.T.; Skubic, C.; Lee, C.M.; Cokan, K.B.; Rozman, D. Regulation of cytochrome P450 enzyme activity and expression by nitric oxide in the context of inflammatory disease. Drug Metab. Rev. 2020, 52, 455–471. [Google Scholar] [CrossRef] [PubMed]
- El-Boraie, A.; Tyndale, R.F. The role of pharmacogenetics in smoking. Clin. Pharmacol. Ther. 2021, 110, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Zarezadeh, M.; Saedisomeolia, A.; Shekarabi, M.; Khorshidi, M.; Emami, M.R.; Müller, D.J. The effect of obesity, macronutrients, fasting and nutritional status on drug-metabolizing cytochrome P450s: A systematic review of current evidence on human studies. Eur. J. Nutr. 2021, 60, 2905–2921. [Google Scholar] [CrossRef]
- Miksys, S.; Lerman, C.; Shields, P.G.; Mash, D.C.; Tyndale, R.F. Smoking, alcoholism and genetic polymorphisms alter CYP2B6 levels in human brain. Neuropharmacology 2003, 45, 122–132. [Google Scholar] [CrossRef]
- Sarparast, M.; Dattmore, D.; Alan, J.; Lee, K.S.S. Cytochrome P450 metabolism of polyunsaturated fatty acids and neurodegeneration. Nutrients 2020, 12, 3523. [Google Scholar] [CrossRef]
- Hayashi, T.; Harada, N. Post-translational dual regulation of cytochrome P450 aromatase at the catalytic and protein levels by phosphorylation/dephosphorylation. FEBS J. 2014, 281, 4830–4840. [Google Scholar] [CrossRef]
- Saito, M.; Okutomi, T.; Shimizu, M.; Matsumoto, Y.; Yamazaki, H.; Hoka, S. Activities of rat cytochrome P450 3A and 2C isoforms are increased in vivo by magnesium sulfate as evidenced by enhanced oxidation of bupivacaine and testosterone in liver microsomes. Drug Metab. Pharmacokinet. 2006, 21, 201–207. [Google Scholar] [CrossRef] [PubMed]
- van Heeswijk, R.P.; Cooper, C.L.; Foster, B.C.; Chauhan, B.M.; Shirazi, F.; Seguin, I.; Phillips, E.J.; Mills, E. Effect of high-dose vitamin C on hepatic cytochrome P450 3A4 activity. Pharmacotherapy 2005, 25, 1725–1728. [Google Scholar] [CrossRef]
- Chun, Y.J.; Shimada, T.; Waterman, M.R.; Guengerich, F.P. Understanding electron transport systems of Streptomyces cytochrome P450. Biochem. Soc. Trans. 2006, 34, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhou, S.Y.; Fabriaga, E.; Zhang, P.H.; Zhou, Q. Food-drug interactions precipitated by fruit juices other than grapefruit juice: An update review. J. Food Drug Anal. 2018, 26, S61–S71. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Waterman, M.R.; Egli, M. Recent structural insights into cytochrome P450 function. Trends Pharmacol. Sci. 2016, 37, 625–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.; Meng, Q. Prediction of cytochrome 450 mediated drug-drug interactions by three-dimensional cultured hepatocytes. Mini Rev. Med. Chem. 2012, 12, 1028–1036. [Google Scholar] [CrossRef]
- Kumar, S.; Sharma, R.; Roychowdhury, A. Modulation of cytochrome-P450 inhibition (CYP) in drug discovery: A medicinal chemistry perspective. Curr. Med. Chem. 2012, 19, 3605–3621. [Google Scholar] [CrossRef]
- Wienkers, L.C.; Heath, T.G. Predicting in vivo drug interactions from in vitro drug discovery data. Nat. Rev. Drug Discov. 2005, 4, 825–833. [Google Scholar] [CrossRef]
- Trieu, L.; Murray, M. Metabolite-intermediate complexation and inhibition of microsomal CYP3A in rat liver by diltiazem. Xenobiotica 2000, 30, 131–140. [Google Scholar] [CrossRef]
- Türk, D.; Hanke, N.; Wolf, S.; Frechen, S.; Eissing, T.; Wendl, T.; Schwab, M.; Lehr, T. Physiologically based pharmacokinetic models for prediction of complex CYP2C8 and (SLCO1B1) drug-drug-gene interactions: A modeling network of gemfibrozil, repaglinide, pioglitazone, rifampicin, clarithromycin and itraconazole. Clin. Pharmacokinet. 2019, 58, 1595–1607. [Google Scholar] [CrossRef] [Green Version]
- Verbeurgt, P.; Mamiya, T.; Oesterheld, J. How common are drug and gene interactions? Prevalence in a sample of 1143 patients with CYP2C9, CYP2C19 and CYP2D6 genotyping. Pharmacogenomics 2014, 15, 655–665. [Google Scholar] [CrossRef]
- Tompkins, L.M.; Wallace, A.D. Mechanisms of cytochrome P450 induction. J. Biochem. Mol. Toxicol. 2007, 21, 176–181. [Google Scholar] [CrossRef]
- Pelkonen, O.; Turpeinen, M.; Hakkola, J.; Honkakoski, P.; Hukkanen, J.; Raunio, H. Inhibition and induction of human cytochrome P450 enzymes: Current status. Arch. Toxicol. 2008, 82, 667–715. [Google Scholar] [CrossRef]
- Smutny, T.; Mani, S.; Pavek, P. Post-translational and post-transcriptional modifications of pregnane X receptor (PXR) in regulation of the cytochrome P450 superfamily. Curr. Drug Metab. 2013, 14, 1059–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elcombe, C.R.; Peffer, R.C.; Wolf, D.C.; Bailey, J.; Bars, R.; Bell, D.; Cattley, R.C.; Ferguson, S.S.; Geter, D.; Goetz, A.; et al. Mode of action and human relevance analysis for nuclear receptor-mediated liver toxicity: A case study with phenobarbital as a model constitutive androstane receptor (CAR) activator. Crit. Rev. Toxicol. 2014, 44, 64–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawajiri, K.; Fujii-Kuriyama, Y. Cytochrome P450 gene regulation and physiological functions mediated by the aryl hydrocarbon receptor. Arch. Biochem. Biophys. 2007, 464, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Meyer, U.A. Induction of drug metabolism: The role of nuclear receptors. Pharmacol. Rev. 2003, 55, 649–673. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, S.; Sobhany, M.; Moore, R.; Perera, L.; Pedersen, L.; Sueyoshi, T.; Negishi, M. Phenobarbital indirectly activates the constitutive active androstane receptor (CAR) by inhibition of epidermal growth factor receptor signaling. Sci. Signal. 2013, 6, ra31. [Google Scholar] [CrossRef] [Green Version]
- Kalra, B.S. Cytochrome P450 enzyme isoforms and their therapeutic implications: An update. Indian J. Med. Sci. 2007, 61, 102–116. [Google Scholar] [CrossRef]
- McGinnity, D.F.; Riley, R.J. Predicting drug pharmacokinetics in humans from in vitro metabolism studies. Biochem. Soc. Trans. 2001, 29, 135–139. [Google Scholar] [CrossRef]
- Pelley, J.W. 4—Enzymes and energetics. In Elsevier’s Integrated Review Biochemistry, 2nd ed.; Pelley, J.W., Ed.; W.B. Saunders: Philadelphia, PA, USA, 2012; pp. 29–37. [Google Scholar]
- Olkkola, K.T.; Aranko, K.; Luurila, H.; Hiller, A.; Saarnivaara, L.; Himberg, J.J.; Neuvonen, P.J. A potentially hazardous interaction between erythromycin and midazolam. Clin. Pharmacol. Ther. 1993, 53, 298–305. [Google Scholar] [CrossRef]
- Palmer, T.; Bonner, P.L. 8—Enzyme inhibition. In Enzymes, 2nd ed.; Palmer, T., Bonner, P.L., Eds.; Woodhead Publishing: Sawston, UK, 2011; pp. 126–152. [Google Scholar]
- Lin, J.H.; Lu, A.Y. Inhibition and induction of cytochrome P450 and the clinical implications. Clin. Pharmacokinet. 1998, 35, 361–390. [Google Scholar] [CrossRef]
- Aldred, E.M.; Buck, C.; Vall, K. Chapter 19—Pharmacodynamics: How drugs elicit a physiological effect. In Pharmacology; Aldred, E.M., Buck, C., Vall, K., Eds.; Churchill Livingstone: Edinburgh, UK, 2009; pp. 137–143. [Google Scholar]
- Zhou, S.; Yung Chan, S.; Cher Goh, B.; Chan, E.; Duan, W.; Huang, M.; McLeod, H.L. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin. Pharmacokinet. 2005, 44, 279–304. [Google Scholar] [CrossRef] [PubMed]
- Tannenbaum, C.; Sheehan, N.L. Understanding and preventing drug-drug and drug-gene interactions. Expert Rev. Clin. Pharmacol. 2014, 7, 533–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malki, M.A.; Pearson, E.R. Drug-drug-gene interactions and adverse drug reactions. Pharm. J. 2020, 20, 355–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storelli, F.; Samer, C.; Reny, J.L.; Desmeules, J.; Daali, Y. Complex drug-drug-gene-disease interactions involving cytochromes P450: Systematic review of published case reports and clinical perspectives. Clin. Pharmacokinet. 2018, 57, 1267–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakkola, J.; Hukkanen, J.; Turpeinen, M.; Pelkonen, O. Inhibition and induction of CYP enzymes in humans: An update. Arch. Toxicol. 2020, 94, 3671–3722. [Google Scholar] [CrossRef]
- Manikandan, P.; Nagini, S. Cytochrome P450 structure, function and clinical significance: A review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef]
- Samer, C.F.; Lorenzini, K.I.; Rollason, V.; Daali, Y.; Desmeules, J.A. Applications of CYP450 testing in the clinical setting. Mol. Diagn. Ther. 2013, 17, 165–184. [Google Scholar] [CrossRef] [Green Version]
- Brousseau, D.C.; McCarver, D.G.; Drendel, A.L.; Divakaran, K.; Panepinto, J.A. The effect of CYP2D6 polymorphisms on the response to pain treatment for pediatric sickle cell pain crisis. J. Pediatr. 2007, 150, 623–626. [Google Scholar] [CrossRef] [Green Version]
- Stamer, U.M.; Lehnen, K.; Höthker, F.; Bayerer, B.; Wolf, S.; Hoeft, A.; Stuber, F. Impact of CYP2D6 genotype on postoperative tramadol analgesia. Pain 2003, 105, 231–238. [Google Scholar] [CrossRef]
- Lima, J.J.; Thomas, C.D.; Barbarino, J.; Desta, Z.; Van Driest, S.L.; El Rouby, N.; Johnson, J.A.; Cavallari, L.H.; Shakhnovich, V.; Thacker, D.L.; et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for CYP2C19 and proton pump inhibitor dosing. Clin. Pharmacol. Ther. 2021, 109, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Karnes, J.H.; Rettie, A.E.; Somogyi, A.A.; Huddart, R.; Fohner, A.E.; Formea, C.M.; Ta Michael Lee, M.; Llerena, A.; Whirl-Carrillo, M.; Klein, T.E.; et al. Clinical Pharmacogenetics implementation consortium (CPIC) guideline for CYP2C9 and HLA-B genotypes and phenytoin dosing: 2020 update. Clin. Pharmacol. Ther. 2021, 109, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Caudle, K.E.; Sangkuhl, K.; Whirl-Carrillo, M.; Swen, J.J.; Haidar, C.E.; Klein, T.E.; Gammal, R.S.; Relling, M.V.; Scott, S.A.; Hertz, D.L.; et al. Standardizing CYP2D6 genotype to phenotype translation: Consensus recommendations from the clinical pharmacogenetics implementation consortium and dutch pharmacogenetics working group. Clin. Transl. Sci. 2020, 13, 116–124. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CYP Family | Primary Functions | Subfamilies | Genes |
|---|---|---|---|
| 1 | drug metabolism | 3 | 3 |
| 2 | drug/steroid metabolism | 13 | 16 |
| 3 | drug metabolism | 1 | 4 |
| 4 | arachidonic acid/ fatty acid metabolism | 5 | 12 |
| 5 | thromboxane synthase | 1 | 1 |
| 7 | steroid 7α-hydroxylase | 2 | 2 |
| 8 | bile acid biosynthesis; prostacyclin synthase | 2 | 2 |
| 11 | steroid biosynthesis | 2 | 3 |
| 17 | steroid 7α-hydroxylase | 1 | 1 |
| 19 | aromatase | 1 | 1 |
| 20 | function not determined | 1 | 1 |
| 21 | steroid biosynthesis | 1 | 1 |
| 24 | vitamin D deactivation | 1 | 1 |
| 26 | retinoic acid hydroxylase | 3 | 3 |
| 27 | bile acid biosynthesis; vitamin D3 activation | 3 | 3 |
| 39 | function not determined | 1 | 1 |
| 46 | cholesterol 24-hydroxylase | 1 | 1 |
| 51 | lanosterol 14α-demethylase | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. https://doi.org/10.3390/ijms222312808
Zhao M, Ma J, Li M, Zhang Y, Jiang B, Zhao X, Huai C, Shen L, Zhang N, He L, et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. International Journal of Molecular Sciences. 2021; 22(23):12808. https://doi.org/10.3390/ijms222312808
Chicago/Turabian StyleZhao, Mingzhe, Jingsong Ma, Mo Li, Yingtian Zhang, Bixuan Jiang, Xianglong Zhao, Cong Huai, Lu Shen, Na Zhang, Lin He, and et al. 2021. "Cytochrome P450 Enzymes and Drug Metabolism in Humans" International Journal of Molecular Sciences 22, no. 23: 12808. https://doi.org/10.3390/ijms222312808
APA StyleZhao, M., Ma, J., Li, M., Zhang, Y., Jiang, B., Zhao, X., Huai, C., Shen, L., Zhang, N., He, L., & Qin, S. (2021). Cytochrome P450 Enzymes and Drug Metabolism in Humans. International Journal of Molecular Sciences, 22(23), 12808. https://doi.org/10.3390/ijms222312808